
Abstract
Cardiac remodeling is the process by which the heart adapts to stressful stimuli, such as hypertension and ischemia/reperfusion; it ultimately leads to heart failure upon long-term exposure. Autophagy, a cellular catabolic process that was originally considered as a mechanism of cell death in response to detrimental stimuli, is thought to be one of the main mechanisms that controls cardiac remodeling and induces heart failure. Dysregulation of the adipokines leptin and adiponectin, which plays essential roles in lipid and glucose metabolism, and in the pathophysiology of the neuroendocrine and cardiovascular systems, has been shown to affect the autophagic response in the heart and to contribute to accelerate cardiac remodeling. The obesity-associated protein leptin is a pro-inflammatory, tumor-promoting adipocytokine whose elevated levels in obesity are associated with acute cardiovascular events, and obesity-related hypertension. Adiponectin exerts anti-inflammatory and anti-tumor effects, and its reduced levels in obesity correlate with the pathogenesis of obesity-associated cardiovascular diseases. Leptin- and adiponectin-induced changes in autophagic flux have been linked to cardiac remodeling and heart failure. In this review, we describe the different molecular mechanisms of hyperleptinemia- and hypoadiponectinemia-mediated pathogenesis of cardiac remodeling and the involvement of autophagy in this process. A better understanding of the roles of leptin, adiponectin, and autophagy in cardiac functions and remodeling, and the exact signal transduction pathways by which they contribute to cardiac diseases may well lead to discovery of new therapeutic agents for the treatment of cardiovascular remodeling.
Introduction
The World Health Organization (WHO) classifies cardiovascular diseases as the leading cause of death worldwide, with an estimated 17.9 million annual deaths attributed to this class of diseases.Citation1 Risk factors of cardiac remodeling, hypertrophy, and heart failure are various; however, hypertension is a crucial player in the pathogenesis of these conditions.Citation2 Hypertension induces increased mechanical stretch of the vascular smooth muscle cells.Citation3 The ability of the cardiac muscle to sense and respond to mechanical stretch occurs through a process known as mechanotransduction, which is the conversion of mechanical triggers into biochemical incidents.Citation4 This, in turn, causes a change in myocardial function and structure as a compensatory mechanism during adaptation to the trigger.Citation5 In the case of prolonged stimuli, signal transduction occurs and the whole process becomes maladaptive, causing a change in the physiology of the heart, and subsequently leading to development of cardiac remodeling/hypertrophy.Citation5,Citation6
Leptin and adiponectin are physiologically essential for glucose and lipid metabolism, as well as neuroendocrine and cardiovascular functions. However, at dysregulated levels, they are considered major contributors to the pathogenesis of heart failure.Citation7 The biological characteristics of the aforementioned adipokines are contradictory. Explicitly, adiponectin triggers potent anti-inflammatory and anti-tumor responses as opposed to the pro-inflammatory and tumor-promoting features of leptin.Citation8 Although the effects of adiponectin and leptin on cardiac remodeling are multifactorial in nature, recent studies have reported that they take place through the induction of autophagy.Citation8 Autophagy, a cellular catabolic process, was originally considered a mechanism of cell death in response to detrimental stimuli.Citation9 However, recent studies have reported a vital role of autophagy in controlling the cardiac hypertrophic response and the pathogenesis of heart failure.Citation9,Citation10 In this review, we summarize the different mechanisms involved in the development and progression of cardiac dysfunctions, highlight the roles of adiponectin, leptin, and autophagy in the pathogenesis of cardiac diseases, and illustrate the interplay between all of the abovementioned pathways.
Cardiac Remodeling
The molecular mechanisms underlying cardiovascular remodeling produce significant changes in the function of the cardiovascular system, leading to cardiovascular dysfunction. Such alterations include cardiac contractility dysfunction, hypertrophy, fibrosis, and inhibition of the autophagic process.
Cardiac remodeling can be described as a physiologic and pathologic condition that affects cardiac function and structure. It occurs in response to several factors, such as inflammation and vascular and cardiomyocyte hypertrophy, and is a complex process that involves cardiomyocyte growth and death, fibrosis and electrophysiological changes.Citation11 It includes cellular and interstitial modifications of the heart, clinically established by changes in cardiac function and structure/size/wall thickness. Cardiac remodeling occurs due to biomechanical stresses and pathological stimuli.Citation12 Scarred tissue, fibrosis, and inflammatory infiltrate are also detectable in a remodeling heart.
Heart failure is the major complication of cardiac remodeling. It starts at cellular-level changes that occur due to a remodeling-inducing stimulus, leading to cellular and molecular alterations; these changes, in turn, contribute to progressive ventricular dysfunction, which is originally asymptomatic but with time, induces heart failure.Citation13 Cardiac dysfunction and failure occur in response to excessive sympathetic nervous system and angiotensin system stimulations, which activate signal transduction pathways that increase the production of cardiac hypertrophic proteins in cardiomyocytes.Citation14
Cardiac remodeling is also associated with a major increase in the production of reactive oxygen species (ROS). ROS can lead to several factors that induce cardiac remodeling; these include DNA damage, protein oxidation, activation of matrix metalloproteinases, lipid peroxidation, cellular dysfunction, and enhancement of cardiomyocyte apoptosis.Citation15
Autophagy (which we discuss in detail below) is a natural catabolic mechanism in which lysosomes degrade any damaged or unnecessary components in the cell. This process can result in the accumulation of defective proteins, leading to proteotoxicity.Citation16 Research has shown that autophagy is another process that leads to cardiac remodeling.Citation17
In addition, microRNA-22 (miR-22) has emerged as an important regulator of cardiovascular remodeling. MiR-22 has been reported as a crucial modulator of the expression of genes involved in cardiac hypertrophy, actin cytoskeleton reorganization, and metabolic pathways leading to cardiovascular remodeling.Citation4
During cardiac remodeling, changes occur in the calcium transport system; these include a decrease in L-type calcium channels, calsequestrin and calmodulin kinase activity, and phospholamban phosphorylation, leading to reduced calcium supply during cardiac contraction and increased supply during diastole. Consequently, the modifications in the calcium transit proteins may contribute to cardiac dysfunction.Citation18
Moreover, collagen content plays an important role in maintaining cardiac architecture and function. During cardiac remodeling, an imbalance may occur between the synthesis and degradation of collagen, resulting in increased myocardial stiffness and decreased coronary flow.Citation18,Citation19
Hypertension, Cardiac Hypertrophy, and Heart Failure
Cardiac hypertrophy is a way by which the heart adapts to hypertension. This response alters the myocardial structure in a way that vastly contributes to cardiovascular mortality, unexpected death, heart failure, and stroke.Citation20 Initially, hypertrophy is beneficial because it induces the production of a stronger contractile force and retrieves the normal wall stress caused by the increased systolic pressure in the heart. However, upon long-term stimulation, hypertrophy not only increases myocytes’ size, but also causes undesirable reprogramming in gene expression, which ultimately leads to heart failure.Citation21
Although cardiac hypertrophy can be induced by many stimulants, including neuronal and hormonal factors, hypertension (mechanical stretch) is considered to be the most common cause of this state.Citation22 Eventually, long-term hypertension leads to heart failure, and in most cases, patients with heart failure have a history of hypertension.Citation21 Before converting the increased mechanical stretch in response to prolonged hypertension into a biological signal, the cardiomyocyte senses this stretch first. Three sensing systems exist in the cardiac cells: Focal adhesion components, specialized proteins found on the plasma membrane, and sensing proteins found in the cardiac Z line.Citation6 In turn, intracellular signal transduction gets activated, resulting in cardiac hypertrophy.
Many studies have investigated the potential e-link between chronic stretch (hypertension), hypertrophy, and heart failure at the molecular level. Herein, we review the possible pathways accounting for cardiac hypertrophy and the role of two adipokines, leptin and adiponectin, in these mechanisms. Myocardial infarction, the irreversible death of the cardiac muscle, is an important factor that enhances the transition from hypertension to heart failure.Citation23,Citation24 In animal models, the cellular pathogenesis through which myocardial infarction leads to heart failure is well studied. The process is characterized by early ischemia, cardiomyocyte remodeling, and edema development. Together, these would initiate progressive cell death within three hours of myocardial infarction.Citation23,Citation24 Oxidative stress and calcium overload also cause reversible acute contractile dysfunction.
Adipokines and Inflammation
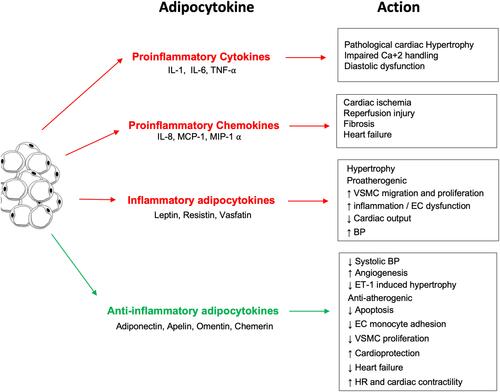
Obesity induces a state of dysfunction in adipose tissue, leading to upregulated secretion of pro-inflammatory adipokines and downregulated production of anti-inflammatory adipokines. The collective effect of obesity-induced adipose tissue dysfunction is the pathogenesis of several diseases, including hypertension and cardiovascular disease.Citation25 Adipokines are secreted by adipocytes and are important proteins of various metabolic processes. Adipocytes secrete bioactive substances related to inflammation (), including tumor necrosis factor-α (TNF-α), interleukin (IL)-6, hapto-globin, nerve growth factor, and macrophage migration factor. Moreover, C-reactive protein (CRP) has been shown to be synthesized by adipose tissue, and its mRNA expression was found to be upregulated in the adipose tissue of adiponectin knockout mice.Citation26 The increased expression of these inflammation-associated adipocytokines has been implicated in the pathogenesis of cardiovascular disease.Citation27 Specifically, these adipocytokines play an important role in the extensive crosstalk between adipose tissue, other organs, and various metabolic systems.
Adipokines play an important role in regulating vascular tone. For instance, the adipokines adiponectin, visfatin, omentin, and the unidentified adipocyte-derived relaxing factor (ADRF) exert vasorelaxing effects on the vascular wall.Citation28 On the other hand, resistin and angiotensin II, which are also released by adipocytes, exert vasoconstricting effects.Citation28 In addition, adipose tissue-secreted leptin, ROS, apelin, tumour necrosis factor α, and interleukin-6 share both vasoconstricting and vasorelaxing properties.Citation29 A dysregulation in the release of pro-inflammatory and vasoactive adipokines is a key factor leading to the pathophysiological vascular reactivity observed in obesity and obesity-related disorders.
Figure 1 The inflammatory and anti-inflammatory effects of the different adipocytokines produced by white adipose tissue.
Leptin as a Pro-Inflammatory Cytokine
Leptin is the first adipokine (obesity-associated protein) identified in 1994.Citation30,Citation31 It is a 16 kDa protein that helps regulate energy balance by controlling food intake via its actions on the hypothalamus. Even though the leptin gene is highly expressed in adipose tissues, it can also be expressed at other sites like mammary epithelial cells, bone marrow, and vascular smooth muscle cells.Citation32,Citation33 In a healthy state, leptin controls blood pressure by regulating sympathetic activity-dependent vasoconstriction and the endothelial release of nitric oxide (NO), as well as angiotensin II (Ang II)-dependent vasoconstriction.Citation34
The physiological activity of leptin strongly depends on its binding to its Ob-R receptors (). These receptors are expressed in six isoforms, and according to their structural differences, they are classified into three classes: Long, short, and secretory. The extracellular domain is common for all isoforms, but the intracellular domain is variable. Ob-Ra, Ob-Rc, Ob-Rd, and Ob-Rf are short isoforms. Ob-Re is soluble and can bind to circulating leptin, thus regulating the free leptin concentration. Ob-Rb is the long isoform with the longest intracellular signaling domain and is the most prevalent isoform in the hypothalamus.Citation35 It is involved in energy homeostasis and regulates the activity of the secretory organs.
Figure 2 Interactions between leptin and the immune system. Leptin upregulates the formation of different inflammatory proteins through activating both innate and adaptive immunity.
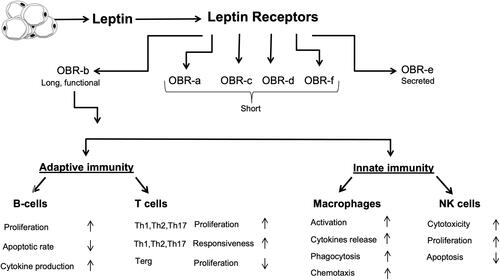
Even though leptin is not a classical cytokine, several immune cells, including polymorphonuclear leukocytes, lymphocytes, monocytes, and macrophages, have receptors for leptin and can be modulated by it.Citation36 Leptin shows structural similarity to the cytokines of the long-chain helical family, which includes IL-6, IL-11, IL-12, and oncostatin M (OSM);Citation37 in addition, most of leptin’s pro-inflammatory actions seem to be mediated by the Ob-Rb receptor, which has structural homology with the members of the class I cytokine receptor (gp130) superfamily, which includes receptors for IL-6, granulocyte colony-stimulating factor (G-CSF), and leukocyte inhibitory factor (LIF).Citation36
When leptin binds to the Ob-Rb receptor on monocytes and macrophages, it improves phagocytosis by regulating oxidative stress.Citation38 It also increases the synthesis of NO and eicosanoids, acts as a chemoattractant, increases the release of cytokines like TNF-α, IL-1, IL-1RA, IL-6, and CC-chemokine ligand, and prevents apoptosis.Citation38 In studies that used ob/ob mice and db/db animal models, mice lacking leptin or with leptin resistance demonstrated numerous pathologies of the immune system.Citation39,Citation40
Leptin and the Immune Response
Obesity causes low-grade chronic inflammation due to modifications in both the innate and adaptive immune systems.Citation41 Due to the excessive activation of pro-inflammatory signal transduction pathways in their adipose tissue, overweight and obese individuals generally have higher levels of the pro-inflammatory cytokines C-reactive protein, IL-6, and TNF-α. In turn, inflammatory stimuli, including IL-1β, IL-6, lipopolysaccharide, and TNF-α, induce the production of leptin, whose plasma levels have been shown to be markedly increased during inflammation, acute infection, and sepsis.Citation42,Citation43 The leptin receptor has been found in both innate and adaptive immunity cells, including macrophages, neutrophils, natural killer (NK) cells, dendritic cells, and lymphocytes.Citation36 In turn, leptin modulates the immune response and inflammation in several ways.
In the innate immune system, leptin exerts various actions. It stimulates monocytes to release pro-inflammatory cytokines, such as IL-6, IL-12, and TNF-α, and upregulates the expression of cell surface markers involved in the activation of resting monocytes.Citation36 Leptin also promotes the proliferation and differentiation of monocytes into macrophages but exerts an anti-apoptotic effect in macrophages by stimulating phagocytosis.Citation44 Notably, neutrophil activation in response to leptin requires TNF-α release from monocytes.Citation45
In the adaptive immune system, leptin induces the differentiation of CD4+ cells into IL-17-secreting Th17 cells, while inhibiting the formation of CD4+CD25+ Treg cells and stress-induced T cell apoptosis.Citation44 Studies have also shown that fa/fa rats, ob/ob mice, and humans with congenital leptin deficiency or leptin receptor mutations have higher infection susceptibility, which is associated with low total lymphocyte and circulating CD4+ T cell levels, thymocyte apoptosis, and a shift towards a Th2 immune response. Administering recombinant leptin to humans with leptin deficiency as well as ob/ob mice significantly increases thymic cellularity and upregulated cytokine secretion by immune cells.Citation46 In B cells, leptin exerts pro-inflammatory actions by inducing the secretion of IL-6, IL-10, and TNF-α.Citation47
Leptin and Cardiovascular Diseases
In humans, hyperleptinemia is associated with acute cardiovascular events and obesity-related hypertension.Citation48 Cardiac hypertrophy occurs in response to a change in load. In conditions like hypertension, the parallel addition of sarcomeres increases the myocyte width, which in turn increases the wall thickness. Hyperleptinemia is observed in patients with left ventricular hypertrophy (LVH), where the myocardium of the left ventricle enlarges. Several factors like hypertension, age, and obesity contribute to LVH. Although its exact role in cardiac hypertrophy is not entirely clear, leptin is believed to maintain the left ventricular wall thickness and cellular structure of myocyte.Citation48 In the leptin-deficient ob/ob mouse model, leptin repletion has been shown to reverse LVH back to normal, a process that is mediated through the activation of janus kinase (JAK)-dependent signaling.Citation49 Hence, leptin or its components in downstream signaling exert an anti-hypertrophic effect on the heart independent of weight loss.
In contrast, in obese people whose leptin levels are significantly increased, this hormone has been shown to be a key factor in the pathogenesis of LVH.Citation50 In vitro studies done by exposing neonatal rat ventricular myocytes to a concentration of leptin that represents average leptin levels in obese people showed an increase in cell surface area by 42%.Citation51 Inhibitors of ERK1/2 and p38 have been shown to reduce leptin-induced cardiomyocyte hypertrophy.Citation51
In addition, leptin decreases the contractility of cardiomyocytes, possibly by increasing NO production.Citation52,Citation53 For instance, acute infusion of leptin in isolated rat ventricular myocytes increases the activity of NO and decreases cardiac contractility.Citation54 With leptin, NO production was increased and intracellular calcium transients were reduced.Citation55 The spatial containment of different NO isoforms within separate subcellular compartments of the cardiac myocyte allows NO signals to have independent effects on cardiac phenotype and contractile response.Citation56
To better understand myocardial dysfunction in obesity, the β-adrenergic response to leptin in cardiomyocytes provides significant insight. Cardiomyocyte contractility is directly depressed by leptin-induced NO increase, an important aspect to consider in obesity. Leptin resistance, an important characteristic of obesity, attenuates the β-adrenergic response.Citation57 In the H9c2 cardiac cell line, 30-minute leptin therapy showed a greater basal and catecholamine-stimulated adenylate cyclase activity, but a reduced adenylate cyclase activity was seen after the 18-hour treatment.Citation58 Therefore, leptin deficiency attenuates the β-adrenergic response, while moderate stimulation of leptin can improve the contractile response.
Moreover, leptin has been shown to shift myocardial metabolism towards fatty acid utilization.Citation59 ATP, which sustains the contractile function of the heart, is mainly produced by the oxidation of fatty acids. In isolated working rat hearts, leptin introduction increased fatty acid oxidation and triacylglycerol lipolysisCitation60 and myocardial oxygen consumption was increased due to an increase in mitochondrial uncoupling protein activity.Citation60 Increased absorption of fatty acids along with reduced oxidation leads to lipotoxicity of cardiomyocytes treated with leptin.
Several actions are exerted by leptin on the vascular system. These include vascular remodeling, hypertrophy, and angiogenesis.Citation61 Leptin aids migration, proliferation, and hypertrophy of vascular smooth muscle cells. Increased platelet aggregation can be induced by leptin as well.Citation61 Moreover, in studies where mechanical stretch was used to mimic hypertension in the rat portal vein, leptin production and secretion were increased, suggesting that leptin expression is upregulated in the vasculature in hypertension.Citation32,Citation62
Molecular Signaling Pathways for Cardiac Hypertrophy
Leptin-Dependent Pathways
Leptin induces cardiac hypertrophy through several pathways. In the next sections, we provide a brief description of the JAK/STAT, MAPK, and Rho pathways that are activated by leptin, which in turn, promote cardiac remodeling and hypertrophy.
Leptin binding to its receptor leads to the dimerization of the receptor and autophosphorylation and subsequent activation of the Janus kinase-2 (JAK2).Citation63 Activated JAK2, in turn, phosphorylates Tyr 985, Tyr 1077, and Tyr 1138 residues on OB-Rb. Activated Tyr 1138 and Tyr 1077 recruit and activate signal transducers and activators of transcription 3 and 5 (STAT3 and STAT5) transcriptional factors, respectively, allowing their detachment from the complex and translocation to the nucleus.Citation64 This, in turn, promotes the transcription of the genes responsible for the production of leptin anorexigenic peptide, an appetite depressing peptide.Citation65 Furthermore, JAK2 activation leads to the phosphorylation of the src homology 2 (SH2) domain of SHP-2, which can activate the MAPK ERK1/2 signaling pathway.Citation66 Leptin is also involved in phosphatidylinositol 3 kinase (PI3K) signaling through activation of insulin receptor substrate (IRS).Citation67
Regulation of the leptin pathway involves protein tyrosine phosphatase 1B (PTP1B), which can dephosphorylate JAK2 and thereby inhibit leptin signaling.Citation68 Another negative regulator of this pathway is mediated by STAT3, which induces expression of the suppressors of the cytokine signaling (SOCS3) family members that exert negative feedback on leptin signaling via binding to Tyr 985 and specific sites on the Ob-Rb/JAK complex.Citation69
The MAPK pathway is mediated by ERK and p38 family members. This pathway is activated by Ob-Ra and Ob-Rb. ERK can be activated either by phosphorylation of Tyr 985 and activation of SHP-2 or independently of tyrosine phosphorylation, where JAK2 couples with the SH2 domain of SHP-2.Citation31 Leptin treatment has been reported to induce phosphorylation of STAT3 and ERK1/2 in leptin-deficient ob/ob mice but not in Ob-Rb-deficient db/db mice,Citation70 suggesting that Ob-Rb is the main signaling receptor or that the activation of ERK1/2 by JAK2 does not significantly occur in cardiac tissues.Citation70 p38 MAPK can also be phosphorylated in response to leptin.Citation64 The alpha and beta isoforms of p38 are abundant in the heart. Leptin has been shown to induce hypertrophy in vascular smooth muscle cells and cardiomyocytes through the p38 MAPK pathway.Citation71
Studies have shown that the Rho pathway plays an important role in mediating the hypertrophic effect of leptin via a MAPK-dependent mechanism and regulation of actin dynamics.Citation72 Inhibiting ROCK and Rho have blocked the activity of ERK1/2 and p38 after leptin treatment in cultured neonatal rat ventricular myocytes, indicating that Rho/ROCK contributes to the phosphorylation of ERK1/2 and p38.Citation73 The same effect on ERK1/2 and p38 activation was observed after disrupting the actin cytoskeleton.Citation74 Collectively, leptin can inactivate cofilin, an actin-binding protein, in a Rho/ROCK-dependent mechanism, causing an increase in G-actin and thereby a decrease in the G-actin to F-actin ratio. In turn, MAPK activation leads to hypertrophy and cardiac remodeling.Citation72
Leptin-Independent Pathways
Various signaling pathways are known to be implicated in cardiac hypertrophy. These pathways are complicated and overlappe, but all are associated with an increase in intracellular calcium ion (Ca2+) concentration upon stimulation.Citation75 Binding of Ang II and endothelin-1 (ET-1) to the cardiomyocytes’ cell surface receptors activates phospholipase C and subsequently generates inositol triphosphate and diacylglycerol, recruiting intracellular Ca2+ and activating protein kinase C.Citation76,Citation77 Thus, Ras and MAP kinase pathways are transducers of hypertrophic signals.Citation78
Moreover, calcineurin and calmodulin are molecules involved in the molecular mechanisms that induce cardiac hypertrophy. Calcineurin is a calcium-calmodulin-dependent serine/threonine protein phosphatase that activates the transcription factor NFAT.Citation79,Citation80 Upon activation, NFAT translocates to the nucleus and activates gene involved in the hypertrophic response in myocytes.Citation81 In the beginning, Ca2+ binds to the regulatory subunit of calcineurin and calmodulin, activating the calcineurin phosphatase activity. NFAT is then dephosphorylated and subsequently activated, rapidly localizing into the nucleus where it binds to GATA-4Citation82 leading to hypertrophy-related gene expression.Citation83,Citation84 Overexpression of calcineurin and NFAT in mice cardiomyocytes has been shown to lead to severe hypertrophy and subsequent heart failure.Citation85
Chronic mechanical stretch (mimicking hypertension) increases calcium levels inside the cell, thereby increasing calcineurin activation and the sequence of events leading to hypertrophy.Citation86 To illustrate that calcineurin is a crucial hypertrophic transducer in cardiomyocytes, several studies have used Cyclosporin A (CsA) or FK506, immunosuppressants and known inhibitors for calcineurin,Citation87 in their hypertrophic experimental models.Citation88,Citation89 Interestingly, various in vitro and in vivo studies demonstrated that calcineurin contributes to hypertrophy in cardiomyocytes.Citation90–Citation92 However, more recent studies that used calcineurin inhibitory agents failed to find a significant inhibition of cardiac hypertrophy in vitro.Citation93–Citation95
Indeed, research has shown that CsA and FK506 may have undesirable effects that affect the specificity of calcineurin inhibition. High doses of CsA can alter sarcoplasmic reticulum Ca2+ release through different direct and indirect mechanisms.Citation96,Citation97 CsA introduction can also inhibit the Na+, K+‐ATPase, resulting in neurotoxicity and nephrotoxicity, and subsequent increase in blood pressure (mechanical stretch).Citation98 To overcome this issue, it has been suggested that calcineurin could be genetically inhibited in different ways (reviewed in ). A recent novel study demonstrated the effect of calcineurin inhibition by CU-natriuretic peptides (CU-NP) on cardiac cell hypertrophy.Citation99 Neonatal rat myocytes treated with Ang II, PE, and ET-1 in the presence of CU-NP exhibited an attenuation of calcineurin activation and thus inhibition of NFAT translocation to the nucleus and hypertrophy induction.Citation99 highlights potential methods for calcineurin genetic inhibition. Combined together, these methods emphasize the important role of calcineurin as a transducer of cardiac hypertrophy.
Table 1 Potential Methods for Calcineurin Genetic Inhibition
Adiponectin and the Cardiovascular System
Adiponectin, an anti-inflammatory adipokine () also termed ACRP30, AdipoQ, apM1, and gelatin-binding protein (GBP), is a 30 kDa protein that is produced by adipocytes. Present abundantly, adiponectin accounts for 0.01% of total plasma proteins and functions in energy metabolism, vascular physiology, and inflammation.Citation104 The gene that encodes adiponectin is present on chromosome 3q27 and consists of three exons and two introns.Citation105 Adiponectin circulates in the body at high concentrations but its levels are lowered in obese individuals compared to lean individuals.Citation106 The primary sequence of the adiponectin protein is homologous to complement protein C1q and has structural homology to tumor necrosis factor-α (TNF-α).Citation107
Figure 3 Anti-inflammatory properties of adiponectin. Adiponectin affects several tissues and promotes anti-inflammatory actions by several mechanisms.
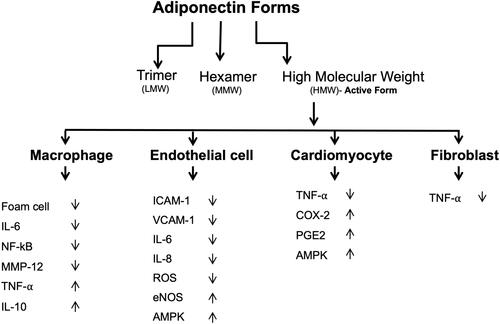
Adiponectin consists of three domains which include a signal sequence at the N terminus, a globular domain at the C terminus, and a collagen-like domain.Citation31 This protein can form a wide range of multimer complexes with these three domains. In the circulation, adiponectin exists in three major oligomeric multimers: A high molecular weight (HMW) multimer of 12–18 monomers, a middle molecular weight hexamer (MMW), and low molecular weight (LMW) trimer.Citation108
Adiponectin receptors are mainly AdipoR1, AdipoR2, and T-cadherin. When AdipoR1 and AdipoR2 genes were deleted from different mice models, adiponectin’s actions became defective, indicating the importance of AdipoR1 and AdipoR2 in mediating adiponectin signaling.Citation109 AdipoR1 is expressed highly in the heart compared to AdipoR2.Citation110 5ʹ adenosine monophosphate-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR) signaling are two major targets of adiponectin signaling. AdipoR1 deletion has resulted in the blocking of adiponectin mediated phosphorylation of AMPK, while AdipoR2 gene deletion has resulted in increased adiposity and glucose intolerance.Citation111 T-cadherin is a surface molecule expressed in cardiomyocytes, vascular smooth muscle cells, and endothelial cells. It regulates their proliferation, migration, and survival.Citation112
The role of adiponectin in the pathogenesis of cardiovascular diseases is unclear. A correlation between low adiponectin levels and cardiovascular diseases has been reported.Citation113,Citation114 The association of adiponectin in the development of cardiovascular disease is still controversial, and it appears that low levels of adiponectin are not a reliable marker for cardiovascular disease. However, cardiac remodeling has been shown to be influenced by adiponectin, which functions to suppress cardiac growth.Citation115 Adiponectin attenuates cardiac hypertrophy in response to pressure overload in adiponectin-knockout, wild, and diabetic db/db mice.Citation116
Adiponectin has been shown to exert a protective action against myocardial ischemia-reperfusion injury, a process that is mediated by its ability to activate cyclooxygenase-2 (COX-2) in cardiac cells.Citation116 When adiponectin binds to T-cadherin, it leads to the attenuation of ROSproduction, TNF-α levels, and AMPK and COX-2 activation.Citation108 Thus, by activating the AMPK-mediated anti-apoptotic actions and COX-2 mediated anti-inflammatory actions, adiponectin protects the ischemic heart from injury.
In addition to its anti-inflammatory properties, adiponectin exerts protective actions against cardiac overload-induced hypertrophy.Citation28 Adiponectin has been shown to attenuate overload-induced and adrenergically-induced cardiomyocyte hypertrophy in mice by inhibiting hypertrophic signaling pathways via AMPK.Citation4 Therefore, the downregulated levels of this adipokine in obesity are linked to increased cardiac risk in at least 2 ways: Myocardial insult and inflammation-induced cardiovascular insufficiency.
Furthermore, adiponectin acts on the vascular system and exerts protective effects in various vascular disorders, such as endothelial dysfunction.Citation117 Adiponectin also protects against the harmful vascular remodeling that occurs in response to excessive mechanical stretch induced by hypertension.Citation32 Mechanical stretch activates ERK1/2 signaling, increases ROS formation, and induces GATA-4 nuclear translocation in vascular smooth muscle cells, whereas exogenous adiponectin at physiological concentrations inhibits all of these effects in mechanically stretched vessels.Citation32 Moreover, exogenous adiponectin has been shown to activate the protective LKB1-AMPK-eNOS signaling axis, which in turn, inhibits hypertension-induced hypertrophy.Citation32,Citation118,Citation119
Autophagy
Autophagy, a catabolic process that is vastly conserved in all eukaryotes and mammals,Citation120 is prompted by one of two processes. The first occurs via physiological processes such as the intracellular accumulation of proteins and organelles that would consequently be degraded into reusable, biologically active monomers like amino acids;Citation120 the second process occurs via pathological triggers like starvation, cellular stress, and ROS.Citation121 The process of autophagy is essential for organism survival. Pyo et al report that transgenic mice overexpressing autophagy-related gene 5 (Atg5), an essential protein for the formation of autophagosomes, have a median life span increase by 17% in addition to their anti-aging phenotypes, which include enhanced leanness, increased insulin sensitivity, and improved motor function.Citation122 Concurrently, Yoshii et al report that a deficiency in Atg5 is lethal.Citation123
Autophagy is categorized into three types: Macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA).Citation124 Macroautophagy is considered the chief pathway for the lysis of large portions of the cytoplasm and cellular contents, such as long-lived proteins, aggregated proteins, damaged organelles, and intracellular pathogens.Citation125 This process is initiated by the nucleation and expansion of the double-membraned structure, known as the phagophore, followed by the fusion of lysosomes, membrane-bound cell organelles that contains digestive enzymes, eventually leading to the formation of autophagolysosomes.Citation124 Microautophagy is the process by which lysosomes digest small quantities of cytosolic substrates; this could be induced by nitrogen starvation or rapamycin via regulatory signaling complex pathways.Citation124 Additionally, the process is initiated by autophagic tubes-mediated direct engulfment of cytoplasmic materials and involves both invagination and vesicle scission into the lumen.Citation126 On the other hand, CMAis initiated following prolonged physiological stress and is mediated through heat shock cognate protein.Citation124 Intriguingly, CMA is a uniquely selective mechanism; proteins that are targeted through this pathway are individually tagged through a recognition motif in their amino acid sequences. This ensures efficient and specific degradation of damaged or abnormal proteins.Citation127
Autophagy and Cardiac Remodeling
A variety of physiological, such as exercise and pregnancy, and pathological, including hypertension, valvular heart disease, and myocardial infarction, stimuli could trigger cardiac compensatory mechanisms.Citation128 Cardiac modulation in terms of chambers’ volume, systolic contraction, diastolic relaxation, heart rate, and muscle mass are common in response to environmental stimulation.Citation128 In pathological cases, cardiac remodeling begins with the compensatory mechanism of increasing cardiac overload.Citation128 Nonetheless, persistent pathological stimuli consequently induce systolic dysfunction and heart failure.Citation128
Autophagy is thought to be one of the main mechanisms controlling the cardiac hypertrophic response.Citation129 Although it seems counterintuitive, the involvement of autophagy, a degradation mechanism, in myocyte proliferation and hypertrophic remodeling is attributed to the complex nature of cardiac myocytes. This warrants more than protein synthesis to extend to the disassembling and remodeling of existing cellular elements throughout the process of cardiac remodeling.Citation128 In fact, a growing body of evidence in both human heart failure and diseased animal models supports the involvement of the lysosomal pathway in the pathogenesis of heart disease;Citation129 indeed, augmented cardiac hypertrophy was found to be associated with autophagy.Citation129
To elaborate on the involvement of autophagy in cardiac remodeling, it is crucial to differentiate between adaptive, which is vital for cellular homeostasis, and maladaptive, that is associated with sustained stimuli.Citation128 In this review, we illustrate the different maladaptive autophagy signaling pathways in cardiac hypertrophy. Taking into account the minute capacity of cardiac myocytes to undergo cell proliferation, hypertrophy is, therefore, their only method of cellular growth in response to various stimuli. Cardiac hypertrophy is originally initiated as an adaptive mechanism in response to elevated cardiac wall tension and to increase cardiac output.Citation130 For instance, Yamaguchi et al report that inducing transient pressure overload through a thoracic transverse aortic constriction (TAC) in wild-type mice leads to cardiac hypertrophy without resulting in cardiac dysfunction or fibrosis.Citation131 To illustrate the role autophagy plays in the described case, the expression of LC3, a soluble protein that is recruited to the autophagosomal membrane and is reflective of starvation-induced autophagic activity, was determined using immunoblotting. The study reports a significant decrease in LC3 expression in TAC-induced hypertrophied hearts at 1-week post-surgery as opposed to placebo wild-type hearts and transgenic mice.Citation131 Similarly, Dammrich et al report a diminished autophagic response in Sprague–Dawley rats following supravalvular aortic constriction.Citation132
Cardiac hypertrophy is a multi-phased process; during the compensatory hypertrophic response, autophagy is downregulated. The noted decrease in autophagy leads to the accumulation of ROS and harmful proteins.Citation133 In the case of a sustained stimulus, such as continuous exposure to an elevated load, cardiac hypertrophy becomes maladaptive.Citation133 This, coupled with the increased accumulation of malfunctional mitochondria and harmful proteins, causes a further increase in ROS generation, thus, inducing cellular necrosis and eventually leading to heart failure.Citation130 Concurrently, Nishida et al report an initial downregulation in autophagy 1-week post-surgery in wild-type mouse hearts treated with TAC, as opposed to the observed left ventricular (LV) dilation and cardiac dysfunction associated with an upregulation of autophagy following 4 weeks of surgery in the same animal model.Citation130 Additionally, in Atg5-deficient mice, a similar stimulus leads to the development of cardiac hypertrophy coupled with LV dilatation and cardiac dysfunction 1-week post surgery.Citation130 This emphasizes the role that autophagy upregulation plays in the transitioning from hypertrophy to heart failure.
The mechanism by which autophagy leads to heart failure is yet to be fully illustrated. Nonetheless, studies have shown that the accumulated malfunctional mitochondria increase the generation of ROS and decrease ATP production.Citation134 This, in turn, triggers energy sensor AMPK and inhibits mammalian target of rapamycin (mTOR), permitting the activation of Unc-51-like kinase (ULK1).Citation135 The formation of Atg1/ULK1 and Class III phosphatidylinositol 3-kinase (PI3K III)/Beclin1 is crucial in the development of the phagophore. The elongation and maturation process of the phagophore is facilitated through two ubiquitin-like conjugation systems; the first involves Atg12-Atg5-conjugate which facilitates localization, while the second involves conjugation of phosphatidylethanolamine (PE) to microtubule-associated protein 1 light chain 3 (LC3)/Atg8, leading to the formation of autophagosome-associated form LC3-III and eventually forming the autolysosomes.Citation136 The described pathway leads to the upregulation of autophagy in heart failure.Citation134 Moreover, Nishida et al and Miyata et al have reported similar findings in the myocardium caused by dilated cardiomyopathy, valvular disease, and ischemic heart diseaseCitation130 and in animal models such as UM-7.1 cardiomyopathic hamsters,Citation137 respectively.
Regulation of Autophagy by Inflammatory Cytokines
As pointed above, this review focuses on the pro-inflammatory cytokine leptin and anti-inflammatory adiponectin. In general, Th1 cytokines, such as IL-1, IL-2, IL-6, interferon gamma (IFN-γ), transforming growth factor beta (TGF-β), and TNF-α, induce autophagy. Conversely, the classical Th2 cytokines, including IL-4, IL-10 and IL-13, generally inhibit autophagy.Citation138
IFN-γ-induced activation of macrophages increases the maturation of mycobacteria-containing phagosomes and stimulates autophagy in an Irgm1/IRGM-dependent manner, leading to increased intracellular killing of pathogens.Citation139 However, phagosome maturation by IFN-γ can be abrogated by TNF blockers, suggesting the involvement of TNF-α in IFN-γ-induced phagosome maturation and autophagy. Interestingly, TNF-α involvement in autophagy induction has also been shown in various cell types; however, the actions and mechanisms differ between the different cell types.Citation138 TNF-α can also stimulate autophagy via the ERK1/2 pathway.Citation140 Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) has been shown to inhibit TNF-α-induced autophagy, a process that is dependent on ROS generation.Citation141
TGF-β induces the formation of autophagosomes and increases the expression of autophagic mRNA, including Atg5 and Atg7.Citation142 However, both CC chemokine CCL2 (monocyte chemoattractant protein-1) and IL-6 can stimulate autophagy and upregulate anti-apoptotic proteins.Citation138 In addition, IL-1 has been shown to stimulate autophagy,Citation143 whereas insulin-like growth factor-1Citation144 and fibroblast growth factor 2Citation145 both exert inhibitory effects on autophagy. However, the detailed mechanisms still need to be elucidated.
On the other hand, Th2 cytokines have been shown to inhibit starvation- or inflammatory stimulation-induced autophagy through different pathways.Citation138 Inhibition of starvation-induced autophagy has been shown to occur via the Akt pathway, whereas inhibition of IFN-γ or rapamycin-induced autophagy occurs through the STAT signaling pathway.Citation138,Citation146
Pro-Inflammatory Leptin and Autophagy
Few studies have discussed the relationship between leptin and autophagy. For instance, Malik et al have shown that leptin injections stimulate the expression of LC3 protein and reduce the expression of p62 protein in the myocardium, as well as in the kidney, skeletal muscle, and liver, indicating the activation of autophagy.Citation147 In contrast, the study also reported that mice with leptin receptor deficiency manifested an increase in autophagy in these tissues.Citation147 Therefore, this study suggests that any modification in leptin levels may lead to autophagy.Citation147 Being a downstream regulator of leptin and an upstream effector of autophagy,Citation148 mTOR, a nutrient-sensitive protein, is a key player in this mechanism.
A recent study suggested that in cardiac endothelial cells, leptin contributes to autophagy during cardiac hypertrophic remodeling.Citation149 In pressure overload-induced hypertrophy, the activation of the Akt/mTOR pathway through leptin receptor inhibited endothelial autophagy and promoted cardiac inflammation, fibrosis, and dysfunction.Citation149 Leptin was shown to impair cardiomyocyte contractile function; however, this effect was reduced after inhibition of autophagy using e-methyladenin (3-MA), demonstrating that leptin affects cardiac contractile function through an autophagy-dependent mechanism.Citation150 In addition, Luo et al have shown that leptin reduces contractile function in rat cardiomyocytes by decreasing oxidative stress and inhibiting autophagy.Citation55
Leptin induces the formation of autophagosomes, as evidenced by leptin-mediated upregulation in LC3-II, Atg5, and beclin 1 expression and downregulation in p62 expression.Citation151 Moreover, it has been shown that peripherally administering recombinant leptin leads to autophagy in peripheral tissues, including the heart, skeletal muscle, and liver.Citation147 In addition, pharmacologically inhibiting ROS by tempol has been shown to attenuate leptin-induced autophagosome formation, demonstrating a crucial role for ROS in leptin-induced autophagy.Citation150
Anti-Inflammatory Adiponectin and Autophagy
Recently, the regulation of cardiac autophagy has been considered as a crucial mechanistic part of the adiponectin cardioprotective effect. In response to oxidative stress, adiponectin has been shown to inhibit excessive autophagic activity in cardiomyocytes mainly through the ERK-mTOR-AMPK signaling pathway. This demonstrates the potential of adiponectin to act as an anti-oxidant in oxidative stress-related cardiovascular diseases including hypertension-mediated heart failure.Citation152 An important role for adiponectin was also reported in high-fat diet-induced obesity and cardiac pathology. Deficiency in adiponectin enhanced high-fat diet-induced obesity, cardiac hypertrophy, and contractile dysfunction, possibly through AMPK/mTOR-dependent reduction in myocardial autophagy.Citation153 Moreover, adiponectin deficiency has been shown to promote Ang II–induced cardiac inflammation and fibrosis and reduce macrophage autophagy in cardiac tissues.Citation154
Research has shown that in response to pressure overload, aged mice with adiponectin deficiency exhibit reduced cardiac autophagy and increased susceptibility to cardiac dysfunction.Citation155 Furthermore, adiponectin directly enhances autophagy flux in cardiac myoblasts.Citation155 Collectively, these data indicate that adiponectin may protect against the development of cardiac dysfunction likely through inducing cardiac autophagy, which is important in preserving normal heart function in response to stress or aging.Citation155 Induction of autophagy is involved in many biological processes regulated by both leptin and adiponectin, although the mechanisms by which these adipokines modulate autophagy are not well defined.
Conclusion
Due to the increased risk of cardiovascular disease development in obesity, there is recently a great interest in identifying new mechanisms that govern the cardiovascular response in diseases related to obesity. The mechanisms by which leptin and adiponectin contribute to cardiovascular diseases are multifactorial in nature. For instance, leptin mediated-activation of JAK/STAT, MAPK, and Rho pathways was found to initiate and promote a complex process involving cardiomyocyte proliferation, fibrosis, vascular rarefaction, electrophysiological changes, and inflammation, eventually leading to cardiac remodeling and hypertrophy. Conversely, the role of adiponectin in the pathogenesis of cardiovascular diseases is yet to be fully elucidated. Adiponectin was reported to possess cardio-protective properties through the activation of AMPK-mediated anti-apoptotic actions and COX-2 mediated anti-inflammatory actions. Equally, autophagic activity is crucial in maintaining cardiovascular balance and function. Alterations in autophagic flux in response to leptin and adiponectin occur in many types of heart disorders, and autophagic activity seems to be essential in maintaining cardiovascular balance and function. Changes in autophagic flux, whether in excessive or inadequate levels, may cause cardiac dysfunction. Thus, fully elucidating the mechanisms through which leptin and adiponectin exert their actions on autophagy and determining their effect on the cardiovascular system would be pivotal in the identification of new therapeutic targets for the treatment of various cardiovascular diseases.
Disclosure
The authors report no conflicts of interest for this work and have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the materials discussed in this paper.
Additional information
Funding
References
- Ruan Y, Guo Y, Zheng Y, et al. Cardiovascular disease (CVD) and associated risk factors among older adults in six low-and middle-income countries: results from SAGE wave 1. BMC Public Health. 2018;18(1):778. doi:10.1186/s12889-018-5653-9
- Kannan A, Janardhanan R. Hypertension as a risk factor for heart failure. Curr Hypertens Rep. 2014;16(7):447. doi:10.1007/s11906-014-0447-7
- Wu J, Thabet SR, Kirabo A, et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114(4):616–625. doi:10.1161/CIRCRESAHA.114.302157
- Ghantous CM, Farhat R, Djouhri L, et al. Molecular mechanisms of adiponectin-induced attenuation of mechanical stretch-mediated vascular remodeling. Oxid Med Cell Longev. 2020;2020:6425782. doi:10.1155/2020/6425782
- Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116(8):1462–1476. doi:10.1161/CIRCRESAHA.116.304937
- Maillet M, Van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38. doi:10.1038/nrm3495
- Andrade-Oliveira V, Camara NO, Moraes-Vieira PM. Adipokines as drug targets in diabetes and underlying disturbances. J Diabetes Res. 2015;2015:681612. doi:10.1155/2015/681612
- Park PH. Autophagy induction: a critical event for the modulation of cell death/survival and inflammatory responses by adipokines. Arch Pharm Res. 2018;41(11):1062–1073. doi:10.1007/s12272-018-1082-7
- Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51(4):584–593. doi:10.1016/j.yjmcc.2011.06.010
- Geng J, Baba M, Nair U, Klionsky DJ. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J Cell Biol. 2008;182(1):129–140. doi:10.1083/jcb.200711112
- Roever L, Palandri Chagas AC. Editorial: cardiac remodeling: new insights in physiological and pathological adaptations. Front Physiol. 2017;8:751. doi:10.3389/fphys.2017.00751
- Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407.
- Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122(25):2727–2735. doi:10.1161/CIRCULATIONAHA.110.942268
- Sayer G, Bhat G. The renin-angiotensin-aldosterone system and heart failure. Cardiol Clin. 2014;32(1):21–32. doi:10.1016/j.ccl.2013.09.002
- Rababa’h AM, Guillory AN, Mustafa R, Hijjawi T. Oxidative stress and cardiac remodeling: an updated edge. Curr Cardiol Rev. 2018;14(1):53–59. doi:10.2174/1573403X14666180111145207
- Wang C, Wang X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim Biophys Acta. 2015;1852(2):188–194. doi:10.1016/j.bbadis.2014.07.028
- Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Curr Hypertens Rep. 2009;11(6):406–411. doi:10.1007/s11906-009-0070-1
- Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. 2016;106(1):62–69.
- Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116(7):1269–1276. doi:10.1161/CIRCRESAHA.116.305381
- Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis. 2006;48(5):326–341. doi:10.1016/j.pcad.2006.02.001
- Force T, Rosenzweig A, Choukroun G, Hajjar R. Calcineurin inhibitors and cardiac hypertrophy. Lancet. 1999;353(9161):1290–1292. doi:10.1016/S0140-6736(99)90016-8
- Doggrell SA, Brown L. Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc Res. 1998;39(1):89–105. doi:10.1016/S0008-6363(98)00076-5
- Drazner MH. The transition from hypertrophy to failure: how certain are we? Am Heart Assoc. 2005;112:936-938. doi:10.1161/CIRCULATIONAHA.105.558734
- Francis GS. Pathophysiology of chronic heart failure. Am J Med. 2001;110(7):37–46. doi:10.1016/S0002-9343(98)00385-4
- Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. 2016;118(11):1786–1807. doi:10.1161/CIRCRESAHA.115.306885
- Ouchi N, Kihara S, Funahashi T, et al. Reciprocal association of C-Reactive protein with adiponectin in blood stream and adipose tissue. Circulation. 2003;107(5):671–674. doi:10.1161/01.CIR.0000055188.83694.B3
- Farkhondeh T, Llorens S, Pourbagher-Shahri AM, et al. An overview of the role of adipokines in cardiometabolic diseases. Molecules. 2020;25:21. doi:10.3390/molecules25215218
- Mattu HS, Randeva HS. Role of adipokines in cardiovascular disease. J Endocrinol. 2013;216(1):T17–T36. doi:10.1530/JOE-12-0232
- Vlasova M, Purhonen AK, Jarvelin MR, Rodilla E, Pascual J, Herzig KH. Role of adipokines in obesity-associated hypertension. Acta Physiologica. 2010;200(2):107–127. doi:10.1111/j.1748-1716.2010.02171.x
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi:10.1038/372425a0
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. doi:10.1038/27376
- Ghantous CM, Kobeissy FH, Soudani N, et al. Mechanical stretch-induced vascular hypertrophy occurs through modulation of leptin synthesis-mediated ROS formation and GATA-4 nuclear translocation. Front Pharmacol. 2015;6:240.
- Soudani N, Ghantous CM, Farhat Z, Shebaby WN, Zibara K, Zeidan A. Calcineurin/NFAT activation-dependence of leptin synthesis and vascular growth in response to mechanical stretch. Front Physiol. 2016;7:433. doi:10.3389/fphys.2016.00433
- Landecho MF, Tuero C, Valenti V, Bilbao I, de la Higuera M, Fruhbeck G. Relevance of leptin and other adipokines in obesity-associated cardiovascular risk. Nutrients. 2019;11:11. doi:10.3390/nu11112664
- Yadav A, Kataria MA, Saini V, Yadav A. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta. 2013;417:80–84. doi:10.1016/j.cca.2012.12.007
- Fernández-Riejos P, Najib S, Santos-Alvarez J, et al. Role of leptin in the activation of immune cells. Mediators Inflamm. 2010;2010:568343. doi:10.1155/2010/568343
- Cava AL, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4(5):371–379. doi:10.1038/nri1350
- Paz-Filho G, Mastronardi C, Franco CB, Wang KB, Wong ML, Licinio J. Leptin: molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq Bras Endocrinol Metabol. 2012;56(9):597–607. doi:10.1590/S0004-27302012000900001
- Chandra RK. Cell-mediated immunity in genetically obese (C57BL/6J ob/ob) mice. Am J Clin Nutr. 1980;33(1):13–16. doi:10.1093/ajcn/33.1.13
- Mandel MA, Mahmoud AAF. Impairment of cell-mediated immunity in mutation diabetic mice (db/db). J Immunol. 1978;120(4):1375.
- McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. 2017;127(1):5–13. doi:10.1172/JCI88876
- Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflammation. 2013;2013:139239. doi:10.1155/2013/139239
- Iikuni N, Lam QLK, Lu L, Matarese G, La Cava A. Leptin and inflammation. Curr Immunol Rev. 2008;4(2):70–79. doi:10.2174/157339508784325046
- Francisco V, Pino J, Campos-Cabaleiro V, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. 2018;9:640. doi:10.3389/fphys.2018.00640
- Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJ. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J Immunol. 2004;172(3):1809–1814. doi:10.4049/jimmunol.172.3.1809
- Poetsch MS, Strano A, Guan K. Role of leptin in cardiovascular diseases. Front Endocrinol (Lausanne). 2020;11:354. doi:10.3389/fendo.2020.00354
- Agrawal S, Gollapudi S, Su H, Gupta S. Leptin activates human B cells to secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J Clin Immunol. 2011;31(3):472–478. doi:10.1007/s10875-010-9507-1
- Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108(6):754–759. doi:10.1161/01.CIR.0000083716.82622.FD
- Tune JD, Considine RV. Effects of leptin on cardiovascular physiology. J Am Soc Hypertens. 2007;1(4):231–241. doi:10.1016/j.jash.2007.04.001
- Hall ME, Harmancey R, Stec DE. Lean heart: role of leptin in cardiac hypertrophy and metabolism. World J Cardiol. 2015;7(9):511–524. doi:10.4330/wjc.v7.i9.511
- Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2003;93(4):277–279. doi:10.1161/01.RES.0000089255.37804.72
- Chaban R, Buschmann K, Ghazy A, et al. In vitro effect of leptin on human cardiac contractility. J Nutr Sci. 2019;8:e12–e12. doi:10.1017/jns.2019.6
- Dong F, Zhang X, Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor–NADPH oxidase pathway. Hypertension. 2006;47(2):222–229. doi:10.1161/01.HYP.0000198555.51645.f1
- Nickola MW, Wold LE, Colligan PB, Wang G-J, Samson WK, Ren J. Leptin attenuates cardiac contraction in rat ventricular myocytes. Hypertension. 2000;36(4):501–505. doi:10.1161/01.HYP.36.4.501
- Luo L-J, Liu Y-P, Yuan X, et al. Leptin attenuates the contractile function of adult rat cardiomyocytes involved in oxidative stress and autophagy. Acta Cardiologica Sinica. 2016;32(6):723–730.
- Barouch L, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi A. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. nature. 2002;416(6878):337–339. doi:10.1038/416337a
- Carter S, Caron A, Richard D, Picard F. Role of leptin resistance in the development of obesity in older patients. Clin Interv Aging. 2013;8:829–844.
- Illiano G, Naviglio S, Pagano M, et al. Leptin affects adenylate cyclase activity in H9c2 cardiac cell line: effects of short- and long-term exposure. Am J Hypertens. 2002;15(7 Pt 1):638–643. doi:10.1016/S0895-7061(02)02925-4
- Ma Y, Li J. Metabolic shifts during aging and pathology. Compr Physiol. 2015;5(2):667–686.
- Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277(33):29424–29430. doi:10.1074/jbc.M203813200
- Nakata N, Soejima N, Maruyama I. Leptin promotes aggregation of human platelets via the long form of its receptor. Diabetes. 1999;48:426–429. doi:10.2337/diabetes.48.2.426
- Zeidan A, Rajapurohitam V, Javadov S, Chakrabarti S, Karmazyn M. Leptin induces vascular smooth muscle cell hypertrophy through angiotensin II- and endothelin-1-dependent mechanisms and mediates stretch-induced hypertrophy. J Pharmacol Exp Thera. 2005;315:1075–1084. doi:10.1124/jpet.105.091561
- Ferrao RD, Wallweber HJ, Lupardus PJ. Receptor-mediated dimerization of JAK2 FERM domains is required for JAK2 activation. Elife. 2018;7:e38089. doi:10.7554/eLife.38089
- Procaccini C, Lourenco EV, Matarese G, La Cava A. Leptin signaling: a key pathway in immune responses. Curr Signal Transduct Ther. 2009;4(1):22–30. doi:10.2174/157436209787048711
- Sobrino Crespo C, Perianes Cachero A, Puebla Jiménez L, Barrios V, Arilla Ferreiro E. Peptides and food intake. Front Endocrinol (Lausanne). 2014;5:58. doi:10.3389/fendo.2014.00058
- Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol Histopathol. 2007;22(11):1251–1267.
- Thon M, Hosoi T, Ozawa K. Possible integrative actions of leptin and insulin signaling in the hypothalamus targeting energy homeostasis. Front Endocrinol (Lausanne). 2016;7:138. doi:10.3389/fendo.2016.00138
- Tsou RC, Bence KK. Central regulation of metabolism by protein tyrosine phosphatases. Front Neurosci. 2013;6:192. doi:10.3389/fnins.2012.00192
- Mullen M, Gonzalez-Perez RR. Leptin-induced JAK/STAT signaling and cancer growth. Vaccines (Basel). 2016;4(3):26. doi:10.3390/vaccines4030026
- Buettner C, Pocai A, Muse ED, Etgen AM, Myers MG Jr, Rossetti L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 2006;4(1):49–60. doi:10.1016/j.cmet.2006.04.014
- Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101(6):545–559. doi:10.1161/CIRCRESAHA.107.156596
- Zeidan A, Javadov S, Karmazyn M. Essential role of Rho/ROCK-dependent processes and actin dynamics in mediating leptin-induced hypertrophy in rat neonatal ventricular myocytes. Cardiovasc Res. 2006;72(1):101–111.
- Zeidan A, Javadov S, Chakrabarti S, Karmazyn M. Leptin-induced cardiomyocyte hypertrophy involves selective caveolae and RhoA/ROCK-dependent p38 MAPK translocation to nuclei. Cardiovasc Res. 2007;77(1):64–72. doi:10.1093/cvr/cvm020
- Hoffman L, Jensen CC, Yoshigi M, Beckerle M. Mechanical signals activate p38 MAPK pathway-dependent reinforcement of actin via mechanosensitive HspB1. Mol Biol Cell. 2017;28(20):2661–2675. doi:10.1091/mbc.e17-02-0087
- Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3(11):a004242–a004242. doi:10.1101/cshperspect.a004242
- Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II–induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ Res. 1993;73(3):424–438. doi:10.1161/01.RES.73.3.424
- Zou Y, Komuro I, Yamazaki T, et al. Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J Biol Chem. 1996;271(52):33592–33597. doi:10.1074/jbc.271.52.33592
- Thorburn A, Thorburn J, Chen S-Y, et al. HRas-dependent pathways can activate morphological and genetic markers of cardiac muscle cell hypertrophy. J Biol Chem. 1993;268(3):2244–2249. doi:10.1016/S0021-9258(18)53988-0
- Kudryavtseva O, Aalkjaer C, Matchkov VV. Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. Febs J. 2013;280(21):5488–5499. doi:10.1111/febs.12414
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi:10.1146/annurev.immunol.15.1.707
- Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110(8):1077–1086. doi:10.1161/CIRCRESAHA.111.260729
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275(5308):1930–1933. doi:10.1126/science.275.5308.1930
- Saito T, Fukuzawa J, Osaki J, et al. Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35(9):1153–1160. doi:10.1016/S0022-2828(03)00234-7
- Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35(12):1385–1394. doi:10.1016/j.yjmcc.2003.10.001
- Molkentin JD, Lu J-R, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–228. doi:10.1016/S0092-8674(00)81573-1
- Zhu J, McKeon F. NF-AT activation requires suppression of Crm1-dependent export by calcineurin. Nature. 1999;398(6724):256. doi:10.1038/18473
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17(18):2205–2232. doi:10.1101/gad.1102703
- Goldspink PH, McKinney RD, Kimball VA, Geenen DL, Buttrick PM. Angiotensin II induced cardiac hypertrophy in vivo is inhibited by cyclosporin A in adult rats. Mol Cell Biochem. 2001;226(1–2):83–88. doi:10.1023/A:1012789819926
- Shimoyama M, Hayashi D, Zou Y, et al. Calcineurin inhibitor attenuates the development and induces the regression of cardiac hypertrophy in rats with salt-sensitive hypertension. Circulation. 2000;102(16):1996–2004. doi:10.1161/01.CIR.102.16.1996
- Sussman MA, Lim HW, Gude N, et al. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281(5383):1690–1693. doi:10.1126/science.281.5383.1690
- Meguro T, Hong C, Asai K, et al. Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circ Res. 1999;84(6):735–740. doi:10.1161/01.RES.84.6.735
- Mende U, Kagen A, Cohen A, Aramburu J, Schoen F, Neer E. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc Natl Acad Sci. 1998;95(23):13893–13898. doi:10.1073/pnas.95.23.13893
- Luo Z, Shyu K-G, Gualberto A, Walsh K. Calcineurin inhibitors and cardiac hypertrophy. Nat Med. 1998;4(10):1092. doi:10.1038/2578
- Zhang W, Kowal RC, Rusnak F, Sikkink RA, Olson EN, Victor RG. Failure of calcineurin inhibitors to prevent pressure-overload left ventricular hypertrophy in rats. Circ Res. 1999;84(6):722–728. doi:10.1161/01.RES.84.6.722
- Fatkin D, McConnell BK, Mudd JO, et al. An abnormal Ca 2+ response in mutant sarcomere protein–mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106(11):1351–1359. doi:10.1172/JCI11093
- Janssen PM, Zeitz O, Keweloh B, et al. Influence of cyclosporine A on contractile function, calcium handling, and energetics in isolated human and rabbit myocardium. Cardiovasc Res. 2000;47(1):99–107. doi:10.1016/S0008-6363(00)00052-3
- Park KS, Kim TK, Kim DH. Cyclosporin A treatment alters characteristics of Ca2+-release channel in cardiac sarcoplasmic reticulum. Am J Physiol-Heart Circulatory Physiol. 1999;276(3):H865–H872. doi:10.1152/ajpheart.1999.276.3.H865
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273(22):13367–13370. doi:10.1074/jbc.273.22.13367
- Kilić A, Rajapurohitam V, Sandberg SM, et al. A novel chimeric natriuretic peptide reduces cardiomyocyte hypertrophy through the NHE-1–calcineurin pathway. Cardiovasc Res. 2010;88(3):434–442. doi:10.1093/cvr/cvq254
- Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97(3):1196–1201. doi:10.1073/pnas.97.3.1196
- Zou Y, Yao A, Zhu W, et al. Isoproterenol activates extracellular signal-regulated protein kinases in cardiomyocytes through calcineurin. Circulation. 2001;104(1):102–108. doi:10.1161/hc2601.090987
- Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T cell development and function in calcineurin A beta -deficient mice. Proc Natl Acad Sci USA. 2002;99(14):9398–9403. doi:10.1073/pnas.152665399
- Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy: where have we been? Where are we going? J Physiol. 2002;541(Pt 1):1–8. doi:10.1113/jphysiol.2002.017129
- Gu HF. Biomarkers of adiponectin: plasma protein variation and genomic DNA polymorphisms. Biomark Insights. 2009;4:123–133. doi:10.4137/BMI.S3453
- Ghoshal K, Bhattacharyya M. Adiponectin: probe of the molecular paradigm associating diabetes and obesity. World J Diabetes. 2015;6(1):151–166. doi:10.4239/wjd.v6.i1.151
- Berk ES, Kovera AJ, Boozer CN, Pi-Sunyer FX, Johnson JA, Albu JB. Adiponectin levels during low- and high-fat eucaloric diets in lean and obese women. Obes Res. 2005;13(9):1566–1571. doi:10.1038/oby.2005.192
- Anders H, Berg T. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89.
- Ghantous CM, Azrak Z, Hanache S, Abou-Kheir W, Zeidan A. Differential role of leptin and adiponectin in cardiovascular system. Int J Endocrinol. 2015;2015:534320. doi:10.1155/2015/534320
- Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17(2):185–196. doi:10.1016/j.cmet.2013.01.001
- Parker-Duffen JL, Nakamura K, Silver M, et al. Divergent roles for adiponectin receptor 1 (AdipoR1) and AdipoR2 in mediating revascularization and metabolic dysfunction in vivo. J Biol Chem. 2014;289(23):16200–16213. doi:10.1074/jbc.M114.548115
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116(7):1784–1792. doi:10.1172/JCI29126
- Resinka TJ, M P, Joshia MB, Kyriakakisa E, Erneb P. Cadherins in cardiovascular disease. J Swiss Soc Infect Dis. 2009;139:0930.
- Amirzadegan A, Shakarami A, Borumand MA, Davoodi G, Ghaffari-Marandi N, Jalali A. Correlation between plasma adiponectin levels and the presence and severity of coronary artery disease. J Tehran Heart Cent. 2013;8(3):140–145.
- Kim-Mitsuyama S, Soejima H, Yasuda O, et al. Total adiponectin is associated with incident cardiovascular and renal events in treated hypertensive patients: subanalysis of the ATTEMPT-CVD randomized trial. Sci Rep. 2019;9(1):16589. doi:10.1038/s41598-019-52977-x
- Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res. 2007;74(1):11–18. doi:10.1016/j.cardiores.2006.10.009
- Teresa A, Hopkins NO, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res. 2007;74:11–18.
- Ebrahimi-Mamaeghani M, Mohammadi S, Arefhosseini SR, Fallah P, Bazi Z. Adiponectin as a potential biomarker of vascular disease. Vasc Health Risk Manag. 2015;11:55–70.
- Zhang M, Dong Y, Xu J, et al. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102(3):328–337. doi:10.1161/CIRCRESAHA.107.163253
- Nour-Eldine W, Ghantous CM, Zibara K, et al. Adiponectin attenuates angiotensin II-induced vascular smooth muscle cell remodeling through nitric oxide and the RhoA/ROCK pathway. Front Pharmacol. 2016;7:86. doi:10.3389/fphar.2016.00086
- Li L, Xu J, He L, et al. The role of autophagy in cardiac hypertrophy. Acta Biochim Biophys Sin (Shanghai). 2016;48(6):491–500. doi:10.1093/abbs/gmw025
- Ucar A, Gupta SK, Fiedler J, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3(1):1078. doi:10.1038/ncomms2090
- Pyo J-O, Yoo S-M, Ahn -H-H, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi:10.1038/ncomms3300
- Yoshii SR, Kuma A, Mizushima N. Transgenic rescue of Atg5-null mice from neonatal lethality with neuron-specific expression of ATG5: systemic analysis of adult Atg5-deficient mice. Autophagy. 2017;13(4):763–764. doi:10.1080/15548627.2017.1280221
- Khandia R, Dadar M, Munjal A, et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8(7):674. doi:10.3390/cells8070674
- Randow F, Münz C. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol. 2012;33(10):475–487. doi:10.1016/j.it.2012.06.003
- Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69(7):1125–1136. doi:10.1007/s00018-011-0865-5
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104. doi:10.1038/cr.2013.153
- Rifki OF, Hill JA. Cardiac autophagy: good with the bad. J Cardiovasc Pharmacol. 2012;60(3):248–252. doi:10.1097/FJC.0b013e3182646cb1
- Miyamoto S. Autophagy and cardiac aging. Cell Death Differ. 2019;26(4):653–664. doi:10.1038/s41418-019-0286-9
- Nishida K, Otsu K. Autophagy during cardiac remodeling. J Mol Cell Cardiol. 2016;95:11–18. doi:10.1016/j.yjmcc.2015.12.003
- Yamaguchi O, Higuchi Y, Hirotani S, et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci. 2003;100(26):15883. doi:10.1073/pnas.2136717100
- Dammrich J, Pfeifer U. Cardiac hypertrophy in rats after supravalvular aortic constriction. II. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;43(3):287–307. doi:10.1007/BF02932962
- Osterholt M, Nguyen TD, Schwarzer M, Doenst T. Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail Rev. 2013;18(5):645–656. doi:10.1007/s10741-012-9346-7
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi:10.1016/j.molcel.2010.09.023
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi:10.1128/MCB.06159-11
- Chiu B, Jantuan E, Shen F, Chiu B, Sergi C. Autophagy-inflammasome interplay in heart failure: a systematic review on basics, pathways, and therapeutic perspectives. Ann Clin Lab Sci. 2017;47(3):243–252.
- Miyata S, Takemura G, Kawase Y, et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am J Pathol. 2006;168(2):386–397. doi:10.2353/ajpath.2006.050137
- Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6(1):24. doi:10.1186/s40169-017-0154-5
- Maphasa RE, Meyer M, Dube A. The macrophage response to mycobacterium tuberculosis and opportunities for autophagy inducing nanomedicines for tuberculosis therapy. Front Cell Infect Microbiol. 2021;10:915. doi:10.3389/fcimb.2020.618414
- Yuan Y, Ding D, Zhang N, et al. TNF-α induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle. 2018;17(11):1390–1402. doi:10.1080/15384101.2018.1482150
- Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Codogno P. Regulation of autophagy by NF-kappaB transcription factor and reactives oxygen species. Autophagy. 2007;3(4):390–392. doi:10.4161/auto.4248
- Ding Y, Choi ME. Regulation of autophagy by TGF-β: emerging role in kidney fibrosis. Semin Nephrol. 2014;34(1):62–71. doi:10.1016/j.semnephrol.2013.11.009
- Khan NM, Ansari MY, Haqqi TM. Sucrose, but not glucose, blocks IL1-β-induced inflammatory response in human chondrocytes by inducing autophagy via AKT/mTOR pathway. J Cell Biochem. 2017;118(3):629–639. doi:10.1002/jcb.25750
- Liu ZQ, Zhao S, Fu WQ. Insulin-like growth factor 1 antagonizes lumbar disc degeneration through enhanced autophagy. Am J Transl Res. 2016;8(10):4346–4353.
- Wang X, Qi H, Wang Q, et al. FGFR3/fibroblast growth factor receptor 3 inhibits autophagy through decreasing the ATG12-ATG5 conjugate, leading to the delay of cartilage development in achondroplasia. Autophagy. 2015;11(11):1998–2013. doi:10.1080/15548627.2015.1091551
- Park HJ, Lee SJ, Kim SH, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. 2011;48(4):720–727. doi:10.1016/j.molimm.2010.10.020
- Malik SA, Mariño G, BenYounes A, et al. Neuroendocrine regulation of autophagy by leptin. Cell Cycle. 2011;10(17):2917–2923.
- Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–1435. doi:10.1152/physrev.00030.2009
- Gogiraju R, Hubert A, Fahrer J, et al. Endothelial leptin receptor deletion promotes cardiac autophagy and angiogenesis following pressure overload by suppressing akt/mtor signaling. Circ Heart Fail. 2019;12(1):e005622.
- Machender RK, Nathan D. Autophagy inhibition rescues against leptin-induced cardiac contractile dysfunction. Curr Pharm Des. 2014;20(4):675–683. doi:10.2174/13816128113199990019
- Nepal S, Kim MJ, Hong JT, et al. Autophagy induction by leptin contributes to suppression of apoptosis in cancer cells and xenograft model: involvement of p53/FoxO3A axis. Oncotarget. 2015;6(9):7166–7181. doi:10.18632/oncotarget.3347
- Essick EE, Wilson RM, Pimentel DR, et al. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS One. 2013;8(7):e68697. doi:10.1371/journal.pone.0068697
- Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta. 2013;1832(8):1136–1148. doi:10.1016/j.bbadis.2013.03.013
- Qi G-M, Jia L-X, Li Y-L, Li -H-H, Du J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology. 2014;155(6):2254–2265. doi:10.1210/en.2013-2011
- Jahng JW, Turdi S, Kovacevic V, Dadson K, Li RK, Sweeney G. Pressure overload-induced cardiac dysfunction in aged male adiponectin knockout mice is associated with autophagy deficiency. Endocrinology. 2015;156(7):2667–2677. doi:10.1210/en.2015-1162