
Abstract
Purpose
During sepsis, an excessive inflammatory immune reaction contributes to multi-organ dysfunction syndrome (MODS), a critical condition associated with high morbidity and mortality; however, the molecular mechanisms driving MODS remain elusive.
Methods
We used RNA sequencing to characterize transcriptional changes in the early phase of sepsis, at 6, 12, 24 hour time points in lung, kidney, liver, and heart tissues, in a cecal ligation and puncture (CLP)-induced polymicrobial sepsis murine model.
Results
The CLP surgery induced significant changes (adj. p-value<0.05) in expression of hundreds of transcripts in the four organs tested, with the highest number exceeding 2,000 differentially expressed genes (DEGs) in all organs at 12 hours post-CLP. Over-representation analysis by functional annotations of DEGs to the Reactome database revealed the immune system, hemostasis, lipid metabolism, signal transduction, and extracellular matrix remodeling biological processes as significantly altered in at least two organs, while metabolism of proteins and RNA were revelaed as being liver tissue specific in the early phase of sepsis.
Conclusion
RNA sequencing across organs and time-points in the CLP murine model allowed us to study the trajectories of transcriptome changes demonstrating alterations common across multiple organs as well as biological pathways altered in an organ-specific manner. These findings could pave new directions in the research of sepsis-induced MODS and indicate new sepsis treatment strategies.
Keywords:
Introduction
Sepsis is a clinical syndrome resulting from a systemic inflammatory response to infection, with a high mortality rate, which ranges between 10–50%, depending on the patient’s age and disease severity.Citation1 The molecular hallmark of sepsis is the release of pathogen-associated molecular patterns (PAMPs) from the infectious agent and host release of damage-associated molecular patterns (DAMPs or alarmins), recognized by Toll-like receptors (TLRs) which lead to a profound increase in cytokine production.Citation2 Dysregulated mechanisms of innate and adaptive immunity result in the early pro-inflammatory phase of sepsis characterized by high production of pro-inflammatory cytokines, followed by an anti-inflammatory phase that translates into functional immunosuppression.Citation3
In 2016, an international consensus conference defined sepsis as life-threatening organ dysfunction caused by dysregulated host response to infection.Citation4 As reviewed recently,Citation5 neither tissue hypoxia, significant cell death, nor adaptive strategy to overwhelming inflammatory injury could be defined as a predominant isolated mechanism of sepsis-related organ dysfunction. Instead, several other mechanisms are probably involved in the pathogenesis of sepsis, including resistance to pathogen load and tolerance to organ injury by infectious agents and the host’s immune response, as well as transient loss of function of the heart, lung, kidney, liver, and intestine (multiple organ dysfunction, MODS) resulting from the alterations in mitochondrial energy generation and cell cycle arrest induction. As a consequence, sepsis-related alterations activate cross talk between dysfunctional organs.Citation5 However, the exact mechanisms underlying development of sepsis, and particularly MODS, remains incompletely understood.
Gene expression profiling is a well-recognized method of searching for the molecular basis of diseases. The human blood leukocyte response to acute systemic inflammation was associated with more than 3,700 differentially expressed genes,Citation6 and systemic administration of LPS altered expression of 8,458 genes in the mouse adrenal gland identifying gene sets and pathways potentially involved in sepsis-induced organ dysfunction.Citation7 While transcriptional signatures induced by LPS stimulation are conserved between humans and mice, substantial species-specific differences in sepsis response signatures were also recognized;Citation8 these may represent, among others, differences in organization of molecular studies that can be conducted in septic patients and animal models. For ethical reasons, organ tissue sampling is impractical in critically ill patients, with the exception of post-mortem examinations, and, therefore, the majority of gene expression studies in septic patients have been conducted using only whole blood samples or isolated white blood cells. Although blood comes into contact with the cells, tissues, and organs of the entire organism, constituting a primary aspect of the immune defense system, comparing blood gene expression profiles from septic patients with organ transcriptomes from animal models can cause possible erroneous conclusions.Citation9 Furthermore, sepsis is a dynamic process within a relatively narrow time window.Citation1 In clinical studies blood samples are usually obtained at a single time point and, as a consequence, may represent different phases of sepsis, while in animal models organ tissue transcriptional alterations can be analyzed repeatedly at precise times to show a trajectory of molecular changes.
In this study using RNA sequencing we analyzed transcriptional changes in the lung, kidney, liver, and heart tissues, in a cecal ligation and puncture (CLP)-induced polymicrobial sepsis murine model. Our objective was to identify both organ specific alterations in gene expression profiles and to characterize pathway changes conserved across multiple organs in the early phase of sepsis.
Materials and Methods
Mice
In this study, 9–12-week-old C57BL/6J male mice were housed in specific-pathogen free conditions with 14–10 hours of light and dark cycles and free access to food and water until the surgery.
Institutional Review Board Statement
Mouse experiments were conducted in accordance with The American Association for Accreditation of Laboratory Animal Care guidelinesCitation10 under approval of The University of Washington Office of Animal Welfare (Protocol #4287-01) at the University of Washington South Lake Union medical research campus.
Cecal Ligation and Puncture (CLP)-Induced Polymicrobial Sepsis
The CLP procedure was performed as previously described and according to the general guidelines.Citation11–Citation13 Briefly, under isoflurane anesthesia, mice were laid supine and a midline incision was made in the lower abdomen after shaving and sterilizing the abdominal skin. The cecum was identified and carefully extracted. Stool in the cecum was squeezed distally and a 3–0 suture was tied around the proximal portion of the cecum and secured tightly, interrupting continuity with the bowel and cutting off blood flow. A 21 g needle was used to make a single puncture in the proximal, antimesenteric aspect of the cecum and a small amount of stool was expressed. The cecum was placed back in the abdominal cavity and the abdominal wall closed with 6–0 suture. Skin was approximated and closed with Gluture tissue adhesive. Immediately post op, mice were given 20 mL/kg normal saline solution and buprenorphine 0.05 mg/kg. Two hours later, mice were given imipenem 25 mg/kg. At 12 hours mice were dosed with normal saline solution 10 mL/kg, and the same doses of imipenem and buprenorphine. Mice were monitored using a clinical severity score every 12 hours, as previously reported.Citation12 Tissue samples were collected at the specified time points. Anesthesia was induced again with isoflurane and blood collected via cardiac puncture. The chest was opened and heart, and lungs collected rapidly into cryovials which were immediately snap frozen in liquid nitrogen. Abdominal organs were collected in a similar fashion immediately thereafter.
RNA Extraction
Total RNA was extracted from tissue using MirVana™ PARIS™ Kit (Ambion) according to manufacturer’s instructions. The concentration of each sample was measured by NanoDrop 2000 (Thermo). Total RNA quality was analyzed with the RNA 6000 Nano Kit on Agilent 2100 Bioanalyzer (Agilent). For RNA-Seq we analyzed RNA samples from 74 organs derived from 35 animals. If possible, the sets of four organs from the same animals were analyzed. Samples from 12 hours had RNA integrity number (RIN) >6.8, while all samples had <10% rRNA contamination following the poly (A) fraction. (Table S1).
Quantitative Reverse Transcription PCR (qRT-PCR)
To synthesize cDNA, 400 ng total RNA was reverse-transcribed with 200 units of MMLV reverse transcriptase (Thermo) and oligo-dT primers in 20-μL reactions. RT reactions were diluted 100-fold with water prior to use in qPCR. GraphGrid Excel-based software was used to acquire, store, and analyze qRT-PCR data. Pair-wise t-test was used to measure statistically significant differences, which are depicted by the size of a circle for each comparison made: p<0.05 by a small circle, p<0.01 by a large circle.Citation14 Primers sequence is provided in Table S2.
The Poly (A) Fraction Purification and Sequencing
Messenger RNA (poly(A)-containing mRNA) was purified from the 1–8 µg of total RNA by magnetic beads coated with oligo (dT) using a Dynabeads® mRNA DIRECT™ Micro Kit (Thermo). To estimate the quality and quantity of mRNA samples we analyzed samples with the RNA 6000 Pico Kit with the 2100 Bioanalyzer (Agilent). The sequencing library of each RNA sample was prepared by using Ion Total RNA-Seq Kit v2 according to the manufacturer’s protocol (Thermo). The libraries were prepared from 1–15 ng of mRNA. Briefly, the mRNA was fragmented using RNaseIII and then purified. The fragmented RNA was hybridized and ligated with ion adapters. The RNA fragments were then reverse transcribed and amplified to double-stranded cDNA. Next, the cDNA was purified by magnetic bead based method. The molar concentration and size selection (50–1,000 bp) of each cDNA library was determined using DNA 1000 Kit on Bioanalyzer 2100 (Agilent). Each library was diluted to ~80 pM concentration prior to template preparation and up to four barcoded libraries were mixed in equal volume and used for automatic template preparation on an Ion Chef (Thermo) instrument using reagents from the Ion PI IC 200 Kit (Thermo) and Ion PI v3 Proton Chip. Samples were sequenced on the Ion Proton System (Thermo) according to the manufacturer’s instructions.
RNA-Seq Data Analysis
Raw reads were mapped to mm10 reference genome with Ion Torrent RNASeqAnalysis Plugin version 5.0 which utilizes STARCitation15 as the primary aligner, and bowtie2 for previously unaligned reads.Citation16 Gene abundance was quantified with htseq-count (HTSeq framework version 0.6Citation17) using Ensembl Gene gtf file from UCSC as reference. Differential gene expression was performed with R package DESeq2 version 1.18, using Wald’s test and Benjamini-Hochberg procedure to control false discovery rate (FDR).
Functional Analyses
Overrepresentation of Reactome pathways in genes which were differentially expressed was performed with R package ClusterProfiler version 3.6, separately for up- and downregulated genes.Citation18 Heatmap visualization was conducted with ComplexHeatmap R package version 2.3.1, using only pathways present in at least two data points and with at least 5% of differential genes present in the pathway.
Data Availability Statement
The RNA-Seq data were deposited as the BAM files in the European Nucleotide Archive under the PRJEB20791 identifier.
Results and Discussion
Cecal Ligation and Puncture Induces Sepsis of Moderate Severity
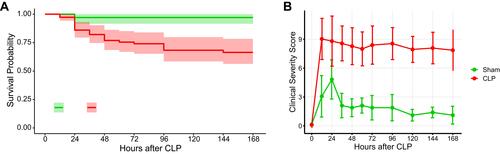
The CLP model of sepsis can be highly variable in that severity with a specific injury in one lab does not necessarily translate to the same severity in another.Citation19 We previously titrated the severity of our model to achieve 25–30% mortality at 7 days in C57BL/6J mice to mimic clinical mortality observed in human septic shock.Citation20,Citation21 This mortality phenotype with the specific injury described here was stable in our hands over multiple experiments for years (). In this model, CLP-operated mice develop significantly elevated clinical severity scores compared to sham-operated mice ().
Figure 1 Multiple CLP experiments in our lab over 3.5 years show a consistent pattern of illness severity. (A) Overall survival of CLP mice had been titrated to about 25–30% in our model. (B) CLP operated mice developed clinical severity scores much worse than the sham operated mice. N=40 and 110 for sham and CLP groups, respectively.
RNA-Seq Survey Across Multiple Organs in Polymicrobial Sepsis Reveals Dynamics of Transcriptional Changes
To establish multi-organ gene expression signatures of the early phase of sepsis polymicrobial sepsis was induced by CLP surgery in male mice and compared to sham-operated animals. At 6, 12, and 24 hours after surgery, three animals from each group were sacrificed and the lungs, kidneys, livers, and hearts were removed and frozen. At 12 hours an additional one CLP and sham-operated kidney tissue sample was included. The organs we have chosen are more frequently affected in the course of sepsis and extent of their dysfunction determines patients’ course from infection to recovery or death.Citation22 Altogether, 74 Poly (A)-enriched mRNA samples, representing four organs removed from CLP and sham-operated animals, were sequenced on an Ion Proton sequencer. The principal component analysis showed that the samples clustered according to the tissue origin during the experiment (). The pairwise comparison between mice with CLP induced sepsis and control (sham-operated) mice identified 1,205, 489, 1,276, and 374 differentially expressed genes (DEGs) (an adjusted p-value<0.05) in liver, kidney, lung and heart samples, respectively, at 6 hours, 3,769, 2,457, 3,828, and 2,442 DEGs, respectively, at 12 hours, and 2,481, 1,470, 1,800, and 154 DEGs, respectively, at 24 hours (; Table S3).
Figure 2 Transcriptome survey reveals dynamics of mRNAs across multiple organs in polymicrobial sepsis. (A) The numbers of differentially expressed genes (adj. p-value<0.05) across the organs and time-points. Red and green arrows indicate up- and downregulated genes, respectively. (B) The Venn diagram shows the overlapping numbers of DEGs for organs at given time point post-CLP.
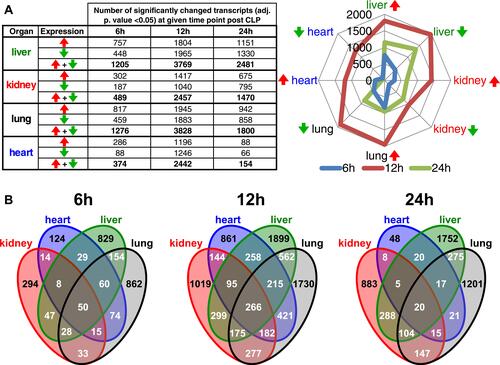
The expression of 294/1,019/883, 124/861/48, 829/1,899/1,752, and 862/1,730/1,201 transcripts were found exclusively altered in CLP induced sepsis at least at one time point (6/12/24 hours) in the kidney, heart, liver, and lung, respectively. In contrast, 50, 266, and 20 DEGs were common to all four organ comparisons for 6, 12, and 24 hour time points, respectively ().
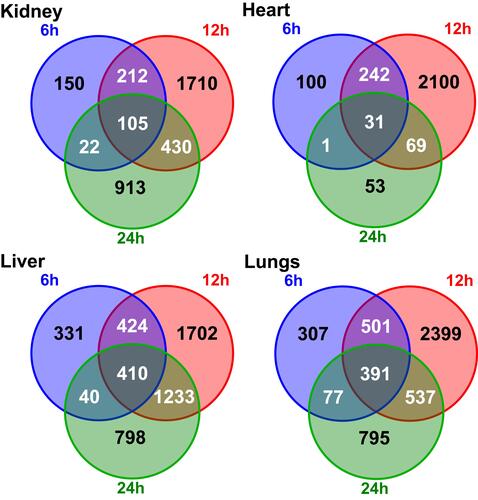
At the earliest time point, the number of significantly changed transcripts passed a 1,000 threshold in the lung and liver only, followed by a burst in expression alteration passing the 2,000 transcripts threshold in every organ at 12 hours. At the experiment endpoint (24 hours) the number of DEGs decreased across all the organs where the reduction of DEGs was most striking in the heart. Of note, the number of DEGs seen at all three time points in a given organ was 105, 31, 410, and 391 in the kidney, heart, liver, and lung, respectively ().
Figure 3 Venn diagram illustrating the overlapping numbers of DEGs for organs during the CLP induced sepsis.
Our RNA-Seq survey uncovered dynamic transcriptional changes across mouse organs with the most pronounced number of DEGs at the 12 hour time point during CLP-induced polymicrobial sepsis. To date, most of the transcriptome studies in sepsis have surveyed expression changes in peripheral blood mononuclear cells. A meta-analysis by Godec et alCitation8 of 389 published transcriptomes studies from mouse and human immune cells induced by LPS or CLP procedures showed that expression profiles between humans and mouse models appeared to be highly conserved. However, no clinical study evaluated sepsis-related gene expression profiles in individual organs as the utility of the biopsies in the critically ill patients are not practical. In turn, only a small number of reports have described organ tissue transcriptional profiles in animal models of sepsis; most of these have studied single organs.Citation7,Citation23–Citation29 Thus, our study, for the first time, reveals septic transcriptional profiles simultaneous analysis across different organs over the time of early sepsis phase, using RNA-seq.
RNA-Seq Data Significantly Correlate with qPCR Measured Abundances of Inflammatory and Endothelial Transcripts
As a way to corroborate RNA-seq data we assayed in qPCR () the same RNA samples as in RNA-Seq survey and included at least three additional RNA samples per group. We used primers to inflammatory (Lcn2, Tnfa, Il-6, Il-1a, Ccl2) and endothelial (Angpt1, Angpt2, Kdr) genes previously studied in sepsis models.Citation14,Citation30 In sepsis endothelial dysfunction results from decreased expression of genes that otherwise maintain microvascular integrity.Citation14 The qPCR confirmed significant alternation of surveyed transcripts across organs in at least one time point and the direction of changes in their expression was in line to other studies utilizing sepsis models.Citation14,Citation30 The Spearman correlation coefficient of qPCR and RNA-Seq data for those genes across the time point and organs is 0.72, and is statistically significant. Of note, the qPCR measured expression of Lcn2, that encodes neutrophil gelatinase-associated lipocalin (Ngal), remained significantly elevated irrespectively of the time-point and organ assayed, confirming previously observed sustained expression of this transcript in CLP-induced sepsis.Citation31
Figure 4 Quantitative polymerase chain reaction (qPCR) gene expression measurements of selected inflammatory (A) and endothelial-related (B) transcripts across multiple organs in CLP-induced polymicrobial sepsis. Total RNA from heart, lungs, kidneys, and livers was used in reverse transcription reactions with oligo-dT primers. cDNA was used in qPCR with gene-specific primers.Citation14 Data are represented as mean±standard error of the mean (n=6 mice in each group). Statistical differences between two means (p-value) are shown by the size of the solid circles and additionally denoted with an asterisk (*): p<0.05 by a small circle, p<0.01 by a large circle.Citation14

Functional Analyses of DEGs Highlight Shared and Organ Specific Biological Pathways Altered in CLP-Induced Polymicrobial Sepsis
To uncover organ and time point specific changes as well as alterations shared across organs and time, we performed functional analyses of significantly up and downregulated transcripts (an adjusted p-value<0.05 and fold change; FC≥1.5) encompassing 12 types of samples using the Reactome database. The functional analysis yielded 26/0, 25/0, 45/3, and 20/8 of significantly enriched pathways for up/downregulated DEGs in heart, kidney, liver, and lungs, respectively, at 6 hours (Table S4), 55/4, 23/0, 49/31, and 11/43 pathways, respectively, at 12 hours (Table S5), and 5/0, 31/12, 43/38, and 23/25 pathways, respectively, at 24 hours (Table S6). To further generalize the results of functional analyses we defined pathways with a high contribution of changed genes by using the threshold of 5% genes altered in a given term while at the same time being altered in at least two separate types of samples, yielding 41 pathways recurrently altered in our sepsis model ().
Figure 5 Functional analyses reveal recurrent Reactome terms across the organs during a polymicrobial sepsis in a murine model.

The CLP upregulated genes categorized under the Immune system consisted of 15 pathways. Pathways overrepresented in all organs for at least one time-point include: Cytokine Signaling in Immune system, Interleukin-1 family signaling, Interleukin-1 signaling, Signaling by Interleukins pathways, Antimicrobial peptides, and Neutrophil degranulation. In contrast, none of the significantly downregulated transcripts could be categorized into the Reactome Immune system category.
As previously reported, the common response at the early phase of polymicrobial sepsis includes activation of inflammatory mediators. Organisms exposed to bacterial endotoxins react with cell surface receptors, including the TLR family, which trigger the release of both proinflammatory and anti-inflammatory mediators through activation of several signal transduction pathways, including nuclear factor κB (NFκB), mitogen activated protein kinase (MAPK), janus kinase (JAK), and signaling transducer and activator of transcription (STAT).Citation1,Citation32 Both concurrent pro-inflammatory and anti-inflammatory phase are considered as the sepsis hallmark.Citation9 Other pathways with a high level of significance were Hemostasis, Platelet activation, signaling and aggregation, Platelet degranulation, and Response to elevated platelet cytosolic Ca2+, which represented the Hemostasis cluster. While all four pathways revealed overrepresentation of upregulated genes at each time point for all organs, with the exception of the lung where, at 12 hours after surgery, they represented significantly downregulated transcripts. Platelets play an important role in sepsis pathophysiology and multi-organ dysfunction as well as in the resolution of inflammation.Citation33
Extensive metabolic dysregulation has been implicated in the pathogenesis of sepsis-induced organ failure, which itself is strongly associates with mortality of septic patients.Citation34 The liver is the central organ for carbohydrate, protein, and fat metabolism and it contributes to the innate immune system.Citation35 Among significantly downregulated genes, Reactome annotation analysis revealed overrepresentation of genes involved in the metabolism category and represented by seven pathways, mostly related to fatty acids metabolism and their oxidation (). The metabolic pathways were already dysregulated at 6 hours post-surgery in the liver, with an increasing level of significance in subsequent time points. The significant signatures of metabolic alterations were also found in septic kidneys (at 12 hours post-surgery) and lungs (at 24 hours post-surgery), but not in the hearts. Interestingly, in the liver at 12 and 24 hour time points the metabolism of proteins related to the asparagine N-linked glycosylation pathway and proteins transport to the Golgi was prominently altered. These two pathways are closely related to other pathway branches that comprise vesicle-mediated transport and were altered at 12 and 24 hours in the liver. Previous studies using the CLP mouse model have highlighted the hepatic changes related to proteins and fatty acids metabolism.Citation36,Citation37
Interestingly, at the 12 hour time-point a parallel increase in RNA metabolism and splicing was observed (Table S5) that could be attributed to the overall elevated transcription and protein translation in the liver. Indeed, the hepatic synthesis of acute phase proteins is a hallmark of infection and inflammation, and N-glycosylation is a main posttranslational modification of these proteins that regulates both their function and lifespan.Citation38 Reactome annotation of significantly downregulated transcripts also revealed hepatic suppression of steroids metabolism, bile acid, and bile salt metabolism pathways (Tables S5 and S6) at 12 and 24 hours post-CLP. The impairment of bile formation in the liver during sepsis promotes hyperbilirubinemia associated with intrahepatic cholestasis.Citation39 It has been shown before that LPS or pro-inflammatory cytokines alter bile acid transporters expression in oxidative stress that results in the inhibition of cAMP-dependent transport function,Citation40 and by the alteration of hepatocytes cytoskeleton and liver tissue morphology, ultimately contributing to altered bile flow.Citation39
Signal transduction by the G protein-coupled receptors (GPCRs) was another major cluster comprised of seven significantly altered pathways. GPCR-related pathways, including WNT signaling, play an important role transducing signals that lead to the production of inflammatory cytokines, and regulation of communications associated with increased blood flow and increased vascular endothelial permeability. Therefore the GPCRs function to both control inflammation and its resolution.Citation41 The expression changes in GPCR-related pathways were most prominent in lungs with five, three, and six out of seven pathways altered at 6, 12, and 24 hour time points, respectively. The pathways altered in the lung comprised both down and upregulated transcripts. On the other hand, in the kidney, three and five GPCR-related pathways were significantly altered at 12 and 24 hour time points, and only represented by overexpressed genes. Signaling by WNT was one of the pathways overrepresented in lungs and negatively regulated at 6 hours while in the heart the downregulation occurred 12 hours post-CLP. Previous studies utilizing experimental mouse models of sepsis highlighted the WNT-pathway deregulation in the adrenal gland,Citation7 heart,Citation27 and lungs.Citation42
The small molecules transport pathway and its derivative term, the Solute Carrier (SLC)-mediated transmembrane transport, represented another major Reactome pathway with alterations in gene expression. The pathways comprised hepatic and pulmonary upregulated transcripts at the 6 hour time point. Conversely, at later time points downregulated transcripts contributed to the significant changes in either of these pathways in the kidney and liver.
Finally, the extracellular matrix (ECM) organization comprised both overexpressed and downexpressed gene sets found in livers and lungs, respectively, 24 hours after surgery. The ECM plays a significant role in the bacterial infection as its adhesive components, including collagens, fibronectin, and laminin, are common binding sites for the invading pathogens. Moreover, the host tissues and immune cells cross-talk as the tissue repair unfolds within the ECM microenvironment. In turn, the ECM undergoes an extensive remodeling both as a result of its degradation by pathogens released proteases and local immune cells response to the insult.Citation43 Importantly, the endothelial cells associated ECM, the glycocalyx, undergoes extensive remodeling, ultimately leading to the disruption of the integrity of the endothelial barrier, a hallmark of sepsis.Citation44 The functional analyses of our transcriptomic data pinpointed over-representation of altered transcripts associated with the hepatic and pulmonary ECM organization. In the liver, the upregulated genes were annotated to the significant terms related to collagen degradation and production at the same time. In the lungs, on the other hand, the downregulated genes were significantly over-represented in the term of collagen and elastin formation. Elastin is a component of a complex fiber structure distributed in compartments of the mammalian lung, including the pulmonary blood vessels.Citation45 These results suggest ongoing tissue remodeling of these organs.
Overall, from the functional analyses it could be concluded that all four organs examined here manifest upregulation of transcripts related to inflammatory response and hemostasis during the early phase of a mouse sepsis model. In contrast, the downregulated genes were over-represented in metabolic pathways related to lipid metabolism, and were observed in the liver at all time points, and in the lungs and kidney at 12 and 24 hours post-CLP, respectively. The changes in proteins and RNAs metabolism were the functional categories that were significantly altered in the liver only.
Post-CLP Organs Tissues Contain DEGs Sets That are Consistent with the Transcriptome Signatures from Septic Peripheral Blood in Mouse Models and Human
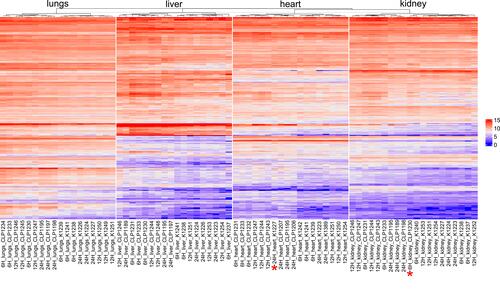
In order to relate the DEGs detected in our experimental model across the organs to available transcriptomic sepsis signatures, we compared our dataset to a set of genes from a compendium of immune signatures derived from multiple sepsis human and mouse peripheral blood transcriptome studies.Citation8 Specifically, we focused on the 667 genes set (Table S7), named the “leading edge” genes, enriched in peripheral blood transcriptome in both species under septic conditions.Citation8 In our dataset (Table S7) grouped as “leading edge” 281 transcripts were altered in at least one sham-operated vs CLP comparison. Based on these transcripts the unsupervised hierarchical clustering of samples from a given organ nearly perfectly separated sham-operated from CLP samples, with an exception of two samples, one from heart and one from kidney tissues ().
Figure 6 The unsupervised hierarchical clustering based on the leading edge gene set from Godec et al.Citation8 Differentiates sham-operated from CLP-operated samples. *Denotes a sample for a given organ that clustered differently than its original treatment.
Conclusion
The CLP induced systemic inflammation evoked dynamic transcriptional changes across heart, kidney, liver, and lung tissues, with the highest number of transcripts altered at 12 hours post-CLP. Moreover, host response set of genes to sepsis was mediated by groups of co-expressed transcripts, regardless of the involved organ. Functional analyses with Reactome database of up- and downregulated DEGs yielded numerous significantly enriched pathways including the Immune system, Hemostasis, Metabolism, Signal transduction, and ECM organization that were shared between at least two organs. Thus, RNA sequencing allows us to study transcriptome dynamics and enables a holistic view of sepsis-altered pathways, several of them previously unexplored, that could pave new directions in research of sepsis-induced organs dysfunction and provide clues for sepsis treatment strategies.
Abbreviations
MODS, multi-organ dysfunction syndrome; CLP, cecal ligation and puncture; DEGs, differentially expressed genes; ECM, extracellular matrix; GPCR, G protein-coupled receptor.
Acknowledgments
This research was funded by Polish National Science Center, grant number 2014/14/M/NZ5/00462 to M.M., by the American Heart Association, grant number 12SDG12040342 to S.S. and by National Institutes of Health R01DK103849 to K.B.
Disclosure
Professor Michal Mikula reports that National Science Centre is a government agency which financed the work. The authors report no other conflicts of interest in this work.
References
- Maslove DM, Wong HR. Gene expression profiling in sepsis: timing, tissue, and translational considerations. Trends Mol Med. 2014;20(4):204–213. doi:10.1016/j.molmed.2014.01.006
- Karmpaliotis D, Kosmidou I, Ingenito EP, et al. Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;283(3):L585–L595. doi:10.1152/ajplung.00048.2002
- Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. doi:10.1111/imr.12499
- Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
- Pool R, Gomez H, Kellum JA. Mechanisms of organ dysfunction in Sepsis. Crit Care Clin. 2015;19(1):63–80. doi:10.1186/s13054-015-0943-4
- Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–1037. doi:10.1038/nature03985
- Chen L-S, Singh SP, Schuster M, Grinenko T, Bornstein SR, Kanczkowski W. RNA-seq analysis of LPS-induced transcriptional changes and its possible implications for the adrenal gland dysregulation during sepsis. J Steroid Biochem Mol Biol. 2019;191:105360. doi:10.1016/j.jsbmb.2019.04.009
- Godec J, Tan Y, Liberzon A, et al. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity. 2016;44(1):194–206. doi:10.1016/j.immuni.2015.12.006
- Průcha M, Zazula R, Russwurm S. Sepsis diagnostics in the era of “omics” technologies. Prague Med Rep. 2018;119(1):9–29. doi:10.14712/23362936.2018.2
- National Research Council Guide for the Care and Use of Laboratory Animals: Eighth Edition. National Academies Press; 2011:12910. doi:10.17226/12910
- Hubbard WJ, Choudhry M, Schwacha MG, et al. Cecal ligation and puncture. Shock. 2005;24(Suppl Supplement 1):52–57. doi:10.1097/01.shk.0000191414.94461.7e
- Standage SW, Waworuntu RL, Delaney MA, et al. Nonhematopoietic peroxisome proliferator-activated receptor-α protects against cardiac injury and enhances survival in experimental polymicrobial sepsis. Crit Care Med. 2016;44(8):e594–e603. doi:10.1097/CCM.0000000000001585
- Standage SW, Bennion BG, Knowles TO, et al. PPARα augments heart function and cardiac fatty acid oxidation in early experimental polymicrobial sepsis. Am J Physiol Heart Circ Physiol. 2017;312(2):H239–H249. doi:10.1152/ajpheart.00457.2016
- Bomsztyk K, Mar D, An D, et al. Experimental acute lung injury induces multi-organ epigenetic modifications in key angiogenic genes implicated in sepsis-associated endothelial dysfunction. Crit Care. 2015;19(1):225. doi:10.1186/s13054-015-0943-4
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi:10.1093/bioinformatics/bts635
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
- Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi:10.1093/bioinformatics/btu638
- Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–36. doi:10.1038/nprot.2008.214
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi:10.1097/00003246-200107000-00002
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest. 2011;140(5):1223–1231. doi:10.1378/chest.11-0352
- Caraballo C, Jaimes F. Organ dysfunction in sepsis: an ominous trajectory from infection to death. Yale J Biol Med. 2019;92(4):629–640.
- Visitchanakun P, Tangtanatakul P, Trithiphen O, et al. Plasma miR-370-3P as a biomarker of sepsis-associated encephalopathy, the transcriptomic profiling analysis of microrna-arrays from mouse brains. Shock. 2020;54(3):347–357. doi:10.1097/SHK.0000000000001473
- Geng H, Bu H-F, Liu F, et al. In inflamed intestinal tissues and epithelial cells, interleukin 22 signaling increases expression of H19 long noncoding RNA, which promotes mucosal regeneration. Gastroenterology. 2018;155(1):144–155. doi:10.1053/j.gastro.2018.03.058
- Wuerth K, Lee AHY, Falsafi R, Gill EE, Hancock REW. Characterization of host responses during pseudomonas aeruginosa acute infection in the lungs and blood and after treatment with the synthetic immunomodulatory peptide IDR-1002. Infection and Immunity. 2019;87:1. doi:10.1128/IAI.00661-18
- Chen L-S, Singh SP, Müller G, Bornstein SR, Kanczkowski W. Transcriptional analysis of sepsis-induced activation and damage of the adrenal endothelial microvascular cells. Front Endocrinol. 2019;10:944. doi:10.3389/fendo.2019.00944
- Chen C, Weng J, Fang D, Chen J, Chen M. Transcriptomic study of lipopolysaccharide-induced sepsis damage in a mouse heart model. Experimental and Therapeutic Medicine. 2020;20(4):3782–3790. doi:10.3892/etm.2020.9086
- Hirbec H, Marmai C, Duroux-Richard I, et al. The microglial reaction signature revealed by RNAseq from individual mice. Glia. 2018;66(5):971–986. doi:10.1002/glia.23295
- Gharib SA, Mar D, Bomsztyk K, et al. System-wide mapping of activated circuitry in experimental systemic inflammatory response syndrome. Shock). 2016;45(2):148–156. doi:10.1097/SHK.0000000000000507
- Sharifian R, Okamura DM, Denisenko O, et al. Distinct patterns of transcriptional and epigenetic alterations characterize acute and chronic kidney injury. Sci Rep. 2018;8(1):17870. doi:10.1038/s41598-018-35943-x
- Vazquez DE, Niño DF, De Maio A, Cauvi DM. Sustained expression of lipocalin-2 during polymicrobial sepsis. Innate Immun. 2015;21(5):477–489. doi:10.1177/1753425914548491
- Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. doi:10.1016/S0140-6736(18)30696-2
- Dewitte A, Lepreux S, Villeneuve J, et al. Blood platelets and sepsis pathophysiology: a new therapeutic prospect in critically [corrected] ill patients? Ann Intensive Care. 2017;7(1):115. doi:10.1186/s13613-017-0337-7
- Lewis AJ, Billiar TR, Rosengart MR. Biology and metabolism of sepsis: innate immunity, bioenergetics, and autophagy. Surg Infect (Larchmt). 2016;17(3):286–293. doi:10.1089/sur.2015.262
- Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55–66. doi:10.1038/nrgastro.2016.168
- Van Wyngene L, Vanderhaeghen T, Timmermans S, et al. Hepatic PPARα function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol Med. 2020;12(2):e11319. doi:10.15252/emmm.201911319
- Whelan SP, Carchman EH, Kautza B, et al. Polymicrobial sepsis is associated with decreased hepatic oxidative phosphorylation and an altered metabolic profile. J Surg Res. 2014;186(1):297–303. doi:10.1016/j.jss.2013.08.007
- Janciauskiene S, Welte T, Mahadeva R. Acute phase proteins: structure and function relationship. Acute Phase Proteins - Regulation and Functions of Acute Phase Proteins. 2011. doi:10.5772/18121
- Nesseler N, Launey Y, Aninat C, Morel F, Mallédant Y, Seguin P. Clinical review: the liver in sepsis. Critical Care. 2012;16(5):235. doi:10.1186/cc11381
- Spirlì C, Fabris L, Duner E, et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology. 2003;124(3):737–753. doi:10.1053/gast.2003.50100
- Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacologica Sinica. 2012;33(3):342–350. doi:10.1038/aps.2011.200
- Villar J, Cabrera-Benítez NE, Ramos-Nuez A, et al. Early activation of pro-fibrotic WNT5A in sepsis-induced acute lung injury. Crit Care. 2014;18(5). doi:10.1186/s13054-014-0568-z
- Tomlin H, Piccinini AM. A complex interplay between the extracellular matrix and the innate immune response to microbial pathogens. Immunology. 2018;155(2):186–201. doi:10.1111/imm.12972
- Tressel SL, Kaneider NC, Kasuda S, et al. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med. 2011;3(7):370–384. doi:10.1002/emmm.201100145
- Mecham RP. Elastin in lung development and disease pathogenesis. Matrix Biol. 2018;73:6–20. doi:10.1016/j.matbio.2018.01.005