
Abstract
Hepatocellular carcinoma (HCC) is the sixth most common cancer globally and the primary cause of death in cancer cases, with significant public health concern worldwide. Despite the overall decline in the incidence and mortality rates of HCC in recent years in recent years, the emergence of metabolic liver disease-related HCC is causing heightened concern, especially in countries like the United States, the United Kingdom, and P.R. China. The escalation of metabolic liver disease-related HCC is attributed to a combination of factors, including genetic predisposition, lifestyle choices, and changes in the living environment. However, the pathogenesis of metabolic liver disease-associated HCC remains imperfect. In this review, we encapsulate the latest advances and essential aspects of the pathogenesis of metabolic liver disease-associated HCC, including alcoholic liver disease (ALD), metabolic dysfunction–associated steatotic liver disease (MASLD), and inherited metabolic liver diseases.
Introduction
Hepatocellular carcinoma (HCC) stands out as the most prevalent and severe form of primary liver cancer globally. It ranks sixth in new cancer cases and third in cancer-related deaths among humans, solidifying its status as one of the deadliest cancers.Citation1 At present, the predominant etiological factors in clinical HCC cases encompass hepatitis B virus (HBV), hepatitis C virus (HCV), and metabolic diseases.Citation2 The historical dominance of HBV and HCV as the main causes of HCC is diminishing due to the widespread use of antiviral drugs, resulting in a declining trend in their associated HCC incidence.Citation3 However, the incidence of metabolic liver disease-associated HCC has shown an upward trend and is gradually supplanting HBV and HCV as the leading cause of HCC.
Metabolic liver disease encompasses a group of diseases causing liver damage due to abnormal metabolism in the body. The liver, being the body’s largest detoxification organ, converts toxic exogenous substances and endogenous metabolites into non-toxic or highly soluble substances, facilitating their excretion through bile or urine. Abnormal metabolism, induced by factors such as prolonged heavy drinking, high-fat diets, medications, and deficiencies or abnormalities in specific enzymes in the body, leads to a substantial accumulation of toxic substances in the liver, causing severe damage.Citation4 Recent years have witnessed a rapid increase in the incidence of metabolic liver disease, driven by changes in lifestyles, environments, and hereditary factors, particularly in countries like the United States, China, and Italy. This heightened incidence elevates the risk of HCC.Citation5 Clinical observations indicate that metabolic liver disease-related HCC has a relatively poorer prognosis and is more aggressive compared to virus-related HCC.Citation6 Therefore, there is an urgent need to deepen our understanding of the pathogenesis of metabolic liver disease-related HCC, to discover new strategies for its treatment.
Alcoholic Liver Disease
Alcoholic liver disease is a widespread chronic liver disease, defined as liver damage caused by prolonged excessive alcohol consumption, typically developing after five consecutive years of average daily alcohol intake of >40 g for men and >20 g for women.Citation7,Citation8 Alcoholic liver disease (ALD) disease progression is progressive. It initially presents as simple hepatic steatosis, known as alcoholic fatty liver (AFL). About 10–35% of AFL cases progress to alcoholic steatohepatitis (ASH), marked by hepatic inflammation, hepatocellular injury, and ballooning. While ASH progresses gradually, sustained heavy alcohol consumption accelerates its advancement, leading to irreversible fibrosis and cirrhosis. Ultimately, this progression may result in HCC.Citation9 Reports indicate that more than 90% of individuals with prolonged high alcohol consumption (>40 g per day) will develop AFL, establishing the groundwork for the high prevalence of AFL-HCC.Citation10
The pathogenesis of metabolic liver disease-related HCC is complex, primarily encompassing metabolism, inflammation, DNA mutation, immunosuppression, and other mechanisms.Citation11 The liver is the main organ involved in metabolizing alcohol intake and is sensitive to the impacts of alcohol consumption. Ethanol dehydrogenase metabolizes alcohol to acetaldehyde in hepatocytes, also inducing the production of cytochrome P450 2E1 (CYP2E1), an enzyme present in hepatocytes that also metabolizes alcohol to acetaldehyde.Citation12,Citation13 Acetaldehyde is highly toxic and carcinogenic, leading to structural and functional changes in proteins, resulting in the production of neoantigens. Moreover, it can form adducts with DNA and interstrand cross-links, potentially leading to DNA mutations when DNA repair is insufficient. Simultaneously, acetaldehyde inhibits the activity of the DNA repair enzyme O6-methylguanine DNA methyltransferase, resulting in inadequate DNA repair and an increased likelihood of DNA mutations.Citation14–16 In addition, CYP2E1 activates procarcinogens to produce carcinogens (eg, nitrosamines).Citation17 At the same time, reactive oxygen species (ROS), another substance produced during alcohol metabolism that accelerates liver damage, can be generated as a byproduct of CYP2E1 or during alcohol-induced inflammation.Citation18–20 ROS-induced production gives rise to the aldehyde metabolites 4-hydroxynonenal (4-HNE) or malondialdehyde (MDA), which can form highly reactive adducts with acetaldehyde, causing DNA mutations.Citation21,Citation22 Finally, activation of the typical WNT/β-catenin pathway promotes CYP2E1 transcription, strongly associating it with the development of alcohol-related HCC.Citation23–25
Alcohol prompts the progression of hepatic inflammation, a pivotal pathophysiological mechanism in HCC. The induction of inflammation can serve as the initiation of tumorigenesis.Citation9 Firstly, chronic heavy alcohol consumption disrupts the homeostasis of the gut microbiota by secreting large amounts of lipopolysaccharides (LPS), defined as pathogen-associated molecular patterns (PAMPs). The toxic effects of alcohol induce cellular stress and damage, leading to the release of factors such as adenosine and ATP, known as damage-associated molecular patterns (DAMPs).Citation26–29 Hepatocytes, Kuffer cells, and hepatic stellate cells (HSCs) in the liver are activated by two modes and secrete numerous cytokines and chemokines, triggering inflammation. The activated HSCs will also support the growth of tumor cells, promoting the development of HCC.Citation30 Simultaneously, Toll-like receptor 4 (TLR4) recognizes two patterns, releasing pro-inflammatory IL-8 and IL-1β.Citation8,Citation31 In summary, CYP2E1 and LPS are vital drivers of alcohol-related tumors, activating HSCs and promoting HCC. Moreover, alcohol directly induces and activates ectopic up-regulation of TLR4, and Nanog, a direct downstream gene of alcohol-induced TLR4, can mediate TLR4-dependent hepatocellular carcinogenesis via hepatic progenitor cells.Citation32–34
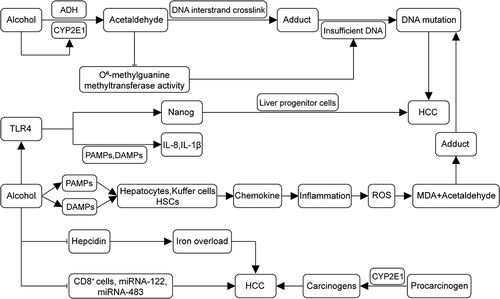
It has been reported that in ALD, liver injury due to alcoholic cirrhosis decreases the level of hepcidin in the liver, a negative regulator of iron metabolism. Consequently, decreased hepcidin levels lead to iron overload.Citation35,Citation36 Iron is a necessary element in the body, but it is also toxic. Significant accumulations of iron in the liver accelerate the development of HCC. Iron overload is an independent risk factor for the development of HCC.Citation37–40 Lastly, alcohol also suppresses the body’s immune system, resulting in a reduction in the number of CD8+ cells and inhibiting the transcriptional regulation of miRNA-122 and miRNA-483, thereby promoting the formation of HCC.Citation41,Citation42 In summary, chronic heavy alcohol consumption promotes the development of HCC by generating aldehyde carcinogens, ROS, PAMPs and DAMPs, inducing inflammation, and causing immunosuppression. shows the pathogenesis of alcohol-related HCC.
Figure 1 Pathogenesis of alcohol-related HCC. Alcohol undergoes metabolism in the liver to acetaldehyde, inducing the production of CYP2E1. Acetaldehyde, a carcinogenic substance, forms adducts with DNA interstrand cross-links. It diminishes the activity of O6-methylguanine DNA methyltransferase, resulting in insufficient DNA repair and, collectively, leading to DNA mutation. Alcohol disrupts gut microbial homeostasis, producing PAMPs and DAMPs. This process activates hepatocytes, Kupffer cells, and hepatic stellate cells (HSCs), leading to the secretion of chemokines, inflammation, and induced production of ROS. The ROS-produced malondialdehyde (MDA) forms adducts with acetaldehyde, resulting in DNA mutations. Alcohol induces ectopic upregulation of TLR4, which recognizes two patterns, releasing pro-inflammatory IL-8 and IL-1β. Moreover, the TLR4 downstream gene Nanog mediates TLR4-dependent hepatocellular carcinogenesis via hepatic progenitor cells. Alcohol also promotes the formation of HCC by down-regulating ferredoxin levels and inhibiting CD8+ cells and transcription of miRNA-122 and miRNA-483. (→, promote; ⊣, inhibit).
Metabolic Dysfunction–Associated Steatotic Liver Disease
It is worth noting that the recent multisociety Delphi consensus statement on a new nomenclature for fatty liver recommends renaming NAFLD (non-alcoholic fatty liver disease) to MASLD (metabolic dysfunction–associated steatotic liver disease).Citation43 MASLD is an acquired metabolic stress liver injury characterized by clinicopathological syndromes of excessive intracellular fat deposition in hepatocytes, resulting from factors unrelated to alcohol and other well-defined hepatic injury factors. It is closely linked to insulin resistance and genetic susceptibility.Citation44 MASLD encompasses a broad spectrum of diseases, including metabolic steatotic liver diseases (MSLD) and metabolic dysfunction–associated steatohepatitis (MASH). These conditions can progress to fibrosis and cirrhosis, increasing the risk of developing HCC.Citation45–47 With changes in people’s lifestyles and environments, obesity and its related metabolic syndromes exhibit a globalized epidemic trend, including insulin resistance, type 2 diabetes mellitus (T2DM), hypertension, and dyslipidemia. The prevalence of MASLD has been rapidly increasing in the last decade, with a current global prevalence rate of about 30%. This explains the rising incidence of HCC.Citation48,Citation49 Hepatitis B and C have been the primary causes of HCC over the years. With the widespread use of antiviral drugs, the incidence of hepatitis virus-related HCC has declined. However, the prevalence of MASLD-related HCC has been increasing, and the global annual incidence of HCC in patients with MASLD is about 0.44 per 1,000 person-years. MASLD has gradually become the primary cause of HCC.Citation50–52
The development of MASLD-related HCC is influenced by various factors, yet a comprehensive investigation is still pending. Key pathogenic mechanisms involve insulin resistance, lipotoxicity, oxidative stress, immune system activation, intestinal microbiota disturbances, autophagy dysregulation, and mitochondrial dysfunction.Citation53,Citation54 Simply put, insulin resistance results from reduced sensitivity of glucose uptake and utilization to the stimulating effects of insulin, necessitating increased insulin secretion to maintain stable blood glucose levels.Citation55,Citation56 Elevated plasma insulin levels lead to a cascade of metabolic disorders. Hyperinsulinemia, T2DM, and obesity are intimately connected to insulin resistance. The insulin signaling pathway is activated by the action of all three, constituting the primary mechanism for the progression of MASLD to HCC.Citation57,Citation58 The synthesis of hyperinsulinemic insulin-like growth factor-1 (IGF-1) increases in MASLD. IGF-1 binds to the insulin-like growth factor-1 receptor (IGF1R), activating insulin receptor substrate-1 (IRS-1). This, in turn, further activates mitogen-activated protein kinase (MAPK), leading to the upregulation of the transcription of proto-oncogenes (eg, c-fos and c-jun). Subsequently, the Wnt/β-catenin pathway is activated, contributing to the progression of MSLD toward hepatic fibrosis and tumor; Citation59–61 IRS-1 also activates the PI3K/AKT pathway, playing a pro-cancer role by promoting cell proliferation and preventing apoptosis.Citation62 Additionally, insulin resistance enhances the expression of the growth hormone receptor (GHR), coupled with growth hormone (GH), promoting the synthesis of IGF-1 and activating the pro-cancer pathway.Citation63 The liver is the primary target of GHR signaling. In cancer cells, GH binding to GHR triggers the activation of intracellular receptor domains such as JAK2, leading to the accelerated activation of oncogenic transcription factors STAT3, and other survival molecules including IRS-1, AKT, and ERK. There is growing evidence suggesting the significant involvement of GHR signaling in the pathogenesis of HCC.Citation64
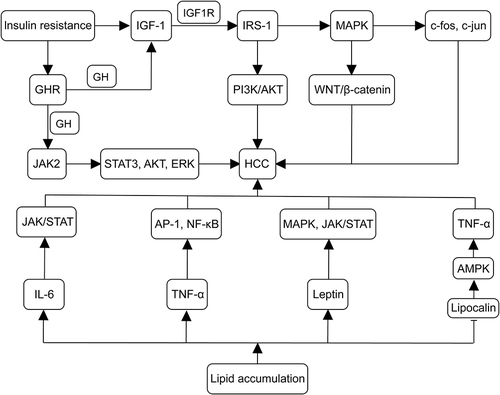
During the progression of MASLD to HCC, a typical pathological change is the accumulation of lipids in hepatocytes. This accumulation leads to a series of pathological changes, including increased mitochondrial oxidation inducing ROS production and chronic inflammation.Citation65,Citation66 In this process, an imbalance of inflammatory cytokines, including interleukin-6(IL-6), tumor necrosis factor-α(TNF-α), leptin, and lipocalin occurs. It has been reported that in MASH, the levels of IL-6, TNF-α, and leptin are elevated, while lipocalin levels are decreased.Citation60,Citation67 IL-6 can induce HCC cell proliferation and tumor formation by activating the JAK/STAT pathway; Citation67,Citation68 TNF-α induces IL-6, which also regulates cancer cell differentiation, metastasis, and angiogenesis.Citation69 TNF-α and IL-6, in turn, activate oncogenic transcription factors, including activator protein-1 (AP-1), nuclear factor-κB (NF-κB), and signal transducer and activator of transcription 3 (STAT3).Citation70,Citation71 Lipocalin inhibits TNF-α expression mainly by triggering AMPK signaling. It is evident that lipocalin levels are decreased in MASH, thereby increasing the risk of developing HCC.Citation72,Citation73 Leptin primarily interacts with PI3K, MAPK, and JAK2/STAT signaling pathways, promoting tumor cell proliferation, invasion, angiogenesis, and metastasis. This accelerates the progression of HCC.Citation74 Wang et alCitation75 discovered that LINC01468 expression was upregulated in patients with MASLD-associated HCC through transcriptomic analysis. They experimentally demonstrated that LINC01468 binds to SHIP2 and promotes cullin 4A (CUL4A)-conjugated ubiquitin degradation. This activation induces the PI3K/AKT/mTOR signaling pathway, promoting lipid synthesis and HCC progression. Furthermore, in MASLD, Kuffer cells in the liver are activated to induce hepatic inflammation and MASLD progression by upregulating TNF-α, IL-1β, IL-6, IL-12, IL-18, IL-10, and IFN-γ.Citation76 Finally, aberrant lipid metabolism leads CD4+ T cells to exert their oncogenic effects through mitochondrial production of more ROS, increased oxidative stress, and endoplasmic reticulum (ER) disruption.Citation77 illustrates the mechanisms of insulin resistance and lipid accumulation in the development of MASLD-associated HCC.
Figure 2 Mechanisms of insulin resistance and lipid accumulation in the development of MASLD-associated HCC. IGF-1 synthesis is enhanced in MASLD, with IGF-1 binding to IGF1R and activating IRS-1. IRS-1 further triggers MAPK, leading to the upregulation of proto-oncogenes (eg, c-fos and c-jun). MAPK activates the Wnt/β-catenin pathway, promoting tumor formation. IRS-1 also activates the PI3K/AKT pathway, contributing to tumorigenic effects. Insulin resistance upregulates GHR expression, binding to GH and promoting IGF-1 synthesis, activating the pro-cancer pathway. In cancer cells, GH binds to GHR to activate JAK2, accelerating the activation of TAT3, IRS-1, AKT, and ERK. Lipid accumulation increases IL-6, TNF-α, and leptin levels while decreasing lipocalin levels. IL-6 activates the JAK/STAT pathway, inducing HCC cell proliferation and tumor formation. TNF-α induced IL-6, regulating cancer cell differentiation, metastasis, and angiogenesis. TNF-α and IL-6 activate AP-1, NF-κ B, and STAT3 oncogenic transcription factors. Lipocalin activates AMPK signaling, inhibiting TNF-α expression, thereby increasing the risk of developing HCC. Leptin interacts with PI3K, MAPK, and JAK2/STAT signaling pathways, hastening HCC progression. (→, promote; ⊣, inhibit).
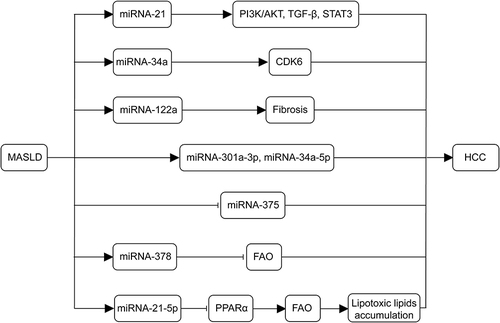
MicroRNAs (miRNAs) play an important role in the development of MASLD-related HCC and are expected to become new therapeutic targets for MASLD-related HCC treatment.Citation78 Firstly, miRNA-21 is one of the most commonly upregulated miRNAs in MASLD and HCC. Lai et alCitation79 found that miRNA-21 is mediated through PTS signaling (PI3K/AKT, TGF- β, and STAT3 Signaling) plays multiple roles in tumor metabolism and activation during the progression of MASLD to HCC. miRNA-122 constitutes 70% of the total miRNA in the liver, and miRNA-122a plays an important role as an intrinsic tumor suppressor gene in the liver. The absence of miRNA-122a increases the incidence of spontaneous HCC; conversely, the re-expression of miRNA-122a will reduces the incidence of liver tumors.Citation80 miRNA-34a is upregulated in a high-fat environment of the liver and induces liver cell aging by targeting CDK6. The miRNA-34a CDK6 signaling axis may promote the development of MASLD in a high-fat environment.Citation81 During the occurrence and development of MASLD, miRNA-301a-3p and miRNA-34a-5p are overexpressed, while the expression level of miRNA-375 decreases.Citation82 This abundance change also exists in HCC species, indicating that these three miRNAs are related to the progression of MASLD-HCC. In addition, Zhang et alCitation83 observed a significant increase in miRNA-378 expression in the fatty liver of high-fat-fed mice and human liver cancer HepG2 cells with lipid accumulation. Liver specific expression of miRNA-378 impaired fatty acid oxidation (FAO) and promoted the development of hepatic steatosis. miRNA-21-5p also drives the progression of MASLD to HCC and regulates peroxisome proliferator-activated receptor alpha (PPARα). Its expression promotes the accumulation of lipophilic lipids, inducing cell cycle activation, proliferation, and tumorigenesis.Citation84 Finally, the absence of miRNA-223, which has anti-inflammatory effects, can induce the progression of MASLD.Citation85 In summary, miRNA is a promising research direction that will play a crucial role in the diagnosis, staging, and treatment of the progression of MASLD to HCC in the future. illustrates the mechanisms of miRNAs in the development of MASLD-associated HCC.
Figure 3 Mechanisms of miRNAs in the development of MASLD-associated HCC. miRNA-21 plays a role in tumor metabolism and activation during the progression of MASLD to HCC through PI3K/AKT, TGF-β and STAT3 signaling. The miR-34a-CDK6 signaling axis promotes MASLD development in high-fat environments. Deletion of the miRNA-122a increases the incidence of spontaneous HCC. The abundance of miRNA-301a-3p, miRNA-34a-5p, and miRNA-375 correlates with the progression of MASLD to HCC. Liver-specific expression of miR-378 inhibits FAO and promotes hepatic steatosis. miR-21-5p regulates PPARα expression, which decreases the oxidation of fatty acids, promoting lipotoxic lipid accumulation, and induces cell cycle activation, proliferation, and tumorigenesis. (→, promote; ⊣, inhibit).
Changes in gut microbiota abundance are also one of the mechanisms underlying the transition from MASLD to HCC, accelerated by dysbiosis, altered mucosal permeability, and bacterial overgrowth in the gut.Citation86 Dysbiosis of the intestinal microbiota disrupts the intestinal mucosal barrier, leading to endogenous ethanol, the release of inflammatory factors, microbial translocation, and immune response activation.Citation87 These changes also result in increased production of the bacterial metabolite lipopolysaccharide (LPS) in the gut, activating the immune system via TLR4, inducing pro-inflammatory factors and oxidative stress, aggravating liver inflammation, and contributing to HCC.Citation88 Elevated LPS levels have been observed in MASLD.Citation89
Autophagy plays a crucial role in MASLD and its progression to HCC, serving as a protective mechanism regulating lipid metabolism, insulin resistance, and mitochondrial health, and protecting hepatocytes. However, impaired autophagy has been found in all pathological changes during MASLD progression to HCC, primarily due to lipid accumulation, hyperinsulinemia, and aberrant secretion of cytokines in the liver. Impaired autophagy, in turn, contributes to abnormal hepatic manifestations, creating a detrimental cycle.Citation71 Autophagy deficiency lead to the accumulation of misfolded proteins, oxidative stress, innate immune activation, and genetic instability, promoting tumorigenesis.Citation90 It was found that upregulation of leucine aminopeptidase 3 (LAP3),Citation91 thyroid hormone receptor-associated protein 3 (Thrap3), tRNA-derived fragment 3001b (tRF-3001b), and autophagy-inhibiting protein RubiconCitation92 in MASLD inhibits autophagy. Thrap3 blocks AMPK activation, inhibiting autophagy and mitochondrial function,Citation93 while tRF-3001b inhibits autophagy primarily by acting on Prkaa1.Citation94 Systemic deletion of Beclin-1, Atg5 and liver-specific deletion of Atg7 in mice result in spontaneous development of liver tumors in animal experiments.Citation95 TGF-β-activated kinase 1 (TAK1) deficiency leads to chronic inflammation, fibrosis, and carcinogenesis in the liver by blocking the AMPK/mTORC1 pathway and autophagy.Citation96 Notably, autophagy has a dual role in MASLD development, playing a protective role against cancer cells and promoting tumor development in the late stages of HCC.Citation97
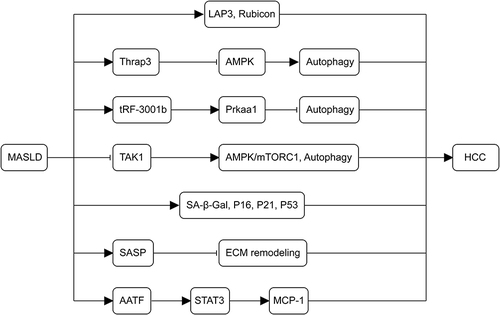
Recent studies emphasize the significant impact of aging on the pathogenesis of MASLD-related HCC, primarily observed in middle-aged and elderly populations. Organ dysfunction due to cellular senescence significantly influences liver disease development with advancing age.Citation98 Hepatocyte senescence is markedly accelerated during the transition from MASLD to HCC, particularly in HCC hepatocytes. Senescence-related markers, such as SA-β-Gal, p16, p21, and p53, are elevated in MASLD, and their levels continue to rise with disease progression, suggesting a close association between senescence and MASLD-HCC.Citation99 Notably, senescent cells exhibit a senescence-associated secretory phenotype (SASP), characterized by the secretion of inflammatory cytokines and chemokines. SASP exacerbates cellular senescence through autocrine and paracrine mechanisms, affecting the hepatocellular microenvironment and causing tissue dysfunction.Citation100,Citation101 SASP contributes to extracellular matrix (ECM) regulation, hindering ECM remodeling. Exosomes, a significant component of the ECM, induce cellular senescence and contribute to tumor formation.Citation102 IL-6, a SASP factor, induces acute-phase protein production, driving senescence and triggering inflammatory signaling, leading to sustained activation of related signaling pathways and ultimately resulting in HCC.Citation103,Citation104 Additionally, nicotinamide adenine dinucleotide (NAD+), silencing information regulator proteins (sirtuins) family, and mechanistic target of rapamycin (mTOR) are crucial targets in the aging process and MASLD development into HCC. They are induced through mechanisms such as DNA damage, cell cycle regulation, gene expression modulation, and oncogenic signaling pathway activation.Citation98 shows the pathogenesis of autophagy and senescence in the development of MASLD-associated HCC.
Figure 4 Mechanisms of autophagy and senescence in the development of MASLD-associated HCC. The upregulation of LAP3, Thrap3, tRF-3001b, and the autophagy-inhibitory protein Rubicon in MASLD inhibits autophagy. The deletion of TAK1 leads to chronic inflammation, fibrosis, and carcinogenesis in the liver by blocking the AMPK/mTORC1 pathway and autophagy. As MASLD advances, senescence-related markers (SA-β-Gal, p16, p21, and p53) persistently increase, contributing to the pathogenesis of MASLD-HCC. SASP worsens cellular senescence through autocrine and paracrine secretion, impedes ECM remodeling, and promotes tumor formation. AATF, overexpressed in MASLD, MASH, and MASLD-HCC via STAT3, enhances MCP-1 expression, promoting HCC.
Finally, apoptosis-antagonizing transcription factor (AATF) levels are upregulated in MASLD, MASH, and MASLD-HCC. SREBP1, an activator of AATF, can be activated by hyperinsulinemia. AATF enhances the expression of MCP-1 via STAT3, contributing to the promotion of HCC.Citation105 The pathogenesis of MASLD-related HCC is highly intricate and has not been comprehensively explored. To summarize, the pathological process of MASLD-HCC involves insulin resistance, lipotoxicity, oxidative stress, immune system activation, gut microbiota disruption, autophagy dysregulation, mitochondrial dysfunction, and cellular senescence.
Inherited Metabolic Liver Disease
Inherited metabolic liver diseases primarily encompass hereditary hemochromatosis (HH), Alpha 1 antitrypsin (AAT) deficiency, and hereditary tyrosinemia type 1(HT1). These genetic disorders give rise to complications, including emphysema, chronic liver disease, and HCC. Here, we focus on the pathogenesis of inherited metabolic liver diseases-related HCC, but relevant studies still need to be completed.
Hereditary Hemochromatosis
HH is an inherited metabolic liver disease common in whites that manifests as increased intestinal iron absorption, resulting in progressive iron accumulation in organs like the liver, heart, and pancreas. This condition is accompanied by reduced secretion of the iron-regulating hormone ferromodulin.Citation106,Citation107 The most common genetic variant is linked to the HFE (high-frequency iron) gene on chromosome 6, primarily the C282Y mutation, identified as a molecular risk factor for HCC in its pure state.Citation108–110 HH gives rise to complications including cirrhosis, HCC, congestive heart failure, diabetes mellitus, and arthropathy, with HCC being a long-term complication of HH that leads to increased mortality.Citation106 An earlier cohort study revealed a 20-fold higher risk of liver cancer development in individuals with hereditary hemochromatosis compared to the general population.Citation108 There are fewer studies on the pathogenesis of HH progressing to HCC. Iron overload, recognized as a crucial factor. The liver as the main iron metabolizing organ, the large accumulation of iron in the liver leads to ROS production and lipid peroxidation, causing DNA damage and mutations in the oncogene p53.Citation111,Citation112 Another study linking iron overload with HCC-specific epigenetic defects reported increased and more extensive aberrant hypermethylation in HH patients compared to non-HH individuals, not associated with cirrhosis.Citation113 Furthermore, hypermethylation of SOCS-1 in affected genes correlated with heightened activity of the JAK/STAT pathway in tumor cells.Citation114 Conversely, inactivation of pro-apoptotic genes, RASSF1A and GSP π1, may induce hepatocyte over-proliferation under iron-overload-induced genotoxic stress, increasing the likelihood of mutations leading to HCC.Citation115,Citation116 Capua et alCitation117 observed that patients with HFE-HCC exhibited a more aggressive disease course and increased co-expression of cancer stem cell markers EpCAM (epithelial cell adhesion molecule) and EpCAM/SALL4 (salt-like transcription factor 4). In conclusion, iron overload, a characteristic pathologic feature of HH, can be an independent carcinogenic factor in HCC.
Alpha 1 Antitrypsin Deficiency
AAT deficiency, one of the most prevalent inherited liver diseases, arises from mutations in the SERPINA1 gene, causing misfolding of the ATZ protein and subsequent polymerization within the endoplasmic reticulum of hepatocytes, initiating liver injury.Citation6,Citation118 The most common defective type is the Protease Inhibitor (Pi) type Z, and evidence suggests that individuals heterozygous for PiZ are at an elevated risk of developing chronic hepatitis, cirrhosis, and HCC in late childhood.Citation119 Reports indicate that HCC occurs in 31–67% of cirrhotic AAT-deficient adults.Citation120 The accumulation of ATZ in the endoplasmic reticulum induces mitochondrial dysfunction, and significant mitochondrial autophagy and damage were observed in the livers of AAT-deficient PiZ mice, accompanied by the presence of markedly activated caspase-3, potentially linked to HCC development.Citation121,Citation122 Beyond mitochondrial dysfunction and autophagy, the aggregation of ATZ activates endoplasmic reticulum stress signaling pathways, particularly NF-κB activation, ER caspases, and BAP31. NF-κB activation is particularly significant for liver injury, as ATZ accumulation can mediate hepatic inflammation via NF-κB, including neutrophil infiltration and NF-κB-targeted interleukin-8. NF-κB activation is closely associated with inflammation-induced carcinogenesis and may play a role in the pathogenesis of AAT deficiency-associated HCC.Citation123 BAP31, involved in protein polymerization in the endoplasmic reticulum, may contribute to mitochondrial dysfunction and the activation of mitochondrial caspase.Citation122 In addition, the aggregation of ATZ results in the absence of UPR signaling, potentially allowing the survival of abnormal cells and contributing to the pathogenesis of HCC.Citation124 Finally, Perlmutter et al found that ATZ accumulation in PiZ mice leads to the proliferation of globule-devoid hepatocytes, heightening the likelihood of HCC development, as adenomas and subsequent carcinomas arise in glomerular livers.Citation125
Hereditary Tyrosinemia Type 1
HT1 is a rare and severe autosomal recessive hereditary liver disease primarily resulting from the accumulation of toxic metabolites due to the deficiency of fumarylacetoacetate hydrolase (FAH), a key enzyme in tyrosine catabolism, and is strongly associated with an elevated risk of cancer (HCC). Angileri et alCitation126 utilizing the fah−/− mouse model observed elevated expression of miRNA-98 and miRNA-200b in HT1 mice, and these miRNAs were demonstrated to be associated with the progression of HT1. An investigation targeting the AKT signaling pathway found that the accumulation of the HT1 metabolite fumaric acid ethyl ester (FAA) in fah−/− mice induced chronic stress in the liver, stabilizing the activation of the AKT survival signaling pathway and inhibiting intrinsic apoptosis, thereby facilitating the development of HCC.Citation127 Moreover, Willenbring et alCitation128 reported severe hepatocellular DNA damage and additional deletions of p21 in FAH-deficient mice, where the proliferation of hepatocytes with DNA damage leads to rapid cancer formation. The deletion of p21 expression is indicative of the transition from hepatocellular dysplasia to HCC.Citation129 Lastly, heat shock proteins (HSPs) also play a pivotal role in the transition from HT1 to HCC. They are overexpressed in FAH-deficient mice, and in conjunction with BCL-2, HSPs promote resistance to apoptosis in tumorigenesis, thereby accelerating HCC.Citation130
Discussion
In recent years, the widespread use of antiviral drugs has led to a gradual decrease in the prevalence of virus-associated HCC, while metabolic liver diseases have emerged as a major etiological factor contributing to the rising incidence of HCC.Citation131 Given the swiftly rising incidence and substantial global mortality associated with metabolic liver disease-related HCC, concerted efforts are needed for prevention, delaying progression, and optimizing treatment strategies. Presently, research on the pathogenesis of metabolic liver disease-related HCC is inconclusive. Ultimately, attaining a comprehensive understanding of the pathogenesis of metabolic liver disease-associated HCC is essential for developing effective prevention strategies, early detection methods, and targeted therapies to mitigate the impact of this lethal cancer. Therefore, directing our attention to the pathogenesis of metabolic liver disease-related HCC is crucial for developing more effective clinical treatment strategies.
Abbreviations
HCC, hepatocellular carcinoma; ALD, alcoholic liver disease; MASLD, metabolic dysfunction–associated steatotic liver disease; HBV, hepatitis B virus; HCV, hepatitis C virus; AFL, alcoholic fatty liver; ASH, alcoholic steatohepatitis; CYP2E1, cytochrome P450 2E1; ROS, reactive oxygen species; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; LPS, lipopolysaccharides; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; TLR4, Toll-like receptor 4; MSLD, metabolic steatotic liver disease; MASH, metabolic dysfunction–associated steatohepatitis; T2DM, type 2 diabetes mellitus; IGF-1, insulin-like growth factor-1; IGF1R, insulin-like growth factor-1 receptor; IRS-1, insulin receptor substrate-1; GHR, growth hormone receptor; GH, growth hormone; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; AP-1, activator protein-1; NF-κB, nuclear factor-κB; STAT3, signal transducer and activator of transcription 3; ER, endoplasmic reticulum; FAO, fatty acid oxidation; PPARα, peroxisome proliferator-activated receptor alpha; LPS, lipopolysaccharide; LAP3, leucine aminopeptidase 3; Thrap3, thyroid hormone receptor-associated protein 3; TRF-3001b, tRNA-derived fragment 3001b; TAK1, TGF-β-activated kinase 1; ECM, extracellular matrix; NAD+, nicotinamide adenine dinucleotide; mTOR, mechanistic target of rapamycin; AATF, apoptosis-antagonizing transcription factor; HH, hereditary hemochromatosis; AAT, Alpha 1 antitrypsin; HT1, hereditary tyrosinemia type 1; EpCAM, epithelial cell adhesion molecule; Pi, Protease inhibitor; FAH, fenugreek acyl acetate hydrolase; HSPs, heat shock proteins.
Author Contributions
All authors contributed significantly to the work reported, participated in drafting or writing, or substantially revising or critically reviewing the article; have agreed on the journal to which the article will be submitted; reviewed and agreed on all versions of the article before submission, during revision, the final version accepted for publication, and any significant changes introduced at the proofing stage; agree to take responsibility and be accountable for the contents of the article.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
We acknowledge Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital for its administrative and technical support.
Additional information
Funding
References
- Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–1362. doi:10.1016/S0140-6736(22)01200-4
- Ito T, Nguyen MH. Perspectives on the underlying etiology of HCC and its effects on treatment outcomes. J Hepatocell Carcinoma. 2023;10:413–428. doi:10.2147/JHC.S347959
- Shin HS, Jun BG, Yi SW. Impact of diabetes, obesity, and dyslipidemia on the risk of hepatocellular carcinoma in patients with chronic liver diseases. Clin Mol Hepatol. 2022;28(4):773–789. doi:10.3350/cmh.2021.0383
- Barchetta I, Cimini FA, Cavallo MG. Vitamin D and metabolic dysfunction-associated fatty liver disease (MAFLD): an update. Nutrients. 2020;12(11):3302. doi:10.3390/nu12113302
- Shiha G, Alswat K, Al Khatry M, et al. Nomenclature and definition of metabolic-associated fatty liver disease: a consensus from the middle east and north Africa. Lancet Gastroenterol Hepatol. 2021;6(1):57–64. doi:10.1016/S2468-1253(20)30213-2
- Alkhouri N, Gawrieh S. A perspective on RNA interference-based therapeutics for metabolic liver diseases. Expert Opin Investig Drugs. 2021;30(3):237–244. doi:10.1080/13543784.2021.1879792
- Neuman MG, Seitz HK, French SW, et al. Alcoholic-hepatitis, links to brain and microbiome: mechanisms, clinical and experimental research. Biomedicines. 2020;8(3):63. doi:10.3390/biomedicines8030063
- Dukić M, Radonjić T, Jovanović I, et al. Alcohol, inflammation, and microbiota in alcoholic liver disease. Int J Mol Sci. 2023;24(4):3735. doi:10.3390/ijms24043735
- Seitz HK, Bataller R, Cortez-Pinto H, et al. Alcoholic liver disease. Nat Rev Dis Primers. 2018;4(1):16. doi:10.1038/s41572-018-0014-7
- Seitz HK, Mueller S. Alcoholic Liver Disease. Clin Hepatol. 2010;2:1111–1151.
- Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7(8):599–612. doi:10.1038/nrc2191
- Lieber CS, Rubin E, DeCarli LM. Hepatic microsomal ethanol oxidizing system (MEOS): differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem Biophys Res Commun. 1970;40(4):858–865. doi:10.1016/0006-291X(70)90982-4
- Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148(1):30–36. doi:10.1053/j.gastro.2014.10.042
- Theruvathu JA. Polyamines stimulate the formation of mutagenic 1, N2-propanodeoxyguanosine adducts from acetaldehyde. Nucleic Acids Res. 2005;33(11):3513–3520. doi:10.1093/nar/gki661
- Mizumoto A, Ohashi S, Hirohashi K, Amanuma Y, Matsuda T, Muto M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int J Mol Sci. 2017;18(9):1943. doi:10.3390/ijms18091943
- Song B-J, Abdelmegeed MA, Cho Y-E, et al. Contributing roles of CYP2E1 and other cytochrome p450 isoforms in alcohol-related tissue injury and carcinogenesis. Adv Exp Med Biol. 2019;1164:73–87.
- Gao J, Wang GJ, Wang Z, et al. High CYP2E1 activity correlates with hepatofibrogenesis induced by nitrosamines. Oncotarget. 2017;8(68):112199–112210. doi:10.18632/oncotarget.22937
- Diesinger T, Buko V, Lautwein A, et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS One. 2020;15(7):e0235990. doi:10.1371/journal.pone.0235990
- Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol. 2013;58(2):395–398. doi:10.1016/j.jhep.2012.08.018
- Mallat A, Lotersztajn S. Glutamate signaling in alcohol-associated fatty liver: ”pas de deux”. Hepatology. 2020;72(1):350–352. doi:10.1002/hep.31194
- Tuma DJ, Thiele GM, Xu D, Klassen LW, Sorrell MF. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology. 1996;23(4):872–880. doi:10.1002/hep.510230431
- Chandrasekaran K, Swaminathan K, Mathan Kumar S, Clemens DL, Dey A. In vitro evidence for chronic alcohol and high glucose mediated increased oxidative stress and hepatotoxicity. Alcohol Clin Exp Res. 2012;36(6):1004–1012. doi:10.1111/j.1530-0277.2011.01697.x
- Zhu L, Yang X, Feng J, et al. CYP2E1 plays a suppressive role in hepatocellular carcinoma by regulating Wnt/Dvl2/β-catenin signaling. J Transl Med. 2022;20(1):194. doi:10.1186/s12967-022-03396-6
- Mercer KE, Hennings L, Sharma N, et al. Alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis in male mice through activation of the Wnt/beta-catenin signaling pathway. Cancer Prev Res. 2014;7(7):675–685. doi:10.1158/1940-6207.CAPR-13-0444-T
- Groll N, Petrikat T, Vetter S, et al. Coordinate regulation of Cyp2e1 by beta-catenin- and hepatocyte nuclear factor 1alpha-dependent signaling. Toxicology. 2016;350–352:40–48. doi:10.1016/j.tox.2016.05.004
- Pandya UM, Egbuta C, Abdullah norman TM, et al. The biophysical interaction of the danger-associated molecular pattern (DAMP) calreticulin with the pattern-associated molecular pattern (pamp) lipopolysaccharide. Int J Mol Sci. 2019;20(2):408. doi:10.3390/ijms20020408
- Gauthier AE, Rotjan RD, Kagan JC. Lipopolysaccharide detection by the innate immune system may be an uncommon defence strategy used in nature. Open Biology. 2022;12(10). doi:10.1098/rsob.220146
- Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130(6):1886–1900. doi:10.1053/j.gastro.2006.01.038
- Wang S, Pacher P, De Lisle RC, Huang H, Ding WX. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol Clin Exp Res. 2016;40(6):1215–1223. doi:10.1111/acer.13078
- Pone EJ. Analysis by flow cytometry of b-cell activation and antibody responses induced by toll-like receptors. Methods Mol Biol. 2016;1390:229–248.
- Amir M, Czaja MJ. Inflammasome-mediated inflammation and fibrosis: it is more than just the IL-1beta. Hepatology. 2018;67(2):479–481. doi:10.1002/hep.29491
- Uthaya Kumar DB, Chen CL, Liu JC, et al. TLR4 signaling via nanog cooperates with STAT3 to activate twist1 and promote formation of tumor-initiating stem-like cells in livers of mice. Gastroenterology. 2016;150(3):707–719. doi:10.1053/j.gastro.2015.11.002
- Yeh DW, Liu C, Hernandez JC, Tahara SM, Tsukamoto H, Machida K. Polycomb repressive complex 2 binds and stabilizes NANOG to suppress differentiation-related genes to promote self-renewal. iScience. 2023;26(7):107035. doi:10.1016/j.isci.2023.107035
- Machida K, Tsukamoto H, Mkrtchyan H, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA. 2009;106(5):1548–1553. doi:10.1073/pnas.0807390106
- Lunova M, Trautwein C, Strnad P, Nahon P. Reply to: “Hepatic hepcidin expression is decreased in cirrhosis and HCC. J Hepatol. 2015;62(4):979–980. doi:10.1016/j.jhep.2014.11.008
- Kohgo Y, Ohtake T, Ikuta K, Suzuki Y, Torimoto Y, Kato J. Dysregulation of systemic iron metabolism in alcoholic liver diseases. J Gastroenterol Hepatol. 2008;23:(Suppl 1):S78–81.
- Nahon P, Nuraldeen R, Rufat P, Sutton A, Trautwein C, Strnad P. In alcoholic cirrhosis, low-serum hepcidin levels associate with poor long-term survival. Liver Int. 2016;36(2):185–188. doi:10.1111/liv.13007
- Ohtake T, Saito H, Hosoki Y, et al. Hepcidin is down-regulated in alcohol loading. Alcohol Clin Exp Res. 2007;31(1 Suppl):S2–8. doi:10.1111/j.1530-0277.2006.00279.x
- Nahon P, Sutton A, Rufat P, et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology. 2008;134(1):102–110. doi:10.1053/j.gastro.2007.10.038
- Yan G, Wang X, Sun C, et al. Chronic alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis via immune disturbances. Sci Rep. 2017;7(1):2567. doi:10.1038/s41598-017-02887-7
- Ambade A, Satishchandran A, Szabo G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1alpha activation. Sci Rep. 2016;6(1):21340. doi:10.1038/srep21340
- Niture S, Gadi S, Qi Q, et al. MicroRNA-483-5p inhibits hepatocellular carcinoma cell proliferation, cell steatosis, and fibrosis by targeting PPARα and TIMP2. Cancers. 2023;15(6):1715. doi:10.3390/cancers15061715
- Rinella ME, Lazarus JV, Ratziu V, et al. A multisociety delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79(6):1542–1556. doi:10.1016/j.jhep.2023.06.003
- Jeong S, Shin WY, Oh YH. Immunotherapy for NAFLD and NAFLD-related hepatocellular carcinoma. Front Endocrinol. 2023;14:1150360. doi:10.3389/fendo.2023.1150360
- Mahmoudi A, Jamialahmadi T, Johnston TP, Sahebkar A. Impact of fenofibrate on NAFLD/NASH: a genetic perspective. Drug Discovery Today. 2022;27(8):2363–2372. doi:10.1016/j.drudis.2022.05.007
- Vachher M, Bansal S, Kumar B, Yadav S, Burman A. Deciphering the role of aberrant DNA methylation in NAFLD and NASH. Heliyon. 2022;8(10):e11119. doi:10.1016/j.heliyon.2022.e11119
- Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):643–654 e641–649; quiz e639–640. doi:10.1016/j.cgh.2014.04.014
- Golabi P, Rhea L, Henry L, Younossi ZM. Hepatocellular carcinoma and non-alcoholic fatty liver disease. Hepatol Internat. 2019;13(6):688–694. doi:10.1007/s12072-019-09995-8
- Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. 2023;77(4):1335–1347. doi:10.1097/HEP.0000000000000004
- Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75(6):1476–1484. doi:10.1016/j.jhep.2021.08.012
- Tovoli F, Ferri S, Piscaglia F. Hepatocellular carcinoma in non alcoholic fatty liver disease. Curr Pharm Des. 2020;26(32):3909–3914. doi:10.2174/1381612826666200429093648
- Fujii H, Kawada N; Japan Study Group Of Nafld J-N. The role of insulin resistance and diabetes in nonalcoholic fatty liver disease. Int J Mol Sci. 2020;21(11):3863. doi:10.3390/ijms21113863
- Gao Y, Zhu R, Dong J, Li Z. Pathogenesis of NAFLD-related hepatocellular carcinoma: an up-to-date review. J Hepatocell Carcinoma. 2023;10:347–356. doi:10.2147/JHC.S400231
- Kim H, Lee DS, An TH, et al. Metabolic spectrum of liver failure in type 2 diabetes and obesity: from NAFLD to NASH to HCC. Int J Mol Sci. 2021;22(9):1.
- Unluhizarci K, Karaca Z, Kelestimur F. Role of insulin and insulin resistance in androgen excess disorders. World J Diabetes. 2021;12(5):616–629. doi:10.4239/wjd.v12.i5.616
- Lebovitz HE. Insulin resistance: definition and consequences. German Soc End. 2001;109(Suppl 2):S135–148. doi:10.1055/s-2001-18576
- Petta S, Amato MC, Di Marco V, et al. Visceral adiposity index is associated with significant fibrosis in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35(2):238–247. doi:10.1111/j.1365-2036.2011.04929.x
- Ramai D, Facciorusso A, Vigandt E, et al. Progressive liver fibrosis in non-alcoholic fatty liver disease. Cells. 2021;10(12):3401. doi:10.3390/cells10123401
- Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semi Cancer Biol. 2013;23(6 Pt B):483–491. doi:10.1016/j.semcancer.2013.07.003
- Bae SDW, George J, Qiao L. From MAFLD to hepatocellular carcinoma and everything in between. Chin Med J. 2022;135(5):547–556. doi:10.1097/CM9.0000000000002089
- Page JM, Harrison SA. NASH and HCC. Clin Liver Dis. 2009;13(4):631–647. doi:10.1016/j.cld.2009.07.007
- Kubota T, Kubota N, Kadowaki T. Imbalanced insulin actions in obesity and type 2 diabetes: key mouse models of insulin signaling pathway. Cell Metab. 2017;25(4):797–810. doi:10.1016/j.cmet.2017.03.004
- Arturi F, Succurro E, Procopio C, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab. 2011;96(10):E1640–E1644. doi:10.1210/jc.2011-1227
- Kaseb AO, Haque A, Vishwamitra D, et al. Blockade of growth hormone receptor signaling by using pegvisomant: a functional therapeutic strategy in hepatocellular carcinoma. Front Oncol. 2022;12:1.
- Santos-Baez LS, Ginsberg HN. Nonalcohol fatty liver disease: balancing supply and utilization of triglycerides. Current Opinion in Lipidology. 2021;32(3):200–206. doi:10.1097/MOL.0000000000000756
- Chrysavgis L, Giannakodimos I, Diamantopoulou P, Cholongitas E. Non-alcoholic fatty liver disease and hepatocellular carcinoma: clinical challenges of an intriguing link. World J Gastroenterol. 2022;28(3):310–331. doi:10.3748/wjg.v28.i3.310
- Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi:10.1016/j.cell.2009.12.052
- Lokau J, Schoeder V, Haybaeck J, Garbers C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers. 2019;11(11):1704. doi:10.3390/cancers11111704
- Chen K, Ma J, Jia X, Ai W, Ma Z, Pan Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: from experimental models to humans. Biochim Biophys Acta Rev Cancer. 2019;1871(1):117–125. doi:10.1016/j.bbcan.2018.11.005
- He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21(1):159–168. doi:10.1038/cr.2010.183
- Wkk W, Zhang L, Chan MTV. Autophagy, NAFLD and NAFLD-Related HCC. Adv Exp Med Biol. 2018;1061:127–138.
- Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nature Med. 2002;8(7):731–737. doi:10.1038/nm724
- Handa P, Maliken BD, Nelson JE, et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology. 2014;60(1):133–145. doi:10.1002/hep.26946
- Jiang N, Sun R, Sun Q. Leptin signaling molecular actions and drug target in hepatocellular carcinoma. Drug Des Devel Ther. 2014;8:2295–2302. doi:10.2147/DDDT.S69004
- Wang H, Wang Y, Lai S, et al. LINC01468 drives NAFLD-HCC progression through CUL4A-linked degradation of SHIP2. Cell Death Discov. 2022;8(1):449. doi:10.1038/s41420-022-01234-8
- Xu FL, You HB, Li XH, Chen XF, Liu ZJ, Gong JP. Glycine attenuates endotoxin-induced liver injury by downregulating TLR4 signaling in Kupffer cells. Am J Surg. 2008;196(1):139–148. doi:10.1016/j.amjsurg.2007.09.045
- Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253–257. doi:10.1038/nature16969
- Takahashi Y, Dungubat E, Kusano H, Fukusato T. Pathology and Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease-Associated Hepatic Tumors. Biomedicines. 2023;11(10):2761. doi:10.3390/biomedicines11102761
- Lai C-Y, Yeh K-Y, Lin C-Y, et al. MicroRNA-21 Plays Multiple Oncometabolic Roles in the Process of NAFLD-Related Hepatocellular Carcinoma via PI3K/AKT, TGF-β, and STAT3 Signaling. Cancers. 2021;13(5):1.
- Tsai W-C, Hsu S-D, Hsu C-S, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Investig. 2012;122(8):2884–2897. doi:10.1172/JCI63455
- Y-e Q, Duan L, He Y, et al. Saturated fatty acids promote hepatocytic senecence through regulation of mir-34a/cyclin-dependent kinase 6. Molecul Nut. 2020;64(23):2000383.
- Guo Y, Xiong Y, Sheng Q, Zhao S, Wattacheril J, Flynn CR. A micro-RNA expression signature for human NAFLD progression. J Gastroenterol. 2016;51(10):1022–1030. doi:10.1007/s00535-016-1178-0
- Zhang T, Zhao X, Steer CJ, Yan G, Song G. A negative feedback loop between microRNA-378 and Nrf1 promotes the development of hepatosteatosis in mice treated with a high fat diet. Metabolism. 2018;85:183–191. doi:10.1016/j.metabol.2018.03.023
- Aspichueta P, Zeisel MB. miR‐21p‐5p coordinates biological pathways to promote MASLD progression. Liver Int. 2023;43(11):2343–2345. doi:10.1111/liv.15740
- He Y, Hwang S, Cai Y, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. 2019;70(4):1150–1167. doi:10.1002/hep.30645
- Luther J, Garber JJ, Khalili H, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol. 2015;1(2):222–232. doi:10.1016/j.jcmgh.2015.01.001
- Mouzaki M, Loomba R. Insights into the evolving role of the gut microbiome in nonalcoholic fatty liver disease: rationale and prospects for therapeutic intervention. Therap Adv Gastroenterol. 2019;12:1756284819858470. doi:10.1177/1756284819858470
- Vespasiani-Gentilucci U, Gallo P, Picardi A. The role of intestinal microbiota in the pathogenesis of NAFLD: starting points for intervention. Arch Med Sci. 2018;14(3):701–706. doi:10.5114/aoms.2016.58831
- Vespasiani-Gentilucci U, Carotti S, Perrone G, et al. Hepatic toll-like receptor 4 expression is associated with portal inflammation and fibrosis in patients with NAFLD. Liver Int. 2015;35(2):569–581. doi:10.1111/liv.12531
- Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47(5):1773–1785. doi:10.1002/hep.22146
- Feng L, Chen Y, Xu K, et al. Cholesterol-induced leucine aminopeptidase 3 (LAP3) upregulation inhibits cell autophagy in pathogenesis of NAFLD. Aging. 2022;14(7):3259–3275. doi:10.18632/aging.204011
- Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64(6):1994–2014. doi:10.1002/hep.28820
- Jang HJ, Lee YH, Dao T, et al. Thrap3 promotes nonalcoholic fatty liver disease by suppressing AMPK-mediated autophagy. Exp Mol Med. 2023;55(8):1720–1733. doi:10.1038/s12276-023-01047-4
- Zhu J, Cheng M, Zhao X. A tRNA-derived fragment (tRF-3001b) aggravates the development of nonalcoholic fatty liver disease by inhibiting autophagy. Life Sci. 2020;257:118125. doi:10.1016/j.lfs.2020.118125
- Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:10.1101/gad.2016211
- Inokuchi-Shimizu S, Park EJ, Roh YS, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124(8):3566–3578. doi:10.1172/JCI74068
- Dash S, Chava S, Chandra PK, Aydin Y, Balart LA, Wu T. Autophagy in hepatocellular carcinomas: from pathophysiology to therapeutic response. Hepat Med. 2016;8:9–20. doi:10.2147/HMER.S63700
- He Y, Su Y, Duan C, et al. Emerging role of aging in the progression of NAFLD to HCC. Ageing Res Rev. 2023;84:101833. doi:10.1016/j.arr.2022.101833
- Baboota RK, Rawshani A, Bonnet L, et al. BMP4 and Gremlin 1 regulate hepatic cell senescence during clinical progression of NAFLD/NASH. Nat Metab. 2022;4(8):1007–1021. doi:10.1038/s42255-022-00620-x
- Rey S, Quintavalle C, Burmeister K, et al. Liver damage and senescence increases in patients developing hepatocellular carcinoma. J Gastroenterol Hepatol. 2017;32(8):1480–1486. doi:10.1111/jgh.13717
- Meijnikman AS, Herrema H, Scheithauer TPM, Kroon J, Nieuwdorp M, Groen AK. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 2021;3(4):100301. doi:10.1016/j.jhepr.2021.100301
- Di Micco R, Krizhanovsky V, Baker D, d’Adda Di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22(2):75–95. doi:10.1038/s41580-020-00314-w
- Li Y, Lu L, Xie Y, et al. Interleukin-6 knockout inhibits senescence of bone mesenchymal stem cells in high-fat diet-induced bone loss. Front Endocrinol. 2020;11:622950. doi:10.3389/fendo.2020.622950
- Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–1415. doi:10.1016/j.jhep.2016.02.004
- Kumar DP, Santhekadur PK, Seneshaw M, Mirshahi F, Uram-Tuculescu C, Sanyal AJ. A regulatory role of apoptosis antagonizing transcription factor in the pathogenesis of nonalcoholic fatty liver disease and hepatocellular carcinoma. Hepatology. 2019;69(4):1520–1534. doi:10.1002/hep.30346
- Mailliard ME, Gollan JL. Metabolic liver disease in the young adult. Best Pract Res Clin Gastro. 2003;17(2):307–322. doi:10.1016/S1521-6918(02)00148-8
- Nowak A, Giger RS, Krayenbuehl P-A. Higher age at diagnosis of hemochromatosis is the strongest predictor of the occurrence of hepatocellular carcinoma in the Swiss hemochromatosis cohort: a prospective longitudinal observational study. Medicine. 2018;97(42). doi:10.1097/MD.0000000000012886
- Britton RS, Fleming RE, Parkkila S, Waheed A, Sly WS, Bacon BR. Pathogenesis of hereditary hemochromatosis: genetics and beyond. Sem gastro dis. 2002;13(2):68–79.
- Adams PC, Jeffrey G, Ryan J. Haemochromatosis. Lancet. 2023;401(10390):1811–1821. doi:10.1016/S0140-6736(23)00287-8
- Jin F, Qu LS, Shen XZ. Association between C282Y and H63D mutations of the HFE gene with hepatocellular carcinoma in European populations: a meta-analysis. J Exper Clinical Cancer Res. 2010;29(1):18. doi:10.1186/1756-9966-29-18
- Funakoshi N, Chaze I, Alary AS, et al. The role of genetic factors in patients with hepatocellular carcinoma and iron overload - a prospective series of 234 patients. Liver Int. 2016;36(5):746–754. doi:10.1111/liv.12984
- Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26(15):2166–2176. doi:10.1038/sj.onc.1210279
- Lehmann U, Wingen LU, Brakensiek K, et al. Epigenetic defects of hepatocellular carcinoma are already found in non-neoplastic liver cells from patients with hereditary haemochromatosis. Hum Mol Genet. 2007;16(11):1335–1342. doi:10.1093/hmg/ddm082
- Brakensiek K, Länger F, Schlegelberger B, Kreipe H, Lehmann U. Hypermethylation of the suppressor of cytokine signalling-1 (SOCS-1) in myelodysplastic syndrome. Br. J. Haematol. 2005;130(2):209–217. doi:10.1111/j.1365-2141.2005.05590.x
- Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65(9):3497–3508. doi:10.1158/0008-5472.CAN-04-4088
- Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-S-transferase family of enzymes. Mutat Res. 2001;482(1–2):21–26. doi:10.1016/S0027-5107(01)00206-8
- Di Capua DM, Shanahan W, Bourke M, et al. Tumour stemness and poor clinical outcomes in haemochromatosis patients with hepatocellular carcinoma. J Clin Pathol. 2023;jcp–2022–208679.doi:10.1136/jcp-2022-208679
- Greene CM, Marciniak SJ, Teckman J, et al. α1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2(1):16051. doi:10.1038/nrdp.2016.51
- Crowther DC, Belorgey D, Miranda E, Kinghorn KJ, Sharp LK, Lomas DA. Practical genetics: alpha-1-antitrypsin deficiency and the serpinopathies. Eur J Hum Genet. 2004;12(3):167–172. doi:10.1038/sj.ejhg.5201127
- Van Thiel DH, Ramadori G. Non-viral causes of hepatocellular carcinoma. J Gastrointest Cancer. 2011;42(4):191–194. doi:10.1007/s12029-010-9195-3
- Schilsky ML, Oikonomou I. Inherited metabolic liver disease. Curr Opin Gastroenterol. 2005;21(3):275–282. doi:10.1097/01.mog.0000159821.78532.21
- Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in α 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286(5):G851–G862. doi:10.1152/ajpgi.00175.2003
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi:10.1016/j.cell.2005.04.014
- Perlmutter DH, Brodsky JL, Balistreri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review. Hepatology. 2007;45(5):1313–1323. doi:10.1002/hep.21628
- Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology. 2004;39(4):1048–1055. doi:10.1002/hep.20118
- Angileri F, Morrow G, Scoazec JY, et al. Identification of circulating microRNAs during the liver neoplastic process in a murine model of hereditary tyrosinemia type 1. Sci Rep. 2016;6:27464. doi:10.1038/srep27464
- Orejuela D, Jorquera R, Bergeron A, Finegold MJ, Tanguay RM. Hepatic stress in hereditary tyrosinemia type 1 (HT1) activates the AKT survival pathway in the fah-/- knockout mice model. J Hepatol. 2008;48(2):308–317. doi:10.1016/j.jhep.2007.09.014
- Willenbring H, Sharma AD, Vogel A, et al. Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis. Cancer Cell. 2008;14(1):59–67. doi:10.1016/j.ccr.2008.05.004
- Plentz RR, Park YN, Lechel A, et al. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology. 2007;45(4):968–976. doi:10.1002/hep.21552
- Angileri F, Morrow G, Roy V, Orejuela D, Tanguay RM. Heat shock response associated with hepatocarcinogenesis in a murine model of hereditary tyrosinemia type I. Cancers. 2014;6(2):998–1019. doi:10.3390/cancers6020998
- Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–238. doi:10.1038/s41575-020-00381-6