
Abstract
Fibrosis is the final stage of the development of chronic inflammation. It is characterized by excessive deposition of the extracellular matrix, leading to tissue structure damage and organ dysfunction, which is a serious threat to human health and life. However, the molecular mechanism of fibrosis is still unclear. Inflammasome is a molecular complex of proteins that has been becoming a key innate sensor for host immunity and is involved in pyroptosis, pathogen infection, metabolic syndrome, cellular stress, and tumor metastasis. Inflammasome signaling and downstream cytokine responses mediated by the inflammasome have been found to play an important role in fibrosis. The inflammasome regulates the secretion of IL-1β and IL-18, which are both critical for the process of fibrosis. Recently, researches on the function of inflammasome have attracted extensive attention, and data derived from these researches have increased our understanding of the effects and regulation of inflammasome during fibrosis. In this review, we emphasize the growing evidence for both indirect and direct effects of inflammasomes in triggering fibrosis as well as potential novel targets for antifibrotic therapies.
Keywords:
Introduction
Inflammasome is an important component of the innate immune response. By directly identifying the pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), it serves as a platform for the activation of the cysteine protease caspase-1 and cleaves the pre-forms of the cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) into their active and secreted forms.Citation1 Inflammasome regulates the secretion of IL-1β and IL-18, both of which are vital for repairing of damaged tissues and play an important role in fibrosis.Citation2,Citation3 Fibrosis is the last stage of a chronic inflammatory response caused by factors such as chemical damage, autoimmune reactions, radiation, allergic reactions, and infections. It can occur in many organs and is characterized by excessive deposition of the extracellular matrix (ECM). Continuous progress can cause damage to structure and function of organ, and even failure, seriously threatening human health.Citation4–Citation6 At present, there is no effective method for the treatment of fibrotic diseases, so further research on the role of inflammasomes in these pathologies is of great significance for the discovery of new therapeutic drugs.Citation7 Thus, in this review, we convey our understanding of the important roles of inflammasomes in the process of fibrotic diseases, assessing the potential of this pathway as a target in fibrotic disease therapies and drug development.
Components of Inflammasomes
Inflammasomes, a group of protein complexes composed of proteins with many different functions, are widely found in cells.Citation4 The inflammasomes are mainly composed of three parts: pattern recognition receptors (PRRs), apoptosis-associated speck-like protein containing a CARD (ASC) and procaspase-1.Citation8,Citation9 The innate immune system has multiple families of germ-line-encoded PRRs. According to the different specific domains contained in PRRs, it is divided into three categories: nucleotide-binding oligomerization domain-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs) and the pyrin receptor, all of these contribute to immune activation in response to various stimuli.Citation10,Citation11
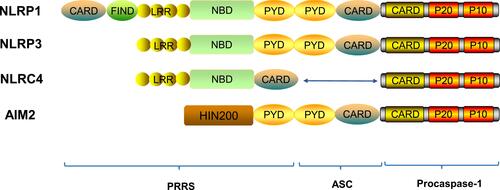
At present, most of researches about PRRs are focusing on NLRs and ALRs. NLRs mainly consists of three parts: the N-terminal contains a pyrin domain (PYD) or a caspase-activating and recruitment domain (CARD), the center contains a nucleotide-binding domain (NBD) and the C-terminal contains a leucine-rich repeat (LRR).Citation12 NLRs can be further classified according to the difference of N domains, the PYD domains structure as NLRPs (NLRP1 ~ 14), the CARD domains structure as NLRCs (NOD1 ~2, NLRC3 ~5). NLRCs contains a CARD that can directly interact with the CARD of caspase-1 in the absence of ASC.Citation13,Citation14 ALRs are mainly composed of N terminal containing PYD domain and C terminal containing a hematopoietic interferon-inducible nuclear protein with a 200-amino–acid repeat (HIN200) domain.Citation15–Citation17 Procaspase-1 is a monomeric zymogen closely related to cell apoptosis,Citation18 which is mainly composed of a CARD domain, a P20 subunit and a P10 subunit containing a cysteine activation site.Citation19 ASC has two domains, an N-terminal PYD domain and a C-terminal CARD domain.Citation20 ASC associates members of the NLR family with procaspase-1, uses its PYD to interact with the PYD of NLR, and uses its CARD to interact with CARD of procaspase-1 to participate in the activation of caspase-1 and the assembly of inflammasomes.Citation21,Citation22 ASC activates caspase-1 and induces the secretion of important pro-inflammatory cytokines such as IL-1β and IL-18 to involve in the initiation and development of inflammation ().Citation20,Citation23
Figure 1 Structures of inflammasome proteins. The NLR family members include NLRP1, NLRP3, NLRC4 etc. They all contain a nucleotide-binding domain (NBD), carboxy-terminal leucine-rich repeat (LRR), and can contain either a pyrin domain (PYD) or a caspase activation and recruitment domain (CARD) or both. The AIM2 is characterized by having, in addition to a PYD, a HIN200 domain, which is involved in ligand binding. FIND, function-to-find domain.
Activation of Inflammasomes
Inflammasomes have been recognized for their crucial role in host defence against pathogens. The maturation and release of IL-1β and IL-18 caused by the activation of inflammasome play a role in many diseases.Citation24,Citation25 Therefore, the strict control of inflammasomes assembly and activation is essential to allow the immune system to initiate inflammatory responses while avoiding tissue damage. Current researches have confirmed the role of NLRP3 and AIM2 inflammasomes in driving collagen deposition in tissues and activating caspase-1 in wound healing.Citation26
Multiple receptors can sense PAMPs and DAMPs to activate inflammatory cascades. Due to the alterations in gut microbiota composition or the increased intestinal permeability, PAMPs are mostly derived from the gut, such as lipopolysaccharide (LPS), bacterial flagellin, peptidoglycan fungal chitin and oomycete glucans.Citation27 DAMPs include adenosine triphosphate, uric acid, cholesterol crystals, DNA fragments and fatty acids, which are mainly derived from damaged cells. The abundance of DAMPs depends on the type of injury and the metabolic event.Citation28 The NLRP3 inflammasome is the most widely studied inflammasomes, which can sense multiple warning signals of PAMPs and DAMPs from endogenous and exogenous sources.Citation29 As the NLRP3 inflammasomes assemble in response to various PAMPs and DAMPs, it is likely that these sense the common cellular signals induced by these molecules, rather than directly interacting with all of these triggers. Recent studies have reported that the activation of NLRP3 inflammasome is mainly through three modes: potassium ion outflow, production of reactive oxygen species (ROS) and lysosomal damage release cathepsin B.Citation30–Citation32 The activation of NLRP3 inflammasome is mainly accomplished in two steps (). The first step, called the priming phase of inflammasome activation, is also the first signal. In this step, microbial molecules or endogenous factors promote the expression of NLRP3, pro-IL-1β, and pro-IL-18 through the intracellular nuclear transcription factor kappa B (NF-κB) signaling pathway.Citation33,Citation34 The second step is the activation stage of inflammasome, also known as the second signal. NLRP3 is activated in response to cell stress and injury, leading to the recruitment of ASC and procaspase-1.Citation35 At the same time, the three parts of the NLRP3 inflammasome are assembled into a stable complex.Citation36 Procaspase-1 generates large P20 subunits and small P10 subunits from proteolysis, as well as removal of the N-terminal CARD domain. The dimerization of P20 and P10 produces the catalytically active caspase-1.Citation37 Inflammasome-activated caspase-1 cleaved its substrates, pro-IL-1β and pro-IL-18, at recognition sites near aspartic acid residues, producing mature IL-1β and IL-18.Citation38
Figure 2 Mechanism of NLRP3 inflammasome activation. NLRP3 inflammasome activation requires two signals to mediate both IL-1β and IL-18 production, including the priming and the activation signals. The NLRP3 inflammasome is activated by a variety of the pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). The first signal activates the NF-κB pathway, leading to upregulation of pro-IL-1β, pro-IL-18 and NLRP3 protein levels. The second signal is activated in response to various stimuli (eg, K+ efflux, ROS and cathepsin B) and promotes the assembly of ASC and pro-caspase-1, leading to activation of the NLRP3 inflammasome complex. The activation drives NLRP3 inflammasome to mediate active caspase-1 production, followed by the maturation and secretion of IL-1β and IL-18. Active caspase-1 also cleaves gasdermin D (GSDMD), which allows the N-terminal domain of GSDMD to form pores in the plasma membrane. This facilitates IL-1β and-18 release and causes a lytic form of cell death (pyroptosis), which supports inflammation.
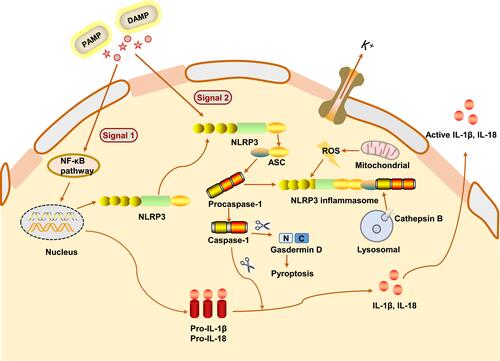
Besides the cytokine cleavage, the assembly of the NLRP3 inflammasome after its activation and consequent caspase-1 autoclavation, resulting in the cleavage of a pore-forming protein called gasdermin D (GSDMD), and leading to a programmed type of cell death called pyroptosis that is characterized by cell swelling followed by lysis and ending with the release of the intracellular contents.Citation39
Additionally, the NLRP3 inflammasome can also be activated through a noncanonical signaling pathway in which caspase-11 recognize LPS from gram-negative bacteria directly and cleave the GSDMD to induce pyroptosis, thereby promoting the release of inflammatory mediators, including IL-1β and IL-18.Citation40 Caspase-11 could also cleave the pannexin 1 channel protein, leading to a drop in intracellular K+ levels, which then induces activation of the NLRP3 inflammasome.Citation41
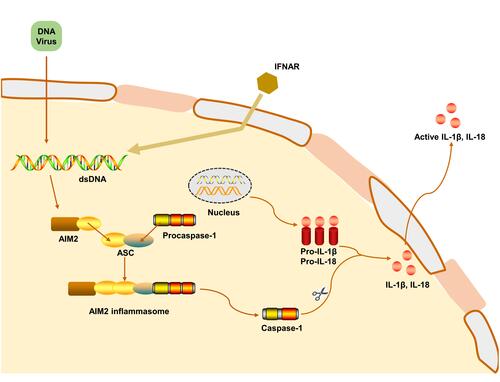
The ability of intracellular DNA to cause immune responses has led to the discovery of AIM2.Citation32 AIM2 is a cytosolic innate immune receptor which recognizes double-stranded DNA (dsDNA) released during cellular perturbation and pathogenic assault.Citation16 AIM2 consists of a HIN200 domain and a PYD.Citation42,Citation43 Structural analysis demonstrated that the positively charged HIN200 domain embraces the dsDNA, while PYD recruits ASC to form a PYD-PYD connection, and allows the CARD domain of ASC to form a complex with procaspase-1.Citation44,Citation45 AIM2 activation initiates the assembly of the AIM2 inflammasome, which is an inherent immune complex that can lead to the activation of inflammatory caspases. AIM2 inflammasome has relatively loose requirements for initiators, such as the cytoplasmic dsDNA of viruses, bacteria or the host itself.Citation15,Citation46 Therefore, AIM2 inflammasome is becoming key guardians of cell integrity, which contributes to the host defense against a subset of bacterial and viral pathogens ().
Figure 3 Mechanism of AIM2 inflammasome activation. AIM2 is activated by cytosolic bacteria and DNA viruses. During bacterial infection, type I interferon provides a feedback loop and activates type I interferon receptor (IFNAR). Type I interferon-signaling aids in the cytosolic access of DNA leading to the activation of AIM2. The mechanism leading to viral DNA recognition by AIM2 is less clear. The HIN-200 domain of AIM2 directly binds dsDNA, while the pyrin domain recruits ASC. Caspase-1 is recruited to ASC inflammasome to mediate pro-IL-1β and pro-IL-18 processing and induce apoptosis.
Effects and Mechanisms of Inflammasome in Fibrosis
The inflammasomes are a complex platform that is responsible for the activation of cytosolic polyprotein caspase, which is involved in the maturation and release of IL-1β and IL-18 to systemically establishes an inflammatory environment. The typical inflammasome/ASC/caspase-1/IL-1β/IL-18 axis promotes the pathophysiology of various diseases by mediating inflammation, and this is likely a critical priming mechanism for fibrosis. As general accepted, besides inducing numerous inflammatory chemokines and cytokines, NF-κB also is important for NLRP3 inflammasome priming and assembly. Likewise, activated inflammasome can regulate NF-κB signaling pathway to establish a circulation in excessive and detrimental inflammation. In addition, the inflammasome is also closely associated with mitochondrial regulation and TGF-β/Smad signaling, which is also related to fibrosis. Therefore, the inflammasome pathway may serve as a valuable therapeutic target for the treatment of fibrosis.
The NF-κB Signaling Pathway
NF-κB is a family of inducible transcription factors responsible for regulating the induction and advancement of inflammatory responses. Increased NF-κB activity is predominant in many diseases, including fibrosis.Citation47–Citation49 Studies have shown that activated NF-κB is translocated to the nucleus to regulate the expression of target genes and the release of inflammatory mediators causes a persistent inflammatory response, leading to excessive ECM accumulation.Citation50 In the process of activating NLRP3 inflammasome, a variety of signals activate cell surface receptors such as toll-like receptors (TLRs), this leads to the upregulation of the transcription and expression of NLRP3 and other key pro-inflammatory components, including pro-IL-1β and pro-IL-18.Citation51 The persistent activation of the NLRP3 inflammasome leads to the abundant secretion of IL-1β and IL-18 and initiation of pyroptosis, all of which are major contributors to the formation of fibrotic lesions.Citation52,Citation53 IL-1β is a potential mediator of crosstalk between inflammation and fibrosis to produce a positive feedback cycle, which controls the distribution of secretory cytokines in cystic fibrosis.Citation54 In addition, the absence or inhibition of IL-18 can alleviate renal and cardiac fibrosis in corresponding mouse models.Citation55,Citation56 IL-1β and IL-18 have also been known as the“prototypic alarm cytokine”, recognizing its pivotal role in innate immunity.Citation57 Once released, IL-1β and IL-18 bind to the IL-1 receptor (IL-1R) and IL-18 receptors (IL-18R), which further activate the NF-κB signaling pathway and induce the expression of a variety of inflammation-related proteins.Citation58 In the fibrosis state, the inflammasome promotes the generation of a considerable amount of IL-1β and IL-18 through NF-κB signaling pathway and then further activate NF-κB signaling pathway to induce the expression of multiple inflammation-related proteins, including upregulating collagen content and ECM production.Citation58,Citation59 Based on these findings, we have reason to believe that the inflammasome may have an important role in the occurrence and development of fibrosis through the NF-κB pathway.
The ROS Signaling Pathway
It is now recognized that oxidative stress and inflammation are closely related to fibrotic diseases, but the mechanisms are not fully understood. Nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) plays an essential role in ROS-mediated innate immunity and host defense against pathogens to induce ROS and mediate downstream signaling in the cell.Citation60 There is further evidence to suggest that NOX4 levels are particularly correlated with the myofibroblast phenotype leading to fibrosis and NOX4–derived ROS plays a significant role in renal, lung, and liver fibrosis.Citation61–Citation64
Almost all NLRP3 activators can induce the production of ROS.Citation65 Therefore, the orchestration between ROS and inflammasome activation needs to be further elucidated in fibrosis. Thioredoxin-interacting protein (TXNIP) is upregulated by ROS and plays an important role in inflammation mediated by oxidative stress. TXNIP has been recognized as a critical regulator of the NLRP3 inflammasome, which leading to conformational changes and activation of the NLRP3 inflammasome.Citation65,Citation66 However, recent studies have demonstrated that NLRP3 seems to have a certain effect on the production of ROS. NLRP3 in renal tubule cells is re-localized from the cytoplasm to mitochondria during hypoxia and bound to mitochondrial antiviral signaling proteins (MAVS). The absence of NLRP3 or MAVS in renal tubule cells under hypoxia reduced the production of mitochondrial ROS and the depolarization of mitochondrial membrane potential.Citation67
To further evaluate the direct role of NLRP3 in triggering fibrogenesis, a recent study found that tubulointerstitial fibrosis was remarkably attenuated in NLRP3 deletion mutants as evidenced by the blockade of ECM deposition. Meanwhile, NLRP3 deletion mutants exhibit normal mitochondrial morphology and DNA copy numbers in renal tubule cells, suggesting that NLRP3 deficiency can improve mitochondrial abnormality.Citation68 In addition, researches have demonstrated that NLRP3 deletion can ameliorate the morphological and functional damage of mitochondria caused by unilateral ureter obstruction (UUO) and alleviate renal fibrosis.Citation69 Mitochondrial dysfunction is associated with increased oxidative stress and inflammation, which results in increased ROS generation and release of proapoptotic products.Citation70 Taken together, these findings indicate that NLRP3 and ROS exhibit a complex functional relationship during the progression of fibrosis.
The TGF-β/Smad Signaling Pathway
It is believed that TGF-β/Smad signaling plays an important role in fibrosis, in which it not only regulates the production and degradation of ECM but also participates in the epithelial-to-mesenchymal transition (EMT) to form myofibroblasts.Citation71 TGF-β exists in three main isoforms (TGF-β1, TGF-β2, and TGF-β3) with TGF-β1 being the most common.Citation72 For instance, mice overexpressing an active form of the TGF-β1 in the liver have shown to develop progressive liver and renal fibrosis.Citation73,Citation74 In TGF-β/Smad signaling, TGF-β1 initiates intracellular signaling by binding to TGF-β type I receptors (TβRI) and promotes a phosphorylation of TGF-β type II receptor (TGF-βRII), resulting in phosphorylation of Smad2 and Smad3. Subsequently, the activated Smad2 and Smad3 form an oligomeric complex with Smad4 to reach the nucleus and start the transcription of genes related to different cellular functions,Citation75 thereby participating in the pathological process of fibrosis.
However, the relationship between TGF-β and the NLRP3 inflammasome is not well understood. The recognition that activation of the inflammasome complex has the function of activating IL-1β, and IL-1β in turn leads to increased TGF-β1 expression, bring the idea that the inflammasome complex triggers EMT in fibrosis through TGF-β1 produced by IL-1β.Citation76,Citation77 Furthermore, in a model of bleomycin-induced lung fibrosis, that the activation of the NLRP3 inflammasome leads to EMT via TGF-β1, and once NLRP3 is silenced there are reduced TGF-β1 levels and the EMT does not occur.Citation78 The same result was reached in a high glucose-induced renal tubular fibrosis.Citation79 Recent findings suggest that NLRP3/ASC expression was increased in renal fibrosis induced by high UA and with activation of both inflammasome-related caspase-1 and inflammasome unrelated p-Smad 2/3 pathways. Ultrastructural co-localization of NLRP3 and Smad 2/3 indicated physical interaction between the two molecules.Citation80 Additionally, the expression of TGF-β1 and connective tissue growth factor (CTGF) and the phosphorylation of Smad2/3 are dramatically reduced in the kidney of NLRP3–/– diabetic mice in a manner that can ameliorate renal function.Citation81 Consistent with these findings, overexpression of NLRP3 in 293T cells resulted in increased Smad3 phosphorylation and activity.Citation82 It was demonstrated that the involvement of the NLRP3 inflammasome in inducing fibrosis may be related to the TGF-β/Smad signaling.
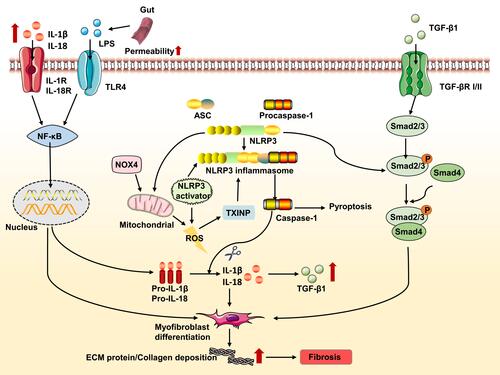
Besides the Smad-dependent pathways, TGF-β1 also activates Smad-independent pathways such as mitogen-activated protein kinase and phosphatidylinositol-3-kinase pathways.Citation83 The interaction between the NLRP3 and the Smad-independent pathways is not fully understood and requires more studies. Based on these findings, we speculate that a complex interaction exists among the NLRP3 inflammasome and these signaling pathways during the progression of fibrosis ().
Figure 4 Effect and regulation of the NLRP3 inflammasome during fibrosis. Lipopolysaccharide (LPS) and other compounds induces TLR4 (toll-like receptor 4)-dependent activation of nuclear transcription factor kappa B (NF-κB), resulting in NLRP3, pro-IL-1β and pro-IL-18 expression. For instance, alcohol consumption increases permeability of the intestine to bacterial endotoxin that in turn, elevates LPS levels during alcoholic liver disease. The increase in NADPH oxidase 4 (NOX4) levels promotes mitochondrial reactive oxygen species (ROS) that leads to inflammasome activation. Thioredoxin-interacting protein (TXNIP) modulates the biological structure of NLRP3 leading to NLRP3 inflammasome assembly. Additionally, NLRP3 may damage the function of mitochondrial and induce mitochondrial ROS production, ultimately promoting NLRP3 inflammasome activation. Increased NLRP3 also involved in the transforming growth factor (TGF-β)-mediated Smad2/3 phosphorylation. IL-1β and IL-18 secreted by the inflammasome can stimulate myofibroblasts to remain activated; with the excessive extracellular matrix (ECM)-producing capacity of these cells eventually contributing to fibrosis. The mature form of IL-1β and IL-18 reaches its receptor to promote the transcription of NF-κB and increase the expression of TGF-β1. The secreted TGF-β1 binds to its receptor, triggering the Smad-dependent pathway and promoting the progression of fibrosis.
Inflammasomes in the Progression of Fibrotic Diseases
Fibrosis is considered as a complex form of tissue damage that is ubiquitous in the late stages of many diseases. It is also associated with a high mortality, which is characterized by deposition of ECM, including appearance of collagen, fibronectin and glycoproteins.Citation84 Fibrosis is the ultimate result of chronic inflammatory reactions caused by various stimuli including persistent infection, autoimmune response, allergic reactions, chemical insults, radiation and tissue damage.Citation85 Strategies to deal with fibrotic diseases rely on attempts to neutralize the aggressor, which forces clinicians to try to find ways to control and reverse fibrosis. In recent years, more and more studies have found that inflammasome plays a role in fibrosis, thus affecting the fibrosis process. The activation of inflammasomes such as NLRP3 and AIM2 play a key role in the process of tissue fibrosis.Citation86 The proinflammatory cytokines such as IL-1β and IL-18 secreted by the inflammasomes can aggravate the development of fibrosis diseases, and eventually lead to organ structure destruction and dysfunction ().Citation87
Table 1 Role of Inflammasomes in Different Types of Fibrosis Development
Liver Fibrosis
After activation of hepatic stellate cells (HSCs), they are transformed into myofibroblasts, leading to the deposition of ECM and the formation of liver fibrosis.Citation53 It is usually caused by persistent chronic liver injury, including viral hepatitis, long-term drinking, autoimmune diseases and non-alcoholic fatty liver disease (NAFLD).Citation54 The morbidity and mortality of liver fibrosis are very high and the trend is increasing year by year, which has become an urgent medical problem.Citation55 Effectively inhibiting the occurrence and development of liver fibrosis is of great significance for the treatment of liver diseases.
Several studies have shown that the expression level of inflammasomes increased in liver fibrosis including NLRP1, NLRP3, and AIM2, especially in HSCs, which regulate a variety of functions of HSCs and are associated with HSCs’ activation.Citation88 The present study suggested platelet-derived growth factor (PDGF), a potent mitogenic molecule responsible for HSCs’ activation, can enhance phosphorylation of PDGF-β receptor (PDGF-βR) and its downstream signal molecules NLRP3 and caspase-1 in HSCs. Therefore, we extrapolated that PDGF increased expression of inflammatory cytokine were associated with activation of the PDGF-βR/NLRP3/caspase-1 pathway in cultured HSCs.Citation89 Additional Studies have shown that NLRP3L351P/+ Lrat Cre mice (a mutant strain of mice in which hepatic expression of NLRP3L351P is restricted to HSC) and NLRP3−/− mice, intraperitoneal injection of lipopolysaccharide (LPS) induces the activation of NLRP3 inflammasome, and the mutant mice showed fibrosis, and the expression of collagen I, IL-1β, and alpha-smooth muscle actin (α-SMA) in the isolated HSC was significantly enhanced, but not the NLRP3−/− mice. It is confirmed that NLRP3 inflammasome has a direct role in the activation of HSCs and the development of liver fibrosis.Citation90
Moreover, after activation of inflammasome, caspase-1 cleaves pro-IL-18 and pro-IL-1β into matured IL-18 and IL-1β. As two downstream cytokines of inflammasome, IL-18 and IL-1β can also promote the proliferation and differentiation of HSCs.Citation91 Studies in vitro have shown that IL-1β promotes proliferation and differentiation of HSCs with a significant increase in the levels of fibrogenic markers, including tissue inhibitor of metalloproteinases-1, collagen I and TGF-β, and decreases the expression of BAMBI, a negative regulator of TGF-β signaling.Citation92–Citation94 In vivo studies using either IL-1β or IL-1α knockout mice have shown that the IL-1 pathway deficiency protects against the progression of steatosis to steatohepatitis and liver fibrosis.Citation95 In addition, pyroptosis can be induced by caspase-1 followed by the activation of NLRP3 inflammasome. After that, cytoplasmic components including IL-1β, IL-18, TGF-β, and CTGF will be released into the extracellular environment, thereby promoting the development of liver fibrosis.Citation96,Citation97
Based on this, we continue to explore the mechanism of NLPR3 inflammasome activation in fibrosis. The renin-angiotensin system (RAS) has regulatory effects on liver fibrosis.Citation98 Ang II is a key molecule of RAS, which can induce the production of NOX4/mitochondrial-derived ROS and aggravate liver fibrosis. Studies have shown that the activation of NLRP3 inflammasome is closely related to NOX4/mitochondrial-derived ROS. Ang II stimulation-induced ECM deposition, HSC proliferation and ROS production, and the inhibition of Ang II can alleviate liver fibrosis.Citation99 Based on these findings, we speculate that a complex interaction exists among ROS and NLRP3 inflammasome during the progress of liver fibrosis.
Furthermore, NF-κB signaling is also important for the process of liver fibrosis, while NLRP3 inflammasome activation is accompanied by the upregulation of NF-κB signaling. Study on the development of NAFLD to fibrosis found that TLR4 inhibitors reversed palmitic acid-induced TLR4 and NF-κB activity and attenuated palmitic acid-mediated NLRP3 inflammasome activation. Inhibition of NF-κB reversed NLRP3 inflammasome activation and HSCs phenotype changes.Citation100 Taken together, TLR4 activation is necessary to induce NLRP3 inflammasome activation in HSCs, and NF-κB is an important downstream effector that promotes this effect. Therefore, the results suggest that blocking TLR4/NF-κB signaling pathway to reduce NLRP3 inflammasome activation may be a strategy for the treatment of NAFLD. Other studies have shown that endoplasmic reticulum stress (ERS) can induce the activation of NLRP3 inflammasome, which ultimately leads to hepatocyte death and NAFLD.Citation101
Alcoholic liver disease (ALD) is a general term used to refer to alcohol-related liver injuries, includes a range of pathologies ranging from mild steatosis to fulminant hepatitis, to the silent development of fibrosis.Citation28 The factors which lead to the development of these pathologies in certain people are not well understood, it is obvious that inflammation and fibrosis play a role in the ultimate organ dysfunction. Ethanol and its metabolic derivatives (especially acetaldehyde) can change the intracellular signal transduction pathway, which leads to the destruction of epithelial tight junctions and the increase in the permeability of the adjacent cells to macromolecules, so that bacterial products (such as LPS, bacteria RNA and DNA) from the intestine to the liver and facilitates inflammasomes activation, then inducing the differentiation of HSCs, thereby promoting the production of hepatic fibrotic factors.Citation102,Citation103 The involvement of NLRP3 inflammasome in ALD has been demonstrated by a robust expression of NLRP3, caspase-1 and IL-1β in alcohol-fed mice. Moreover, NLRP3 deficiency prevents the development of alcohol-induced liver inflammation and significantly ameliorated liver damage and steatosis.Citation104,Citation105 Therefore, targeting the inflammasome and IL-1β might represent promising strategies towards novel treatment for ALD.
These results indicate that NLRP3 inflammasome activation plays a potential role in the activation of HSCs and accelerates the pathological process of liver fibrosis. Therefore, inhibiting the activity of NLRP3 inflammasome may become a new potential target for the treatment of liver fibrosis. The relationship between NLRP3 inflammasome and liver fibrosis has not been thoroughly studied. Further research in future is essential to understand the relationship between NLRP3 inflammasome and progression of liver fibrosis.
Renal Fibrosis
Renal fibrosis is the basic pathological change of most chronic kidney disease progressing to end-stage renal disease and its pathogenesis is regulated by multiple signaling pathways. The various types of chronic renal diseases, which affect 8–16% of the global population, can all lead to fibrosis.Citation106,Citation107 Increasing evidence strongly indicates that the expression levels of NLRP3 and caspase-1 are significantly elevated in the renal fibrosis patients, suggesting that NLRP3 inflammasome may be activated and participate in the process of renal fibrosis.Citation108 Previous studies have suggested that renal tubular epithelial cells can express and release IL-18, NLRP3 inflammasome and caspase-1 have also been found in renal tubular epithelial cells, indicating that NLRP3 and ASC are expressed in glomerular podocytes.Citation109,Citation110 And in vitro studies have shown that IL-1β secreted by inflammasome could induce the development of chronic kidney disease and the transformation of renal tubular epithelial cells into myofibroblasts.Citation111 Studies of a UUO mouse model and 5/6 nephrectomy mice showed that these mice had increased renal matrix accumulation, increased phosphorylated NF-κB levels, and activated NLRP3 inflammasome.Citation112 Artemisinin may down-regulate the NF-κB/NLRP3 signaling pathway to reduce renal fibrosis in 5/6 nephrectomy rats.Citation113 Taken together, these findings indicate that NLRP3 and NF-κB exhibit a complex functional relationship in renal fibrosis.
ROS can dissociate TXNIP from thioredoxin, and then TXNIP binds to NLRP3 to activate the NLRP3 inflammasome. Accordingly, studies have found that Ginsenoside Rg1 could delay renal aging and inhibit glomerular fibrosis by reducing NOX4-derived ROS generation and down-regulating NLRP3 inflammasome expression.Citation114 Some researchers have found that in UUO renal fibrosis model, the levels of caspase-1, IL-1β and IL-18 in renal are abnormally increased, while at the same time causing the elevated ERS, suggesting that inhibiting the ERS may reduce the activation of NLRP3 inflammasome, thereby improving the development of renal fibrosis.Citation115 The NLRP3 inflammasome is a complex platform, which is involved in the maturation and release of IL-1β and IL-18 and systemically establishes an inflammatory environment within the renal under stimulation appropriately. In addition, certain cytokines can also regulate the activation of NLRP3 inflammasome to participate in renal inflammation and fibrosis. For instance, IL-22 can down-regulate the NLRP3/caspase-1/IL-1β pathway and reduce the expression of fibronectin and collagen IV in mesangial cells induced by high glucose, indicating that IL-22 plays an anti-fibrosis role by suppressing the activation of the NLRP3 inflammasome.Citation116 Therefore, the NLRP3 inflammasome pathway may serve as a valuable prophylactic and therapeutic target for the treatment of renal fibrosis and may also provide a potential target that can be used for the prevention and treatment of this disease.
Additionally, recent findings suggest that expression of AIM2 inflammasome is increased in renal tubular epithelial cells and infiltrating leukocytes derived from patients with diabetic nephropathy or hypertensive nephrosclerosis.Citation117 AIM2 inflammasome deficiency attenuates renal injury, inflammation, and fibrosis in UUO mouse models.Citation118 Findings by these indicate that AIM2 inflammasome may also be involved in the pathogenesis of renal fibrosis.
Lung Fibrosis
Lung fibrosis is manifested by the excessive proliferation of fibroblasts and the deposition of a large number of ECM, which eventually leads to the destruction of lung structure and respiratory failure.Citation119 It is known that inflammation plays an important role in the initiation and development of lung fibrosis. It has been reported that stimulants such as silica asbestos, and bleomycin can activate the NLRP3 inflammasome and damage lung epithelial cells, resulting IL-1β secretion subsequently.Citation120 Sohn et alCitation121 prepared a model of lung fibrosis by using an image-guided small-animal irradiator (the X-RAD 320). After irradiation, study found that radiation can damage the cells that contribute to lung structures (eg, fibroblasts and epithelial cells) and activate NLRP3 inflammasome in macrophages, which is marked by cytokines (IL-4, IL-5 and IL-13) secretion. Ultimately, these cytokines induce pneumonia and fibrosis. There is increasing concern about the role of EMT in lung fibrosis. Many studies have provided controversial inferences about EMT in fibrosis, which may make us suspect the role of inflammasome and EMT in lung fibrosis.Citation122 One study demonstrated that the level of IL-1β and TGF-β1 decreased significantly when NLRP3 inflammasome was silenced. In addition, the E-cadherin level was significantly decreased and the α-SMA expression was markedly increased. Thus, NLRP3 inflammasome was activated in the alveolar epithelial cells and may regulate EMT through TGF-β1.Citation78 Therefore, we believed that targeted inhibition of these pro-inflammatory pathways could significantly reduce fibrosis in normal lung tissues, thereby further improving the therapeutic index of lung cancer treatment.
Currently, accumulating evidence suggests that AIM2 inflammasome-mediated pyroptosis both in macrophages and epithelial cells plays a key role in the development of lung tissue damage.Citation123 Study revealed that andrographolide ameliorates radiation-induced lung injury and fibrosis by suppressing AIM2 inflammasome mediated-pyroptosis in macrophage, identifying AIM2 inflammasome leads to caspase-1 activation and resulting DNA damage induced by radiation.Citation124 Although our knowledge of AIM2 inflammasome is growing, further study still needs to be undertaken to fully understand how the AIM2 inflammasome assembles and activates, as well as its roles in lung fibrosis.
Dermal Fibrosis
Dermal fibrosis is characterized by thickening of the dermis and obstruction of some appendages such as hair follicles, sweat glands, and cutaneous blood vessels, which generally occur in systemic sclerosis (SSc). SSc is an autoimmune disease characterized by inflammation and extensive fibrosis.Citation125,Citation126 We recently found that activation of the inflammasome orchestrates the increased collagen synthesis in SSc fibroblasts. We also found that inhibition of inflammasome signaling significantly abolished myofibroblast phenotype.Citation127 Therefore, we conclude that inflammasome activation plays an important role in the pathogenesis of SSc fibrosis. Based on these findings, Artlett et alCitation128 shown that expression of genes associated with the inflammasome signaling molecules is increased in SSc fibroblasts. In vitro inhibition of innate immunity via caspase-1 leads to a decrease in collagen secretion and expression of α-SMA from myofibroblasts. Furthermore, NLRP3−/− mice and ASC−/− mice were resistant to bleomycin-induced skin fibrosis. Thus, innate immune response contributes to SSc fibrosis via activation of the inflammasome and caspase 1. Martínez-Godínez et alCitation129 collected 42 patients with SSc (21 limited SSc and 21 diffuse SSc) and 13 healthy individuals for skin biopsy. The results proved that the skin of the two subtypes of SSc showed significantly increased expression of NLRP3, caspase-1, IL-1β, IL-18 and endothelin-1 compared to healthy individuals. This study identified NLRP3 over-expression in skin of patients with SSc, suggesting that NLRP3 inflammasome plays an important role in the pathophysiology of skin fibrosis in human SSc. In conclusion, these studies demonstrate that the inflammasome platform in the skin of patients with SSc is an important sensor of fibrosis and vascular damage. The NLRP3 inflammasome and related cytokine agonists/antagonists could be therapeutic targets for limiting fibrosis in SSc.
Myocardial Fibrosis
Myocardial fibrosis is cardiac interstitial remodeling, characterized by excessive proliferation of cardiac interstitial fibroblasts, ECM deposition and abnormal distribution of collagen.Citation130 Established fibrosis is a key factor leading to tissue stiffness and dysfunction, which ultimately leads to heart failure. It has increasingly been recognized that the ability of the NLRP3 inflammasome to continuously secrete pro-inflammatory IL-1β and IL-18 from infiltrating and resident cells in the damaged heart, resulting in abnormal tissue remodeling and the fibrosis progression.Citation131,Citation132
Expression of the myofibroblast inflammasomes have been shown to be mediate the initial inflammatory response following myocardial I/R.Citation133 Ang II-induced hypertension was found to associated with increased cardiac inflammation, tissue remodeling and fibrosis induced by myofibroblasts, and increased expression of NLRP3 and activated caspase-1 in the myocardium.Citation134 NLRP3−/– mice were protected from Ang II-induced cardiac fibrosis demonstrating preserved cardiac architecture and reduced levels of collagen I, further highlighting the critical role of NLRP3 in the pathophysiology of myocardial fibrosis.Citation135 NLRP3 has been shown to promote human cardiac fibroblasts proliferation and ECM production directly, contributing to adverse tissue remodeling.Citation52 Taken together, these studies suggest that many pro-inflammatory and pro-fibrotic mediators contribute to the progression of cardiac fibrosis by prolonging the expression and activation of NLRP3 inflammasomes in cardiac myofibroblast.
Previous studies have indicated that neurohumoral factors induce cardiovascular diseases through the NLRP3 inflammasome-related pathway. Pan et alCitation136 reported that low-dose triptolide could attenuate mouse cardiac fibrosis via inhibition of the NLRP3 inflammasome-related profibrotic pathway, including down-regulating the expression of NLRP3 inflammasome components and interrupting NLRP3 inflammasome assembly. Together, these effects decrease the activation of the NLRP3/TGF-β1/Smad pathway. Therefore, we speculate that activation of the NLRP3/TGF-β1/Smad pathway may be related to myocardial fibrosis. Furthermore, findings showed that serelaxin (RLX) can inhibit the TGF-β1/IL-18/TLR-4/NLRP3 pathway via a neuronal NOS-dependent mechanism on cardiac myofibroblasts to suppress NLRP3 inflammasome priming and activity, which in turn disrupts the profibrotic influence. Proposing that RLX is more efficacious than MCC950 in suppressing the contribution of the NLRP3 inflammasome in myofibroblasts to fibrosis progression.Citation137 Accordingly, these studies indicated that NLRP3 inflammasome is closely related to the occurrence and development of myocardial fibrosis, and the regulation of activity in different signaling pathways needs more experimental studies to prove.
In addition to NLRP3 inflammasome, AIM2 inflammasome is also related to some types of myocardial fibrosis. Increased ROS production activates a series of cytokines and inflammatory-related factors, which promote cardiomyocyte death and fibrosis.Citation138,Citation139 Based on these, Wang et alCitation140 found that AIM2 may be activated in diabetic rats and the high levels of ROS play a promoting role in the activation of AIM2. It was confirmed that AIM2 regulates the process of cell death and fibrosis in the heart tissue of diabetic rats via the GSDMD pathway, which further affects the heart function. The underlying mechanism remains unclear, and further studies are needed to investigate the effects of AIM2 inflammasome in the progression of myocardial fibrosis.
Primary Myelofibrosis
Primary myelofibrosis is a myeloproliferative neoplasm (MPN) caused by the clonal proliferation of abnormal hematopoietic stem cells, which leads to progressive myelofibrosis. Some researchers have found that the gain-of-function mutation JAK2V617F is commonly seen in Philadelphia-chromosome-negative MPN patients. Janus Kinase 2 (JAK2) is an intracellular tyrosine protein kinase that changes the first base GTC the 617th codon from G to T, causing its encoded valine to phenylalanine. Liew et alCitation141 established a novel human cell line D9 using the pluripotent blood cell line ut-7/GM to express JAK2V617F with the addition of tetracycline. The results indicated that the associated inflammatory molecules AIM2, IL-1β and caspase 1- apoptosis-related cysteine peptidase, which were significantly up-regulated in JAK2V617F induced cells. Based on this, we speculate that AIM2 inflammasome is involved in the development of primary myelofibrosis patients with JAK2V617F mutation. To explore a possible association of NLRP3 inflammasome signaling with MPN, Zhou et alCitation142 investigated the expression of selected inflammasome-related genes from bone marrow (BM) cells of 67 MPN patients as well as gene polymorphisms in NLRP3, NF-κB, IL-1β and IL-18. It showed that inflammasome-related genes were highly expressed in BM cells from MPN patients. This research provided NLRP3 inflammasome may be as novel biomarkers and potential targets for MPN.
Conclusions and Perspectives
In summary, we have gained important insights into the role of inflammation and in the development of fibrosis in recent years. The discovery of inflammasomes as protein platforms that control the processing of IL-1β and IL-18 constitutes a milestone in the innate immunology field. When various organs are exposed to various danger signals, inflammasome as key regulators and translation agents, inducing the activation of pro-inflammatory caspase 1, the release of IL-1β and IL-18, as well as triggering the death of pyroptotic cell death. Accumulating evidence from various studies has revealed the new role of NLRP3 inflammasome and AIM2 inflammasome as regulators of biology and fibrosis, suggesting that this pathway may represent an attractive target for anti-fibrotic therapy. In most fibrotic diseases, as a pattern recognition receptor, inflammasomes can participate in the development of fibrosis by recognizing various abnormal signals, activating TGF-β1 in inflammation, and promote α-SMA and collagen, which contribute to deposits of ECM. As a result, it may be effective to enhance or reduce the activity of inflammasomes or their effector molecules by tailoring therapies for specific types of fibrosis. Antagonists and monoclonal antibodies against inflammasomes are being developed and can be used in the treatment of fibrosis. However, inappropriate use of inflammasomes therapies may increase the maturity of fibrosis, leading to spontaneous inflammation.
Thus, regulation of inflammasomes is essential for determining health and fibrosis conditions. It is necessary to evaluate the role of inflammasomes in different cells at different stages of fibrotic diseases and promote the development of new treatments for various fibrotic diseases. However, many questions remain unanswered, including the biochemical and genetic regulation of these protein complexes, as well as their role in fibrotic diseases. We believe that these may be the focus of inflammasome research in the coming years.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81770605), Program for Young Excellent Talents in Universities of Anhui Province (No. gxyqZD2018024).
Disclosure
The authors report no conflicts of interest in this work.
References
- Evavold CL, Kagan JC. How inflammasomes inform adaptive immunity. J Mol Biol. 2018;430(2):217–237. doi:10.1016/j.jmb.2017.09.019
- Fernandes FP, Leal VNC, Souza de Lima D, et al. Inflammasome genetics and complex diseases: a comprehensive review. Eur J Hum Genet. 2020;28(10):1307–1321. doi:10.1038/s41431-020-0631-y
- Cordero MD, Alcocer-Gomez E. Editorial: inflammasome complex in health and disease: new pharmacological perspectives. Curr Drug Targets. 2017;18(9):996. doi:10.2174/1389450118666161123153230
- Artlett CM. Inflammasomes in wound healing and fibrosis. J Pathol. 2013;229(2):157–167. doi:10.1002/path.4116
- Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. doi:10.1016/j.cell.2016.03.046
- Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta. 2013;1832(7):979–988. doi:10.1016/j.bbadis.2013.03.020
- Gu YJ, Sun WY, Zhang S, Wu JJ, Wei W. The emerging roles of β-arrestins in fibrotic diseases. Acta Pharmacol Sin. 2015;36(11):1277–1287. doi:10.1038/aps.2015.74
- Xiong W, Meng XF, Zhang C. Inflammasome activation in podocytes: a new mechanism of glomerular diseases. Inflamm Res. 2020;69(8):731–743. doi:10.1007/s00011-020-01354-w
- Gong T, Jiang W, Zhou R. Control of inflammasome activation by phosphorylation. Trends Biochem Sci. 2018;43(9):685–699. doi:10.1016/j.tibs.2018.06.008
- Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol. 2018;50:32–38. doi:10.1016/j.coi.2017.10.011
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi:10.1016/j.cell.2014.04.007
- Evavold CL, Kagan JC. Inflammasomes: threat-assessment organelles of the innate immune system. Immunity. 2019;51(4):609–624. doi:10.1016/j.immuni.2019.08.005
- Lupfer C, Kanneganti TD. Unsolved mysteries in NLR biology. Front Immunol. 2013;4:285. doi:10.3389/fimmu.2013.00285
- Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. 2017;130(23):3955–3963. doi:10.1242/jcs.207365
- Yang Q, Liu R, Yu Q, Bi Y, Liu G. Metabolic regulation of inflammasomes in inflammation. Immunology. 2019;157(2):95–109. doi:10.1111/imm.13056
- Wang B, Tian Y, Yin Q. AIM2 inflammasome assembly and signaling. Adv Exp Med Biol. 2019;1172:143–155.
- Cridland JA, Curley EZ, Wykes MN, et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol. 2012;12:140. doi:10.1186/1471-2148-12-140
- Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50(6):1352–1364. doi:10.1016/j.immuni.2019.05.020
- Shi C, Yang H, Zhang Z. Involvement of nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 inflammasome in the pathogenesis of liver diseases. Front Cell Dev Biol. 2020;8:139. doi:10.3389/fcell.2020.00139
- de Alba E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch Biochem Biophys. 2019;670:15–31. doi:10.1016/j.abb.2019.05.023
- Jin T, Curry J, Smith P, Jiang J, Xiao TS. Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins. 2013;81(7):1266–1270. doi:10.1002/prot.24287
- Guey B, Bodnar M, Manié SN, Tardivel A, Petrilli V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci U S A. 2014;111(48):17254–17259. doi:10.1073/pnas.1415756111
- Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi:10.1038/ncomms4209
- Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi:10.1038/nm.3893
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi:10.1146/annurev-immunol-031210-101405
- Alegre F, Pelegrin P, Feldstein AE. Inflammasomes in Liver Fibrosis. Semin Liver Dis. 2017;37(2):119–127. doi:10.1055/s-0037-1601350
- Richardson MB, Williams SJ. MCL and mincle: c-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front Immunol. 2014;5:288. doi:10.3389/fimmu.2014.00288
- Magdaleno F, Blajszczak CC, Nieto N. Key events participating in the pathogenesis of alcoholic liver disease. Biomolecules. 2017;7(4):9. doi:10.3390/biom7010009
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(9):688. doi:10.1038/nrd.2018.149
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
- Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–1589. doi:10.1038/sj.cdd.4402195
- Wolf AJ, Reyes CN, Liang W, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell. 2016;166(3):624–636. doi:10.1016/j.cell.2016.05.076
- Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi:10.4049/jimmunol.0901363
- Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. doi:10.3390/ijms20133328
- Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
- Ranson N, Kunde D, Eri R. Regulation and sensing of inflammasomes and their impact on intestinal health. Int J Mol Sci. 2017;18(11):2379. doi:10.3390/ijms18112379
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8(6):471–483. doi:10.1016/j.chom.2010.11.007
- Roth S, Ruland J. Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation. Trends Immunol. 2013;34(6):243–250. doi:10.1016/j.it.2013.02.006
- Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
- Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. 2015;45(10):2927–2936. doi:10.1002/eji.201545772
- Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323(5917):1057–1060. doi:10.1126/science.1169841
- Jin T, Perry A, Jiang J, et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36(4):561–571. doi:10.1016/j.immuni.2012.02.014
- Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi:10.1038/nature07725
- Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2018;281(1):99–114. doi:10.1111/imr.12618
- Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402. doi:10.1038/ni.1864
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
- White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
- Luedde T, Schwabe RF. NF-κB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–118. doi:10.1038/nrgastro.2010.213
- Jiang Q, Liu P, Wu X, et al. Berberine attenuates lipopolysaccharide-induced extracelluar matrix accumulation and inflammation in rat mesangial cells: involvement of NF-κB signaling pathway. Mol Cell Endocrinol. 2011;331(1):34–40. doi:10.1016/j.mce.2010.07.023
- Shao BZ, Wang SL, Pan P, et al. Targeting NLRP3 inflammasome in inflammatory bowel disease: putting out the fire of inflammation. Inflammation. 2019;42(4):1147–1159. doi:10.1007/s10753-019-01008-y
- Pinar AA, Scott TE, Huuskes BM, Tapia Cáceres FE, Kemp-Harper BK, Samuel CS. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol Ther. 2020;209:107511.
- Otto G. IL-1β switches on kidney fibrosis. Nat Rev Nephrol. 2018;14(8):475. doi:10.1038/s41581-018-0026-2
- Chen G, Sun L, Kato T, et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest. 2019;129(10):4433–4450. doi:10.1172/JCI125669
- Liang H, Xu F, Zhang T, et al. Inhibition of IL-18 reduces renal fibrosis after ischemia-reperfusion. Biomed Pharmacother. 2018;106:879–889. doi:10.1016/j.biopha.2018.07.031
- Xiao H, Li H, Wang JJ, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur Heart J. 2018;39(1):60–69. doi:10.1093/eurheartj/ehx261
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL–1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol. 2009;124(6):1141–1149; quiz 1150–1141. doi:10.1016/j.jaci.2009.11.016
- Ding N, Wei B, Fu X, Wang C, Wu Y. Natural products that target the NLRP3 inflammasome to treat fibrosis. Front Pharmacol. 2020;11:591393. doi:10.3389/fphar.2020.591393
- Hannum CH, Wilcox CJ, Arend WP, et al. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343(6256):336–340. doi:10.1038/343336a0
- Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014;2:267–272. doi:10.1016/j.redox.2014.01.012
- Sampson N, Berger P, Zenzmaier C. Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease. Biomed Res Int. 2014;2014:131737. doi:10.1155/2014/131737
- Gorin Y, Block K, Hernandez J, et al. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005;280(47):39616–39626. doi:10.1074/jbc.M502412200
- Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15(9):1077–1081. doi:10.1038/nm.2005
- Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53(2):289–296. doi:10.1016/j.freeradbiomed.2012.05.007
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi:10.1038/ni.1831
- Oslowski CM, Hara T, O’Sullivan-Murphy B, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16(2):265–273. doi:10.1016/j.cmet.2012.07.005
- Kim SM, Kim YG, Kim DJ, et al. Inflammasome-independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol. 2018;9:2563. doi:10.3389/fimmu.2018.02563
- Gong W, Mao S, Yu J, et al. NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am J Physiol Renal Physiol. 2016;310(10):F1081–F1088. doi:10.1152/ajprenal.00534.2015
- Guo H, Bi X, Zhou P, Zhu S, Ding W. NLRP3 deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm. 2017;2017:8316560. doi:10.1155/2017/8316560
- Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88(5):529–535. doi:10.1161/01.RES.88.5.529
- Alyaseer AAA, de Lima MHS, Braga TT. The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol. 2020;11:883. doi:10.3389/fimmu.2020.00883
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388(6637):82–87. doi:10.1038/40424
- Sanderson N, Factor V, Nagy P, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92(7):2572–2576. doi:10.1073/pnas.92.7.2572
- Kopp JB, Factor VM, Mozes M, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74(6):991–1003.
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi:10.1038/nature02006
- Zhang LL, Huang S, Ma XX, et al. Angiotensin(1-7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radic Biol Med. 2016;97:531–543. doi:10.1016/j.freeradbiomed.2016.07.014
- Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10(1):100. doi:10.1186/1465-9921-10-100
- Tian R, Zhu Y, Yao J, et al. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res. 2017;357(2):328–334. doi:10.1016/j.yexcr.2017.05.028
- Song S, Qiu D, Luo F, et al. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. J Mol Endocrinol. 2018;61(3):101–113. doi:10.1530/JME-18-0069
- Romero CA, Remor A, Latini A, De Paul AL, Torres AI, Mukdsi JH. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J Mol Histol. 2017;48(3):209–218. doi:10.1007/s10735-017-9720-9
- Wu M, Han W, Song S, et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol. 2018;478:115–125. doi:10.1016/j.mce.2018.08.002
- Wang W, Wang X, Chun J, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190(3):1239–1249. doi:10.4049/jimmunol.1201959
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–139. doi:10.1038/cr.2008.328
- Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42.
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi:10.1038/nm.2807
- Luan J, Ju D. Inflammasome: a double-edged sword in liver diseases. Front Immunol. 2018;9:2201. doi:10.3389/fimmu.2018.02201
- Artlett CM. The IL-1 family of cytokines. Do they have a role in scleroderma fibrosis? Immunol Lett. 2018;195:30–37. doi:10.1016/j.imlet.2017.11.012
- Boaru SG, Borkham-Kamphorst E, Tihaa L, Haas U, Weiskirchen R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J Inflamm (Lond). 2012;9(1):49. doi:10.1186/1476-9255-9-49
- Wu X, Zhang F, Xiong X, et al. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life. 2015;67(4):312–321. doi:10.1002/iub.1348
- Inzaugarat ME, Johnson CD, Holtmann TM, et al. NLR family pyrin domain-containing 3 inflammasome activation in hepatic stellate cells induces liver fibrosis in mice. Hepatology. 2019;69(2):845–859. doi:10.1002/hep.30252
- Yaping Z, Ying W, Luqin D, Ning T, Xuemei A, Xixian Y. Mechanism of interleukin-1β-induced proliferation in rat hepatic stellate cells from different levels of signal transduction. APMIS. 2014;122(5):392–398. doi:10.1111/apm.12155
- Reiter FP, Wimmer R, Wottke L, et al. Role of interleukin-1 and its antagonism of hepatic stellate cell proliferation and liver fibrosis in the Abcb4(-/-) mouse model. World J Hepatol. 2016;8(8):401–410. doi:10.4254/wjh.v8.i8.401
- Yan C, Zhou L, Han YP. Contribution of hepatic stellate cells and matrix metalloproteinase 9 in acute liver failure. Liver Int. 2008;28(7):959–971. doi:10.1111/j.1478-3231.2008.01775.x
- Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin–1beta in mice. Gastroenterology. 2010;139(1):323–334.e327. doi:10.1053/j.gastro.2010.03.052
- Kamari Y, Shaish A, Vax E, et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55(5):1086–1094. doi:10.1016/j.jhep.2011.01.048
- Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS One. 2013;8(2):e56100. doi:10.1371/journal.pone.0056100
- Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 2012;92(5):713–723. doi:10.1038/labinvest.2012.45
- Sansoè G, Aragno M, Wong F. Pathways of hepatic and renal damage through non-classical activation of the renin-angiotensin system in chronic liver disease. Liver Int. 2020;40(1):18–31. doi:10.1111/liv.14272
- Cai SM, Yang RQ, Li Y, et al. Angiotensin-(1-7) improves liver fibrosis by regulating the NLRP3 inflammasome via redox balance modulation. Antioxid Redox Signal. 2016;24(14):795–812. doi:10.1089/ars.2015.6498
- Dong Z, Zhuang Q, Ning M, Wu S, Lu L, Wan X. Palmitic acid stimulates NLRP3 inflammasome activation through TLR4-NF-κB signal pathway in hepatic stellate cells. Ann Transl Med. 2020;8(5):168. doi:10.21037/atm.2020.02.21
- Han CY, Rho HS, Kim A, et al. FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep. 2018;24(11):2985–2999. doi:10.1016/j.celrep.2018.07.068
- Dunagan M, Chaudhry K, Samak G, Rao RK. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2012;303(12):G1356–G1364. doi:10.1152/ajpgi.00526.2011
- Wang Y, Tong J, Chang B, Wang B, Zhang D, Wang B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep. 2014;9(6):2352–2356. doi:10.3892/mmr.2014.2126
- Szabo G, Petrasek J, Bala S. Innate immunity and alcoholic liver disease. Dig Dis. 2012;30(Suppl s1):55–60. doi:10.1159/000341126
- Petrasek J, Iracheta-Vellve A, Saha B, et al. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J Leukoc Biol. 2015;98(2):249–256. doi:10.1189/jlb.3AB1214-590R
- Chen H, Zhu J, Liu Y, et al. Lipopolysaccharide induces chronic kidney injury and fibrosis through activation of mTOR signaling in macrophages. Am J Nephrol. 2015;42(4):305–317. doi:10.1159/000441506
- Tang PC, Zhang YY, Chan MK, et al. The emerging role of innate immunity in chronic kidney diseases. Int J Mol Sci. 2020;21(11):4018. doi:10.3390/ijms21114018
- Ke B, Shen W, Fang X, Wu Q. The NLPR3 inflammasome and obesity-related kidney disease. J Cell Mol Med. 2018;22(1):16–24. doi:10.1111/jcmm.13333
- Zhang C, Boini KM, Xia M, et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. 2012;60(1):154–162. doi:10.1161/HYPERTENSIONAHA.111.189688
- Shahzad K, Bock F, Dong W, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87(1):74–84. doi:10.1038/ki.2014.271
- Masola V, Carraro A, Granata S, et al. In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J Transl Med. 2019;17(1):12. doi:10.1186/s12967-019-1770-1
- Seo JB, Choi YK, Woo HI, et al. Gemigliptin attenuates renal fibrosis through down-regulation of the NLRP3 inflammasome. Diabetes Metab J. 2019;43(6):830–839. doi:10.4093/dmj.2018.0181
- Wen Y, Pan MM, Lv LL, et al. Artemisinin attenuates tubulointerstitial inflammation and fibrosis via the NF-κB/NLRP3 pathway in rats with 5/6 subtotal nephrectomy. J Cell Biochem. 2019;120(3):4291–4300. doi:10.1002/jcb.27714
- Shen X, Dong X, Han Y, et al. Ginsenoside Rg1 ameliorates glomerular fibrosis during kidney aging by inhibiting NOX4 and NLRP3 inflammasome activation in SAMP8 mice. Int Immunopharmacol. 2020;82:106339. doi:10.1016/j.intimp.2020.106339
- Ling L, Yang M, Ding W, Gu Y. Ghrelin attenuates UUO-induced renal fibrosis via attenuation of Nlrp3 inflammasome and endoplasmic reticulum stress. Am J Transl Res. 2019;11(1):131–141.
- Wang S, Fan J, Mei X, et al. Interleukin-22 attenuated renal tubular injury in aristolochic acid nephropathy via suppressing activation of NLRP3 inflammasome. Front Immunol. 2019;10:2277. doi:10.3389/fimmu.2019.02277
- Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol. 2019;15(8):501–520. doi:10.1038/s41581-019-0158-z
- Komada T, Chung H, Lau A, et al. Macrophage uptake of necrotic cell DNA activates the AIM2 inflammasome to regulate a proinflammatory phenotype in CKD. J Am Soc Nephrol. 2018;29(4):1165–1181. doi:10.1681/ASN.2017080863
- Meyer KC. Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med. 2017;11(5):343–359. doi:10.1080/17476348.2017.1312346
- Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther Adv Respir Dis. 2015;9(4):188–197. doi:10.1177/1753465815586335
- Sohn SH, Lee JM, Park S, et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol. 2015;39(2):917–926. doi:10.1016/j.etap.2015.02.019
- Wang X, Sun B, Liu S, Xia T. Structure activity relationships of engineered nanomaterials in inducing NLRP3 inflammasome activation and chronic lung fibrosis. NanoImpact. 2017;6:99–108. doi:10.1016/j.impact.2016.08.002
- Hu B, Jin C, Li HB, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354(6313):765–768. doi:10.1126/science.aaf7532
- Gao J, Peng S, Shan X, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019;10(12):957. doi:10.1038/s41419-019-2195-8
- Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–567. doi:10.1172/JCI31139
- Desbois AC, Cacoub P. Systemic sclerosis: an update in 2016. Autoimmun Rev. 2016;15(5):417–426. doi:10.1016/j.autrev.2016.01.007
- Ihn H. Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis. J Dermatol Sci. 2008;49(2):103–113. doi:10.1016/j.jdermsci.2007.05.014
- Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011;63(11):3563–3574. doi:10.1002/art.30568
- Martínez-Godínez MA, Cruz-Domínguez MP, Jara LJ, et al. Expression of NLRP3 inflammasome, cytokines and vascular mediators in the skin of systemic sclerosis patients. Isr Med Assoc J. 2015;17(1):5–10.
- Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27(3):341–348. doi:10.1093/cvr/27.3.341
- Van Tassell BW, Arena R, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. 2014;113(2):321–327. doi:10.1016/j.amjcard.2013.08.047
- Sandanger Ø, Gao E, Ranheim T, et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Commun. 2016;469(4):1012–1020. doi:10.1016/j.bbrc.2015.12.051
- Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604. doi:10.1161/CIRCULATIONAHA.110.982777
- Zhang X, Pan L, Yang K, et al. H3 relaxin protects against myocardial injury in experimental diabetic cardiomyopathy by inhibiting myocardial apoptosis, fibrosis and inflammation. Cell Physiol Biochem. 2017;43(4):1311–1324. doi:10.1159/000481843
- Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. 2014;289(28):19571–19584. doi:10.1074/jbc.M114.550624
- Pan XC, Liu Y, Cen YY, et al. Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis. Int J Mol Sci. 2019;20(2):360. doi:10.3390/ijms20020360
- Cáceres FT, Gaspari TA, Samuel CS, Pinar AA. Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB J. 2019;33(12):14717–14733. doi:10.1096/fj.201901079RR
- Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacol Ther. 2017;172:50–62. doi:10.1016/j.pharmthera.2016.11.013
- Roul D, Recchia FA. Metabolic alterations induce oxidative stress in diabetic and failing hearts: different pathways, same outcome. Antioxid Redox Signal. 2015;22(17):1502–1514. doi:10.1089/ars.2015.6311
- Wang X, Pan J, Liu H, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019;221:249–258. doi:10.1016/j.lfs.2019.02.035
- Liew EL, Araki M, Hironaka Y, et al. Identification of AIM2 as a downstream target of JAK2V617F. Exp Hematol Oncol. 2015;5:2. doi:10.1186/s40164-016-0032-7
- Zhou Y, Yan S, Liu N, et al. Genetic polymorphisms and expression of NLRP3 inflammasome-related genes are associated with Philadelphia chromosome-negative myeloproliferative neoplasms. Hum Immunol. 2020;81(10–11):606–613. doi:10.1016/j.humimm.2020.09.001