
Abstract
Idiopathic pulmonary fibrosis is an etiologically complex interstitial lung disease characterized by progressive scarring of the lungs with a subsequent decline in lung function. While much of the pathogenesis of IPF still remains unclear, it is now understood that genetic variation accounts for at least one-third of the risk of developing the disease. The single-most validated and most significant risk factor, genetic or otherwise, is a gain-of-function promoter variant in the MUC5B gene. While the functional impact of these IPF risk variants at the cellular and tissue levels are areas of active investigation, there is a growing body of evidence that these genetic variants may influence disease pathogenesis through modulation of innate immune processes.
Introduction
Idiopathic pulmonary fibrosis (IPF) is an etiologically complex interstitial lung disease (ILD) characterized by progressive lung scarring with a subsequent decline in function.Citation1 There have been major efforts over the past decade investigating IPF which have resulted in a more specific classification of the diseaseCitation2 and the approval of two disease-specific agents, pirfenidoneCitation3 and nintedanib;Citation4 however, IPF diagnosis remains challenging and disease prognosis remains poor even with pharmacologic intervention.
While the pathogenesis of IPF remains an area of active investigation, it is generally accepted that disease pathobiology results from repeated injury to the airway and alveolar epithelia. These injuries result in an exaggerated and cyclical repair response with subsequent hallmark fibroblast activation and matrix deposition.Citation5–Citation7 It is also now well understood that genetic susceptibility in addition to environmental risk factors, including cigarette smoking, has a major contribution to the risk of developing IPF.Citation8–Citation10 Indeed, the single-most validated and strongest risk factor for IPF, genetic or otherwise, is the single nucleotide polymorphism rs35705950 in the promoter region of the mucin 5B (MUC5B) gene.Citation11–Citation21
What remains more debated in IPF pathogenesis are the roles of inflammation and immune-driven mechanisms. Initially, IPF was thought to be a disease primarily driven by chronic inflammation similar to other immunologic or autoimmune lung diseases. With accumulating evidence that immunomodulatory therapies were ineffective and potentially harmful in the treatment of IPF,Citation22 this viewpoint gradually shifted towards the model of aberrant tissue repair that is currently more accepted. However, there is emerging evidence that innate and adaptive immune processes may also contribute to pulmonary fibrogenesis.Citation23–Citation27 Understanding the interface between immunologic dysfunction and known genetic risk variants may deepen the understanding of the complexity of IPF.
In this review, we provide a brief overview of the intrinsic immune mechanism in the distal airway. We describe how MUC5B and other genetic risk variants for IPF may modulate host defense and innate immune mechanisms towards furthering IPF pathology.
Host Defense and Innate Immunity in the Airway
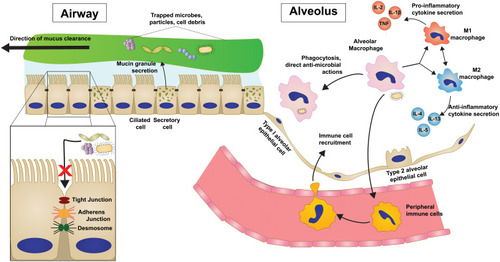
The human airway is constantly faced with threats in the way of inhaled pathogens and particles and thus has evolved a multilayered set of primary innate defensesCitation28 ().
Figure 1 Innate immune and host defense mechanisms in the distal airway and alveolus are multi-tiered.
Mucus Barrier
The mucus barrier is a viscoelastic gel layer designed to trap and remove offending agents and is composed of water, salts, and macromolecules held together by membrane-bound and secreted glycoproteins called mucins.Citation29 Airway mucins are secreted onto the apical surface of the epithelial layer by secretory cells, and through the coordinated beating of cilia on multi-ciliated airway cells, the mucus layer and trapped pathogens are swept proximally until ultimately being cleared from the airway.Citation30,Citation31 This process of mucociliary clearance (MCC) requires precise regulation of factors including mucin production, mucus composition, and coordinated ciliary movements in order to maintain effective defense.
Additionally, airway cells secrete products with antimicrobial properties,Citation32 such as lactoferrin, lysozyme, defensins, and surfactant proteins, into the lumen which remain in the mucus layer to combat potential pathogens during clearance.
Epithelial Barrier
Beneath the mucus barrier, the airway and alveolar epithelial cells themselves participate in the innate airway defense by forming a physical barrier against harmful external substances.Citation33 The epithelial layer accomplishes this selective permeability through the expression and regulation of multiple cell-cell adhesion complexes.Citation33–Citation35 Tight junctions are composed of claudins and occludins and are most involved in preventing paracellular passage of luminal material. Adherens junctions and desmosomes are involved in linking the actin cytoskeleton and intermediate filaments, respectively, on neighboring cells allowing the epithelium to remain intact. Loss or dysregulation of any of these complexes can result in disintegrity of the protective epithelial barrier.
In addition to serving as a physical barrier, epithelial cells are closely involved in the innate response through recognition of uncleared pathogens via the expression of pattern recognition receptors (PRRs).Citation28,Citation36–Citation39 These PRRs, including Toll-like receptors (TLRs), are involved in surveying the environment for the presence of pathogens or dying cells by binding so-called pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) respectively. This vast diversity of PRRs allows the lung to recognize and differentially respond to innumerous environmental cues.
Macrophages and Immune Cell Recruitment
Macrophages are the most predominant immune cells in the healthy lung and organize immune defense through a variety of mechanisms including direct antimicrobial and phagocytic activity.Citation38–Citation40 Like other macrophage populations, AMs are traditionally classified as one of the two phenotypic states, classically activated (M1 macrophages) and alternatively activated (M2 macrophages), which are a result of their exposure to a milieu of cytokines, growth factors, and other signals. M1 macrophages are polarized by Th1 pro-inflammatory cytokines (TNF, IL-2, IFN-γ) and generally induce further inflammatory cytokine signaling and immune cell recruitment. M2 macrophages are induced by Th2 cytokines (IL-4, IL-5, IL-13) and are thought to have anti-inflammatory characteristics seen in chronic repair processes. AMs serve as scavengers in the airways that endocytose inhaled particles, cellular debris, and pathogens that cannot be cleared by the mucociliary escalator. AMs then play a major role in coordinating the subsequent immune response through the secretion of cytokines and chemokines to differentially activate or recruit immune cells. Additionally, AMs can bridge innate and adaptive responses through their role as antigen-presenting cells in the lung periphery.
Genetic Variants Associated with IPF
Numerous familial studies and several larger genome-wide linkage and association studies have identified rare and common genetic variants associated with both familial interstitial pneumonia (FIP), which is characterized by familial clustering of IPF, and sporadic IPF risk (). These variants include the single nucleotide polymorphism (SNP) in the MUC5B geneCitation11–Citation17,Citation19–Citation21 described in detail below as well as genes related to innate immune function (TOLLIP, TLR3, IL1RN, IL8, TGFB1), and epithelial barrier function (DSP, DPP9). Additional gene ontologies represented in those identified variants include telomere maintenance (TERT, TERC, OBFC1, TINF2, DKC1, RTEL1, PARN), surfactant production (SFTPC, SFTPA2, ABCA3), and cell cycle regulation (KIF15, MAD1L1, CDKN1A).
Table 1 Common and Rare Genetic Variants Associated with IPF
MUC5B Promoter Variant
The rs35705950 variant in the promoter region of the mucin 5B (MUC5B) gene was first identified in a 2011 genome-wide linkage study and is associated with an approximately 7-fold increased risk of developing IPF.Citation11 Since then this MUC5B variant has been validated in multiple independent studies and is still considered to be the most significant risk, genetic or otherwise, for IPF.Citation12–Citation17,Citation19–Citation21 In addition to its being the strongest risk factor for disease, the rs35705950 risk allele is also strikingly common in both healthy and IPF populations (mean allele frequency of 9% and 38%, respectively). Interestingly, IPF patients who are heterozygous carriers of the minor allele have been reported to have a paradoxical survival benefit compared to noncarriers,Citation41,Citation42 although this result has not been unanimously confirmed.Citation43,Citation44 In other ILD populations, the rs35705950 variant has also been demonstrated to confer worse survival in interstitial pneumonia with autoimmune featuresCitation45 and a trend towards worse survival in connective tissue disease associated-ILDCitation45 and chronic hypersensitivity pneumonitis.Citation46 The interplay between MUC5B variant and transplant-free survival across ILDs and how these differences are specific to the IPF-related disease processes remains to be investigated.
The minor, disease-associated T allele at rs35705950 is a gain-of-function variant has been suggested to drive differential methylation of and transcription factor binding to the MUC5B leading to increased MUC5B expression.Citation11,Citation47 Interestingly, this variant-dependent increase in MUC5B production has been demonstrated to be specific to IPFCitation48 and localized to the distal airway regions including terminal airwaysCitation49,Citation50 and honeycomb cysts,Citation51 which are thought to be the end result of chronic airway. Examining the influences of a multitude of factors, including epigenetic and environmental factors, on the rs35705950 variant and MUC5B expression continues to be a focus of ongoing studies.
Relationship Between Genetic Variants and IPF Immunopathogenesis
Altered Mucociliary Clearance and Host Defense
As one of the two human primary mucins secreted in the airway, along with mucin 5AC (MUC5AC), MUC5B plays a critical role in maintaining effective mucociliary clearance (MCC) which serves in removing inhaled debris and pathogens.Citation29,Citation52,Citation53 Effective MCC involves the coordinated actions of mucus secretion and ciliary beating. This process is critical for the maintenance of normal lung physiology by acting as the first-line innate defense mechanism in the airway. MCC dysfunction is seen in many chronic lung diseases including chronic obstructive pulmonary disease (COPD),Citation54,Citation55 cystic fibrosis (CF),Citation55–Citation57 and asthma.Citation52
Genetic manipulation of Muc5b expression in mice has revealed pathologic consequences of both deficient and excessive Muc5b in the airways. Complete knockout of Muc5b in mice (Muc5b−/-) results in impaired MCC and chronic lower respiratory infections. Muc5b−/- mice also demonstrate dramatic changes in resident alveolar macrophage (AM) populations with increased AM apoptosis, reduced activation, and impaired phagocytosis.Citation53 Conversely, transgenic Muc5b overexpression, specifically when localized to the distal airway, results in diminished MCC and enhanced fibrosis after intra-tracheal bleomycin instillation.Citation58 Collectively these results demonstrate that host defense and MCC rely on the well-regulated expression of MUC5B and that disturbances can lead to significant pathologic sequela including increased fibroproliferative responses to injury.
As the rs35705950 SNP leads to increases in MUC5B accumulation and presumably mucociliary stasis, this could result in chronic exposure of the distal airway to inhaled particles and pathogens. Environmental and occupational factors including cigarette smoke, silica, and metal dusts have repeatedly been shown to be associated with IPF and other ILDs strongly suggesting that repeated exposure of the airway to inhaled particles is implicated in fibrotic lung diseases.Citation59–Citation61 In genetically susceptible individuals with impaired MCC, it might be expected that exposure to inhaled pro-fibrotic particles could have an even more drastic effect in initiating epithelial cell injury and the fibroproliferative response.
In addition to the clearance of particulate matter, MCC is essential for effectively removing pathogens from distal airspaces. Recent evidence has implicated the lung microbiome in IPF pathogenesis.Citation62–Citation64 Lung dysbiosis characterized by increased bacterial burden and loss of microbiotic diversity has been reported in bronchoalveolar lavage (BAL) specimens from both IPF patients and bleomycin-treated mice.Citation62,Citation64,Citation65 Interestingly, IPF patients with the MUC5B variant had a lower bacterial burden than noncarriers. This may in part help to explain the paradoxical survival benefit that MUC5B variant carriers have, as a bacterial burden has been shown to have an independent correlation with mortality in IPF patients.Citation66
Disrupted Epithelial Barrier Function
In conjunction with the mucous layer, the airway and alveolar epithelial cells themselves serve as a key defense mechanism in the lung by forming a protective barrier from luminal pathogens, cellular debris, and inhaled particles.
A 2013 genome-wide association study (GWAS) identified variants in two cell-cell adhesion-related genes, DSP (desmoplakin) and DPP9 (dipeptidyl peptidase 9), associated with IPF.Citation13 The DSP variant rs2076395 has been further shown to cause a reduction in desmoplakin expression in epithelial cells.Citation67,Citation68 Furthermore, complete knockout of DSP in an airway epithelial cell line was associated with impaired epithelial barrier function and an aberrantly increased wound repair response.Citation68
While the in vivo consequences of DSP loss are areas of active investigation, loss-of-function mutations in other desmosomal genes including DSG1 have been shown to upregulate NF-κB signaling with increased production of pro-inflammatory cytokines and phagocyte recruitment.Citation69 Cytokines including IL-1β, IL-6, and IL-8, which are produced by both injured epithelial cells and activated alveolar macrophages, have been shown to further disrupt epithelial barrier integrity potentiating this process of cyclic damage.Citation70,Citation71 Additionally, as the epithelial layer loses its ability to maintain a barrier either through genetic predisposition or inflammatory signals, innate immune receptors that are normally localized to the protected basolateral membrane, such as TLR2 and TLR6, are exposed to PAMPs and DAMPs.Citation72 Active TLR2 signaling in mouse airway cells has been shown to induce TGF-β expression, which is the most well-studied pro-fibrotic mediator.Citation73
Collectively, these associations between epithelial disintegrity as a consequence of genetic variants and the pro-inflammatory and fibroproliferative environment created by impaired epithelial barriers could reveal important insights into IPF pathobiology.
Autoinflammatory Toll-Like Receptor Family Signaling
As a link between innate and adaptive immune responses, TLR signaling dysregulation has been described in IPF patientsCitation74,Citation75 though the exact contribution of these signaling cascades to the fibroproliferative response remains mostly undefined.
In humans, 10 functional TLRs have been identified which have distinct receptor-ligand associations.Citation76 TLRs are either localized to the cell membrane (TLR1, 2, 4, 5, 6) or endosomal compartments (TLR3, 7, 8, 9) in order to recognize various extracellular and intracellular signals, respectively. The majority of TLRs signaling through a MyD88-dependent pathway with the ultimate result of NF-κB activation and proinflammatory cytokine gene transcription. TLR3 signaling, as well as alternative TLR4 signaling, occurs through a MyD88-independent mechanism whereby recruitment of TRIF subsequently leads to transcription of type I interferon (IFN) genes by IRF3 or IRF7.Citation76
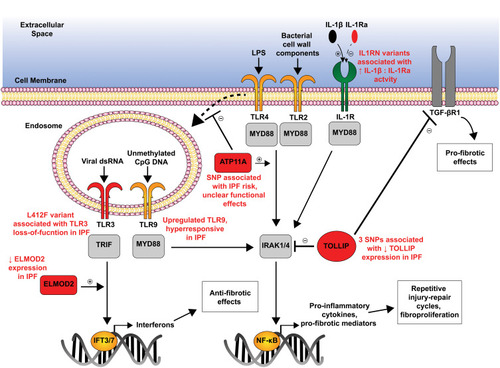
The genetic risk variants affecting TLR family signaling which are associated with IPF are described below ().
Figure 2 Genetic variants disrupt TLR/IL-1R family signaling in IPF.
Pro-Fibrotic TLR Signaling
A 2013 genome-wide association study (GWAS) identified three common variants (rs111521887, rs5743894, rs574389) in the Toll-interacting protein (TOLLIP) gene which were associated with IPF and one of which (rs5743894) is associated with decreased risk of IPF but increased mortality in those with the disease.Citation14 Expression of TOLLIP occurs primarily in lung macrophages and epithelial cells,Citation77 and each of these variants is associated with a 20–50% reduction in TOLLIP mRNA expression.Citation14 Since TOLLIP and MUC5B are adjacent genes at the chromosome 11p15.5 region, there has been conflicting evidence as to whether variants in these are in linkage disequilibrium or provide independent associations with IPF.Citation20,Citation21 Regardless, TOLLIP has been shown to suppress TLR activation, particularly TLR2 and TLR4, by inhibiting IL-1 receptor-associated kinase (IRAK) phosphorylation.Citation77–Citation80 Both TLR2 and TLR4 activation occur in response to many different microbial signals and have been shown to be elevated in IPF epithelia, possibly due to chronic exposure to pathogenic microbes.Citation81 Reduced TOLLIP expression has been shown to increase pro-inflammatory cytokine (IL-6 and TNF) production by macrophages after TLR stimulation.Citation79 TOLLIP induces the production of IL-10,Citation80 which has protective effects against a bleomycin-induced fibrosis model in mice via TLR4-dependent signaling.Citation82 Additionally, TOLLIP antagonizes TGF-β signaling by degrading TGF-β1 receptors through SMAD7-dependent interactions.Citation83 Together, these data suggest that TOLLIP expression may be protective against IPF through dampening of pro-inflammatory, pro-fibrotic cascades.
While less functionally defined, the rs1278769 variant in ATP11A identified in the 2013 GWAS may impact TLR4 signaling.Citation13 ATP11A codes for a phospholipid flippase and has been shown to enhance MyD88-dependent NF-kB activation and production of proinflammatory cytokines through inhibition of TLR4 endocytosis.Citation84 It remains unclear if this IPF-associated variant enhances this signaling cascade.
While not specifically associated with any genetic variant, dysregulated TLR9 activation drives fibroblast-to-myofibroblast differentiation and has been associated with a more aggressive IPF phenotype.Citation85 TLR9 recognizes hypomethylated CpG nucleotides typically from microbial genomic material.Citation76,Citation86 As mitochondrial DNA released from injured cells is also recognized through TLR9, it is possible that cell injury and non-apoptotic cell death that is seen in the IPF distal airway could also be a potent driver of TLR9-mediated fibrosis. Understanding the full extent of these associations between irregular TLR signaling and etiology and progression of IPF will undoubtedly be pivotal in understanding the overlap between innate immunity and fibroproliferation.
The IL-1 receptor (IL-1R) is a member of the same receptor family as TLRs with a shared intracellular Toll/IL-1R (TIR) domain that activates MyD88-dependent signaling.Citation87,Citation88 IL-1R is activated by multiple ligands, of which IL-1α and IL-1β are the best studied, and competitively inhibited by IL-1 receptor antagonist (IL-1Ra). While several variants in the IL-1Ra (IL1RN) gene have been investigated, the most convincing evidence from a meta-analysis demonstrated that a haplotype block of a variable number tandem repeat (VNTR*2) and two minor alleles (rs408392 and rs419598) in IL1RN gene was significantly associated with IPF disease.Citation89 These risk alleles result in a reduction in IL-1Ra expression, leading to unrestricted IL-1R activation in airway/alveolar epithelial cells and innate immune cells. IL-1R signaling has a well-established role in animal models of pulmonary fibrosis with adenoviral-mediated IL-1β overexpression being used as a comparative model to the traditional bleomycin model.Citation90 Alveolar macrophages from IPF patients have an elevated IL-1β:IL-1Ra ratio indicative of this proinflammatory state.Citation91 Both genetic and pharmacologic targeting of the IL-1R/MyD88 axis has been shown to attenuate fibrosis in bleomycin- and silica-induced fibrosis.Citation92,Citation93
Many of these overactive TLR signaling pathways, including TLR2 and TLR4, are directly responsive to both nonpathogenic and pathogenic bacterial signals. As discussed previously, dysregulation of commensal bacterial populations has been suspected to play a contributing role in IPF pathogenesis.Citation62–Citation64,Citation66 TLR-dependent signaling has been shown across multiple systems to tightly maintain homeostasis between host and commensal organisms, and disruption of these signaling pathways can drive disease processes.Citation94,Citation95 In the mouse lung, in particular, it has been demonstrated that dysregulation of commensal bacterial populations can promote fibrosis through TLR2- and TLR4-dependent production of IL-17B; however, this pro-fibrotic phenotype can be diminished through genetic ablation of these pathways.Citation96 The full spectrum of these relationships between pathogen-induced TLR signaling remains unstudied in the context of pulmonary fibrosis.
Taken together these genetic variants along with the general environment in the IPF distal airway may create a pro-inflammatory environment that disrupts the normal homeostasis necessary for repair through hyper-responsiveness to small chronic insults.
Anti-Fibrotic TLR Signaling
A loss-of-function variant in the TLR3 gene (L412F) has been associated with more rapid lung function decline and greater mortality in IPF patients.Citation97 This variant was shown to increase human lung fibroblast proliferation and resistance to apoptosis in culture. Additionally, a TLR3-knockout mouse (TLR3-/-) model of pulmonary fibrosis demonstrated an increase in TGF-β and pro-inflammatory cytokine production.
A TLR3-related gene ELMOD2 has also been identified as a potential candidate gene in FIP and IPF pathobiology with decreased gene and protein expression seen in alveolar epithelial tissue and pulmonary macrophages in IPF patients.Citation98,Citation99 ELMOD2 is necessary for TLR3 signal transduction with targeted knockdown of ELMOD2 resulting in a decreased IFN-dependent response.Citation99 IFNs have been shown to have strong anti-fibrotic characteristics in animal models,Citation100 yet these results have not been observed in patients with established IPF,Citation101 thus suggesting their role in affecting the initiation of the fibrotic cascade rather than reversing established fibrosis.
TLR3 is classically associated as a receptor for viral dsRNA, and conflicting evidence exists as to the role of viral infections, including Epstein-Barr virus (EBV),Citation102,Citation103 herpes simplex virus 1 (HSV-1),Citation104 and cytomegalovirus (CMV)Citation105 as a contributing source driving the progressive fibrosis. A multitude of viruses have been shown to increase fibroproliferative responses both in vitro and in vivo models, particularly in the setting of absent IFN-dependent responses.Citation106–Citation110 Therefore, viral-related TLR signaling likely acts as a blockade to fibroproliferation, and underlying TL3-signaling deficiencies could prime the airway for deficient responses to viral pathogens priming the distal lung for chronic injury-repair cycles that are thought to be central to IPF pathology.
Surfactant Protein Changes
Pulmonary surfactant is a phospholipid-rich substrate produced in the distal airway and alveolus which provides essential roles in preventing alveolar collapse and host defense.Citation111,Citation112 Approximately 10% of surfactant is composed of surfactant proteins which are produced and secreted by alveolar epithelial type II (AE2) cells and terminal airway secretory cells. Of the four surfactant proteins, surfactant proteins A (SP-A) and D (SP-D) are members of a specific family of innate immune protein termed collectins, named for calcium-binding C-terminal lectin domain that recognizes motifs on microbial surfaces.Citation111,Citation112 SP-A and SP-D have both been shown to opsonize common bacterial and viral pathogens and enhance phagocytic-killing by innate immune cells including macrophages and neutrophils.
Rare variants in the two adjacent SP-A coding genes, SFTPA1 and SFTPA2, have been described in several cases of FIP.Citation113–Citation115 While the role that these and other surfactant-related variants play in contribution to sporadic IPF remains less clear, IPF patients have been reported to have reduced BAL concentrations of SP-A compared to healthy patients, and SP-A levels are inversely correlated with survival.Citation116,Citation117 Mice deficient in SP-A (SP-A−/-) show dramatically decreased bacterial clearance, impaired macrophage phagocytosis, and increased production of pro-inflammatory cytokines IL-1β, IL-6, and TNF which are also associated with fibroblast activation.Citation118 SP-A−/- mice have also increased mortality and collagen deposition after induction of fibrosis with intratracheal bleomycin, suggesting a direct link between the pro-inflammatory state created by SP-A deficiency and subsequent fibroproliferative remodeling.Citation119 Additionally, it has been shown that an FIP-associated loss-of-function mutation in SFTPA1 leads to increased necroptosis in AE2 cells in mice.Citation113 Necroptotic cell death leads to the release of highly immunogenic intracellular proteins and has been increasingly been implicated in IPF pathogenesis.Citation120,Citation121 Treatment of mice with exogenous SP-A has been shown to reduce the expression of Th2 cytokines including IL-4 and IL-5, which are involved in chronic tissue repair responses and are linked to the development of fibrosis.Citation122,Citation123 Taken together these findings suggest that the defective host defense and highly inflammatory state created by a lack of SP-A, either due to SPFTA2 variants or generally in IPF, may directly contribute to pro-fibrotic environment in the alveolar space.
Other rare variants in surfactant-related genes, including genes encoding surfactant protein C (SFTPC) and surfactant transporters (ABCA3), are linked to FIP and IPF and are extensively reviewed elsewhere.Citation9,Citation10
MUC5B has been shown to be present at increased levels in the distal airway in IPF patients and at even more significant levels with the presence of the rs35705950 variant. Additionally, it has been reported that MUC5B is co-expressed with other surfactant proteins in AE2 cells to some degree in IPF lungs.Citation58 These data suggest that mucin-surfactant mixing is nearly inevitable in the IPF alveolus, and consequently may interfere with the normal antimicrobial effects of SP-A and SP-D. Furthermore, as MUC5B expression has been shown to occur in both surfactant-producing cell populations in the airway, AE2 cells and club cells, it remains unexplored whether the MUC5B variant negatively impacts the ability of these cells to produce surfactant proteins leaving the airway more susceptible to pathogen-related damage.
Summary
Our working model of IPF pathogenesis involves chronic epithelial injury in the terminal airway which ultimately leads to an uncontrolled response that overwhelms the repair mechanisms of the distal lung. The interplay between inflammation, immune mechanisms, and this aberrant cascade of epithelial–mesenchymal crosstalk remains elusive, yet genetic variants clearly suggest a role of impaired innate defense in this process. While innate immune responses are a critical component to the lung’s defense against offending agents and some level of the inflammatory response is necessary for many repair mechanisms, these processes need to be tightly regulated in order to restore the lung to homeostatic conditions. We suggest that these genetic variants associated with IPF which cause mucociliary dysfunction, impaired removal of pathogenic substances, and persistent inflammatory signaling create an environment in which proper repair becomes impossible ().
Figure 3 Genetic-driven innate immunity changes contribute to IPF pathogenesis.
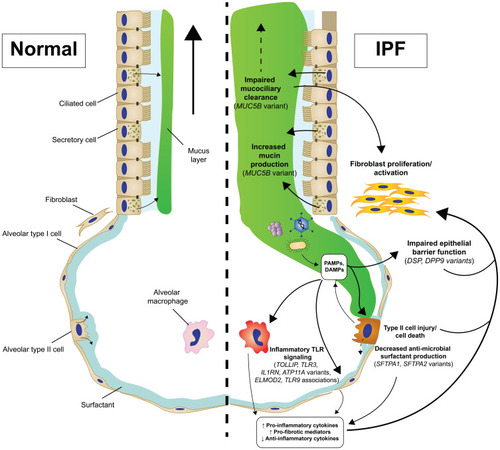
Genetic variants, such as the rs35705950 MUC5B variant, almost certainly interplay with other features of IPF pathobiology aside from impacts on innate host defense including distal airway cell homeostasis and repair, yet these exact relationships continued to be explored and are outside of the scope of this review. Additionally, studying the interactions between these genetic variants and both other genetic and environmental factors remains a key step in understanding IPF. Importantly, little is understood about the impact these genetic risk variants have on each other and how the presence of multiple genetic variants influences disease pathogenesis.
Together, our current understanding of IPF as a disease driven by environment injuries to a genetically susceptible lung raises the possible role of clinical genetic testing. While there are no current recommendations for the role of genome sequencing for sporadic IPF patients, further research into the relationship that genetic variants play on clinical outcomes is certainly necessary.
Disclosure
Dr David A Schwartz reports grants from NIH-NHLBI (UG3/UH3-HL151865), DoD Focused Program Project (W81XWH-17-1-0597), NIH-NHLBI (R01-HL149836), VAMC Merit Review (IO1BX005295), NIH-NHLBI (P01-HL0928701), NIH-NHLBI (UH2/3-HL123442), and NIH-NHLBI (X01-HL134585), during the conduct of the study; personal fees from Eleven P15, Inc. (a company with the mission of early detection and early intervention of IPF), outside the submitted work; In addition, Dr David A Schwartz has patents 14/813.559, 62/624500 pending, and a patent 8,673,565 issued. The authors report no other conflicts of interest in this work.
References
- Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra170575129742380
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.21471066
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A Phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi:10.1056/NEJMoa140258224836312
- Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa140258424836310
- Evans CM, Fingerlin TE, Schwarz MI, et al. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev. 2016;96(4):1567–1591. doi:10.1152/physrev.00004.201627630174
- Wuyts WA, Agostini C, Antoniou KM, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41(5):1207–1218. doi:10.1183/09031936.0007301223100500
- Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19(1):32. doi:10.1186/s12931-018-0730-229471816
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc. 2006;3(4):293–298. doi:10.1513/pats.200512-131TK16738192
- Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(6):1717–1727. doi:10.1183/09031936.0016381425837031
- Kaur A, Mathai SK, Schwartz DA. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front Med. 2017;4:154. doi:10.3389/fmed.2017.00154
- Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. doi:10.1056/NEJMoa101366021506741
- Zhang Y, Noth I, Garcia JGN, Kaminski N. A Variant in the Promoter of MUC5B and Idiopathic Pulmonary Fibrosis. N Engl J Med. 2011;364(16):1576–1577. doi:10.1056/NEJMc101350421506748
- Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. doi:10.1038/ng.260923583980
- Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–317. doi:10.1016/s2213-2600(13)70045-624429156
- Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68(5):436–441. doi:10.1136/thoraxjnl-2012-20178623321605
- Borie R, Crestani B, Dieude P, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8 (8):e70621. doi:10.1371/journal.pone.007062123940607
- Peljto AL, Selman M, Kim DS, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest. 2015;147(2):460–464.25275363
- Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc. 2015;12 Suppl 2(Suppl 2):S193–S199. doi:10.1513/AnnalsATS.201503-110AW26595739
- Horimasu Y, Ohshimo S, Bonella F, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology. 2015;20(3):439–444. doi:10.1111/resp.1246625581455
- Moore C, Blumhagen RZ, Yang IV, et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;200(2):199–208. doi:10.1164/rccm.201810-1891OC31034279
- Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2020;201(5):564–574. doi:10.1164/rccm.201905-1017OC31710517
- Idiopathic Pulmonary Fibrosis Clinical Research. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. doi:10.1056/NEJMoa111335422607134
- Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2008;10(2):287–301. doi:10.1089/ars.2007.189717961066
- Desai O, Winkler J, Minasyan M, Herzog EL. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front Med. 2018;5:43. doi:10.3389/fmed.2018.00043
- Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. 2019;147:79–91. doi:10.1016/j.rmed.2018.12.01530704705
- Warheit-Niemi HI, Hult EM, Moore BB. A pathologic two-way street: how innate immunity impacts lung fibrosis and fibrosis impacts lung immunity. Clin Transl Immunology. 2019;8(6):e1065. doi:10.1002/cti2.106531293783
- Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune Mechanisms in Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2016;55(3):309–322. doi:10.1165/rcmb.2016-0121TR27149613
- Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45(2):189–201. doi:10.1165/rcmb.2011-0011RT21330463
- Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi:10.1056/NEJMra091006121121836
- Adler KB, Tuvim MJ, Dickey BF. Regulated mucin secretion from airway epithelial cells. Front Endocrinol (Lausanne). 2013;4:129. doi:10.3389/fendo.2013.0012924065956
- Kuek LE, Lee RJ. First contact: the role of respiratory cilia in host-pathogen interactions in the airways. Am J Respir Cell Mol Biol. 2020;319(4):L603–L619. doi:10.1152/ajplung.00283.2020
- Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. 2002;109(6):693–697. doi:10.1172/JCI1521811901174
- Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More Than Just a Barrier: the Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front Immunol. 2020;11(761). doi:10.3389/fimmu.2020.00761
- Davies DE. Epithelial barrier function and immunity in asthma. Ann Am Thorac Soc. 2014;11(Suppl 5):S244–251. doi:10.1513/AnnalsATS.201407-304AW25525727
- Hiemstra PS, McCray PB Jr, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. 2015;45(4):1150–1162. doi:10.1183/09031936.0014151425700381
- Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.02220303872
- Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31(3):358–364. doi:10.1165/rcmb.2003-0388OC15191912
- Hartl D, Tirouvanziam R, Laval J, et al. Innate Immunity of the Lung: from Basic Mechanisms to Translational Medicine. J Innate Immun. 2018;10(5–6):487–501. doi:10.1159/00048705729439264
- Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. 2005;2(5):403–411. doi:10.1513/pats.200508-090JS16322590
- Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70(12):1189–1196. doi:10.1136/thoraxjnl-2015-20702026286722
- Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–2239. doi:10.1001/jama.2013.582723695349
- Biondini D, Balestro E, Rigobello C, et al. MUC5B genotype affects survival of patients with idiopathic pulmonary fibrosis (IPF) on antifibrotic treatment. Eur Respir J. 2019;54(suppl63):PA1352. doi:10.1183/13993003.congress-2019.PA1352
- Dudbridge F, Allen RJ, Sheehan NA, et al. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat Commun. 2019;10(1):1561. doi:10.1038/s41467-019-09381-w30952951
- van der Vis JJ, Snetselaar R, Kazemier KM, Ten Klooster L, Grutters JC, van Moorsel CH. Effect of Muc5b promoter polymorphism on disease predisposition and survival in idiopathic interstitial pneumonias. Respirology. 2016;21(4):712–717. doi:10.1111/resp.1272826699835
- Newton CA, Oldham JM, Ley B, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. 2019;53(4):1801641. doi:10.1183/13993003.01641-201830635297
- Ley B, Newton CA, Arnould I, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5(8):639–647. doi:10.1016/S2213-2600(17)30216-328648751
- Helling BA, Gerber AN, Kadiyala V, et al. Regulation of MUC5B Expression in Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2017;57(1):91–99. doi:10.1165/rcmb.2017-0046OC28272906
- Stock CJ, Conti C, Montero-Fernandez Á, et al. Interaction between the promoter MUC5B polymorphism and mucin expression: is there a difference according to ILD subtype? Thorax. 2020;75(10):901–903. doi:10.1136/thoraxjnl-2020-21457932580994
- Nakano Y, Yang IV, Walts AD, et al. MUC5B Promoter Variant rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;193(4):464–466. doi:10.1164/rccm.201509-1872LE26871673
- Okuda K, Chen G, Subramani DB, et al. Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am J Respir Crit Care Med. 2019;199(6):715–727. doi:10.1164/rccm.201804-0734OC30352166
- Seibold MA, Smith RW, Urbanek C, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucociliary pseudostratified epithelium. PLoS One. 2013;8(3):e58658–e58658. doi:10.1371/journal.pone.005865823527003
- Bonser LR, Erle DJ. Airway Mucus and Asthma: the Role of MUC5AC and MUC5B. J Clin Med. 2017;6:12. doi:10.3390/jcm6120112
- Roy MG, Livraghi-Butrico A, Fletcher AA, et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412–416. doi:10.1038/nature1280724317696
- Bhowmik A, Chahal K, Austin G, Chakravorty I. Improving mucociliary clearance in chronic obstructive pulmonary disease. Respir Med. 2009;103(4):496–502. doi:10.1016/j.rmed.2008.10.01419091536
- Mall MA. Unplugging Mucus in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc. 2016;13(Suppl 2):S177–185. doi:10.1513/AnnalsATS.201509-641KV27115954
- Robinson M, Bye PT. Mucociliary clearance in cystic fibrosis. Pediatr Pulmonol. 2002;33(4):293–306. doi:10.1002/ppul.1007911921459
- Kreda SM, Davis CW, Rose MC. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb Perspect Med. 2012;2(9):a009589–a009589. doi:10.1101/cshperspect.a00958922951447
- Hancock LA, Hennessy CE, Solomon GM, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9(1):5363. doi:10.1038/s41467-018-07768-930560893
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242–248. doi:10.1164/ajrccm.155.1.90013199001319
- Baumgartner KB, Samet JM, Coultas DB, et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Am J Epidemiol. 2000;152(4):307–315.10968375
- Paolocci G, Folletti I, Torén K, et al. Occupational risk factors for idiopathic pulmonary fibrosis in Southern Europe: a case-control study. BMC Pulm Med. 2018;18(1):75. doi:10.1186/s12890-018-0644-229784045
- Molyneaux PL, Cox MJ, Willis-Owen SA, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–913. doi:10.1164/rccm.201403-0541OC25184687
- Han MK, Zhou Y, Murray S, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2(7):548–556. doi:10.1016/s2213-2600(14)70069-424767767
- O’Dwyer D, Ashley N, Gurczynski SL, et al. Lung Microbiota Contribute to Pulmonary Inflammation and Disease Progression in Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;199(9):1127–1138. doi:10.1164/rccm.201809-1650OC30789747
- Invernizzi R, Wu BG, Barnett J, et al. The Respiratory Microbiome in Chronic Hypersensitivity Pneumonitis is Distinct from that of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2020. doi:10.1164/rccm.202002-0460OC
- Invernizzi R, Barnett J, Rawal B, et al. Bacterial burden in the lower airways predicts disease progression in idiopathic pulmonary fibrosis and is independent of radiological disease extent. Eur Respir J. 2020;1901519. doi:10.1183/13993003.01519-2019
- Mathai SK, Pedersen BS, Smith K, et al. Desmoplakin Variants Are Associated with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;193(10):1151–1160. doi:10.1164/rccm.201509-1863OC26669357
- Hao Y, Bates S, Mou H, et al. GWAS Functional Variant rs2076295 Regulates Desmoplakin (DSP) Expression in Airway Epithelial Cells. Am J Respir Crit Care Med. 2020. doi:10.1164/rccm.201910-1958OC
- Polivka L, Hadj-Rabia S, Bal E, et al. Epithelial barrier dysfunction in desmoglein-1 deficiency. J Allergy Clin Immunol. 2018;142(2):702–706.e707. doi:10.1016/j.jaci.2018.04.00729705242
- Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13(9):3218–3234. doi:10.1091/mbc.e02-03-013412221127
- Jevnikar Z, Östling J, Ax E, et al. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. 2019;143(2):577–590. doi:10.1016/j.jaci.2018.05.02629902480
- Ioannidis I, Ye F, McNally B, Willette M, Flaño E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol. 2013;87(6):3261–3270. doi:10.1128/JVI.01956-1223302870
- Beisswenger C, Lysenko ES, Weiser JN. Early Bacterial Colonization Induces Toll-Like Receptor-Dependent Transforming Growth Factor β Signaling in the Epithelium. Infect Immun. 2009;77(5):2212. doi:10.1128/IAI.01224-0819255194
- Karampitsakos T, Woolard T, Bouros D, Tzouvelekis A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur J Pharmacol. 2017;808:35–43. doi:10.1016/j.ejphar.2016.06.04527364757
- Samara KD, Antoniou KM, Karagiannis K, et al. Expression profiles of Toll-like receptors in non-small cell lung cancer and idiopathic pulmonary fibrosis. Int J Oncol. 2012;40(5):1397–1404. doi:10.3892/ijo.2012.137422344343
- Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi:10.3389/fimmu.2014.0046125309543
- Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277(9):7059–7065. doi:10.1074/jbc.M10953720011751856
- Li X, Kim SE, Chen T-Y, et al. Toll interacting protein protects bronchial epithelial cells from bleomycin-induced apoptosis. FASEB J. 2020;34(8):9884–9898. doi:10.1096/fj.201902636RR32596871
- Didierlaurent A, Brissoni B, Velin D, et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol. 2006;26(3):735–742. doi:10.1128/MCB.26.3.735-742.200616428431
- Shah JA, Vary JC, Chau TT, et al. Human TOLLIP regulates TLR2 and TLR4 signaling and its polymorphisms are associated with susceptibility to tuberculosis. J Immunol. 2012;189(4):1737–1746. doi:10.4049/jimmunol.110354122778396
- Go H, Koh J, Kim HS, Jeon YK, Chung DH. Expression of toll-like receptor 2 and 4 is increased in the respiratory epithelial cells of chronic idiopathic interstitial pneumonia patients. Respir Med. 2014;108(5):783–792. doi:10.1016/j.rmed.2013.12.00724613046
- Kurosaki F, Uchibori R, Sehara Y, et al. AAV6-Mediated IL-10 Expression in the Lung Ameliorates Bleomycin-Induced Pulmonary Fibrosis in Mice. Hum Gene Ther. 2018;29(11):1242–1251. doi:10.1089/hum.2018.02429598007
- Zhu L, Wang L, Luo X, et al. Tollip, an intracellular trafficking protein, is a novel modulator of the transforming growth factor-β signaling pathway. J Biol Chem. 2012;287(47):39653–39663. doi:10.1074/jbc.M112.38800923027871
- van der Mark VA, Ghiboub M, Marsman C, et al. Phospholipid flippases attenuate LPS-induced TLR4 signaling by mediating endocytic retrieval of Toll-like receptor 4. Cell Mol Life Sci. 2017;74(4):715–730. doi:10.1007/s00018-016-2360-527628304
- Trujillo G, Meneghin A, Flaherty KR, et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med. 2010;2(57):57ra82. doi:10.1126/scitranslmed.3001510
- Bueno M, Zank D, Buendia-Roldán I, et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS One. 2019;14(6):e0218003–e0218003. doi:10.1371/journal.pone.021800331170232
- Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. 2016;38(4):517–534. doi:10.1007/s00281-016-0559-z27001429
- Cohen P. The TLR and IL-1 signalling network at a glance. J Cell Sci. 2014;127(11):2383–2390. doi:10.1242/jcs.14983124829146
- Korthagen NM, van Moorsel CHM, Kazemier KM, Ruven HJT, Grutters JC. IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics. 2012;64(5):371–377. doi:10.1007/s00251-012-0604-622322675
- Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107(12):1529–1536. doi:10.1172/JCI1256811413160
- Kline JN, Schwartz DA, Monick MM, Floerchinger CS, Hunninghake GW. Relative Release of lnterleukin-1β and lnterleukin-1 Receptor Antagonist by Alveolar Macrophages: A Study in Asbestos-Induced Lung Disease, Sarcoidosis, and Idiopathic Pulmonary Fibrosis. Chest. 1993;104(1):47–53. doi:10.1378/chest.104.1.478325116
- Piguet PF, Vesin C, Grau GE, Thompson RC. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine. 1993;5(1):57–61. doi:10.1016/1043-4666(93)90024-y7683505
- Cavalli G, Fallanca F, Dinarello CA, Dagna L. Treating pulmonary silicosis by blocking interleukin 1. Am J Respir Crit Care Med. 2015;191(5):596–598. doi:10.1164/rccm.201412-2150LE25723826
- Sommariva M, Le Noci V, Bianchi F, et al. The lung microbiota: role in maintaining pulmonary immune homeostasis and its implications in cancer development and therapy. Cell Mol Life Sci. 2020;77(14):2739–2749. doi:10.1007/s00018-020-03452-831974656
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of Commensal Microflora by Toll-Like Receptors Is Required for Intestinal Homeostasis. Cell. 2004;118(2):229–241. doi:10.1016/j.cell.2004.07.00215260992
- Yang D, Chen X, Wang J, et al. Dysregulated Lung Commensal Bacteria Drive Interleukin-17B Production to Promote Pulmonary Fibrosis through Their Outer Membrane Vesicles. Immunity. 2019;50(3):692–706.e697. doi:10.1016/j.immuni.2019.02.00130824326
- O’Dwyer DN, Armstrong ME, Trujillo G, et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(12):1442–1450. doi:10.1164/rccm.201304-0760OC24070541
- Hodgson U, Pulkkinen V, Dixon M, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79(1):149–154. doi:10.1086/50463916773575
- Pulkkinen V, Bruce S, Rintahaka J, et al. ELMOD2, a candidate gene for idiopathic pulmonary fibrosis, regulates antiviral responses. FASEB J. 2010;24(4):1167–1177. doi:10.1096/fj.09-13854519966137
- Azuma A, Li YJ, Abe S, et al. Interferon-{beta} inhibits bleomycin-induced lung fibrosis by decreasing transforming growth factor-{beta} and thrombospondin. Am J Respir Cell Mol Biol. 2005;32(2):93–98. doi:10.1165/rcmb.2003-0374OC15557019
- Raghu G, Brown KK, Bradford WZ, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350(2):125–133. doi:10.1056/NEJMoa03051114711911
- Vergnon JM, Vincent M, de Thé G, Mornex JF, Weynants P, Brune J. Cryptogenic fibrosing alveolitis and Epstein-Barr virus: an association? Lancet. 1984;2(8406):768–771. doi:10.1016/s0140-6736(84)90702-56148520
- Kelly BG, Lok SS, Hasleton PS, Egan JJ, Stewart JP. A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002;166(4):510–513. doi:10.1164/rccm.210305812186829
- Wootton SC, Kim DS, Kondoh Y, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1698–1702. doi:10.1164/rccm.201010-1752OC21471095
- Yonemaru M, Kasuga I, Kusumoto H, et al. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur Respir J. 1997;10(9):2040–2045. doi:10.1183/09031936.97.100920409311499
- Malizia AP, Keating DT, Smith SM, Walls D, Doran PP, Egan JJ. Alveolar epithelial cell injury with Epstein-Barr virus upregulates TGFbeta1 expression. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L451–460. doi:10.1152/ajplung.00376.200718621908
- Lok SS, Haider Y, Howell D, Stewart JP, Hasleton PS, Egan JJ. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J. 2002;20(5):1228–1232. doi:10.1183/09031936.02.0027290212449178
- Vannella KM, Luckhardt TR, Wilke CA, van Dyk LF, Toews GB, Moore BB. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(5):465–477. doi:10.1164/rccm.200905-0798OC20185751
- Ebrahimi B, Dutia BM, Brownstein DG, Nash AA. Murine gammaherpesvirus-68 infection causes multi-organ fibrosis and alters leukocyte trafficking in interferon-gamma receptor knockout mice. Am J Pathol. 2001;158(6):2117–2125. doi:10.1016/s0002-9440(10)64683-411395389
- Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79(11):6890–6899. doi:10.1128/JVI.79.11.6890-6899.200515890928
- Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc. 2007;4(3):252–257. doi:10.1513/pats.200701-018AW17607008
- Han S, Mallampalli RK. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann Am Thorac Soc. 2015;12(5):765–774. doi:10.1513/AnnalsATS.201411-507FR25742123
- Takezaki A, Tsukumo S, Setoguchi Y, et al. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J Exp Med. 2019;216(12):2724–2735. doi:10.1084/jem.2018235131601679
- Doubková M, Staňo Kozubík K, Radová L, et al. A novel germline mutation of the SFTPA1 gene in familial interstitial pneumonia. Hum Genome Var. 2019;6(1):12. doi:10.1038/s41439-019-0044-z30854216
- Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–59. doi:10.1016/j.ajhg.2008.11.01019100526
- McCormack FX, King TE Jr, Voelker DR, Robinson PC, Mason RJ. Idiopathic pulmonary fibrosis. Abnormalities in the bronchoalveolar lavage content of surfactant protein A. Am Rev Respir Dis. 1991;144(1):160–166. doi:10.1164/ajrccm/144.1.1602064123
- McCormack FX, King TE Jr, Bucher BL, Nielsen L, Mason RJ. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1995;152(2):751–759. doi:10.1164/ajrccm.152.2.76337387633738
- LeVine AM, Whitsett JA, Gwozdz JA, et al. Distinct Effects of Surfactant Protein A or D Deficiency During Bacterial Infection on the Lung. J Immunol. 2000;165(7):3934. doi:10.4049/jimmunol.165.7.393411034401
- Goto H, Ledford JG, Mukherjee S, Noble PW, Williams KL, Wright JR. The role of surfactant protein A in bleomycin-induced acute lung injury. Am J Respir Crit Care Med. 2010;181(12):1336–1344. doi:10.1164/rccm.200907-1002OC20167853
- Lee JM, Yoshida M, Kim MS, et al. Involvement of Alveolar Epithelial Cell Necroptosis in Idiopathic Pulmonary Fibrosis Pathogenesis. Am J Respir Cell Mol Biol. 2018;59(2):215–224. doi:10.1165/rcmb.2017-0034OC29444413
- Park SW, Lee JM, Yoshida M, et al. Involvement of RIPK3-regulated necroptosis in IPF pathogenesis. Eur Respir J. 2017;50(suppl61):OA4440. doi:10.1183/1393003.congress-2017.OA4440
- Ren J, Deng Y, Xiao B, Wang G, Tao Z. Protective effects of exogenous surfactant protein A in allergic rhinitis: a mouse model. Ann Otol Rhinol Laryngol. 2013;122(4):240–246. doi:10.1177/00034894131220040523697321
- Giannoni E, Sawa T, Allen L, Wiener-Kronish J, Hawgood S. Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34(6):704–710. doi:10.1165/rcmb.2005-0461OC16456184
- Ahn M-H, Park B-L, Lee S-H, et al. A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir Res. 2011;12(1):73. doi:10.1186/1465-9921-12-7321649933
- Vasakova M, Sterclova M, Matej R, et al. IL-4 polymorphisms, HRCT score and lung tissue markers in idiopathic pulmonary fibrosis. Hum Immunol. 2013;74(10):1346–1351. doi:10.1016/j.humimm.2013.07.01123911740
- Kishore A, Žižková V, Kocourková L, et al. Association Study for 26 Candidate Loci in Idiopathic Pulmonary Fibrosis Patients from Four European Populations. Front Immunol. 2016;7:274. doi:10.3389/fimmu.2016.0027427462317
- Xin L, Jiang M, Su G, et al. The association between transforming growth factor beta1 polymorphism and susceptibility to pulmonary fibrosis: A meta-analysis (MOOSE compliant). Medicine. 2018;97(37):e11876–e11876. doi:10.1097/MD.000000000001187630212926
- Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45(10):654–656.18835860
- Korthagen NM, van Moorsel CHM, Barlo NP, Kazemier KM, Ruven HJT, Grutters JC. Association between variations in cell cycle genes and idiopathic pulmonary fibrosis. PLoS One. 2012;7(1):e30442–e30442. doi:10.1371/journal.pone.003044222291954
- Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–579. doi:10.1056/nejm20010222344080511207353
- Lawson WE, Grant SW, Ambrosini V, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977–980. doi:10.1136/thx.2004.02633615516475
- Campo I, Zorzetto M, Mariani F, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res. 2014;15(1):43. doi:10.1186/1465-9921-15-4324730976
- Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi:10.1056/NEJMoa06615717392301
- Alder JK, Chen JJL, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi:10.1073/pnas.080428010518753630
- Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7557. doi:10.1073/pnas.070100910417460043
- Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147(5):1361–1368. doi:10.1378/chest.14-194725539146
- Kropski JA, Mitchell DB, Markin C, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014;146(1):e1–e7. doi:10.1378/chest.13-222424504062
- Cogan JD, Kropski JA, Zhao M, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191(6):646–655. doi:10.1164/rccm.201408-1510OC25607374
- Kannengiesser C, Borie R, Ménard C, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015;46(2):474–485. doi:10.1183/09031936.0004011526022962
- Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–517. doi:10.1038/ng.327825848748