
Abstract
Purpose
MicroRNAs (miRNAs) are emerging as essential regulators in the development of cerebral ischemia/reperfusion (I/R) injury. This study aimed to explore the regulation of miR-9-3p on FGF19-GSK-3β/Nrf2/ARE signaling in cerebral I/R injury.
Materials and Methods
A mouse model with I/R injury was constructed by middle cerebral artery occlusion (MCAO) and an HT22 cell model was established by oxygen-glucose deprivation/reperfusion (OGD/R). The expression of miR-9-3p was detected by RT-qPCR. Protein expression of fibroblast growth factor 19 (FGF19), cleaved caspase-3, and GSK-3β signaling-related proteins (p-GSK-3β and Nrf2) were detected by Western blot. Cell viability was assessed by MTT assay. Oxidative stress was detected by commercial kits. The target of miR-9-3p was predicted by TargetScan and confirmed by luciferase reporter assay. The effects of miR-9-3p on GSK-3β/Nrf2/ARE signaling were assessed by rescue experiments.
Results
MiR-9-3p was significantly upregulated in brain tissues of MCAO/R-treated mice and OGD/R-treated HT22 cells. Downregulation of miR-9-3p attenuated infarct volume and neurological outcomes of MCAO/R-treated mice in vivo and OGD/R-induced cell injury and oxidative stress in vitro, while overexpression of miR-9-3p showed the opposite effects. MiR-9-3p directly bound to the 3′-untranslated region of FGF19 and negatively regulated its expression. Inhibition of miR-9-3p enhanced GSK-3β/Nrf2/ARE signaling-mediated antioxidant response, while this effect was partially eliminated by FGF19 or Nrf2 silencing.
Conclusion
Our study suggests that inhibition of miR-9-3p protects against cerebral I/R injury through activating GSK-3β/Nrf2/ARE signaling-mediated antioxidant responses by targeting FGF19, providing a potential therapeutic target for ischemic stroke.
Introduction
Worldwide, ischemic stroke, caused by a clot that blocks blood flow to the brain, can be severely disabling and sometimes fatal.Citation1 A well-known therapy for ischemic stroke is cerebral ischemia/reperfusion (I/R), which is also a crucial risk factor resulting in poor prognosis for patients with ischemic stroke and exacerbating brain injury.Citation2,Citation3 Cerebral I/R injury has been reported to result in serious clinical manifestations such as myocardial hibernation, acute heart failure, cerebral dysfunction, and multiple organ dysfunction syndrome.Citation4 Increasing evidence has revealed that cerebral I/R injury could lead to the production of a large number of reactive oxygen species (ROS), which might further damage the nervous system.Citation5 Therefore, thorough understanding of the specific molecular mechanisms involved in cerebral I/R injury or ROS resistance contributes to identifying new and effective therapeutic targets for ischemic stroke.
MicroRNAs (miRNAs), found in most eukaryotes, are a class of small non-coding RNA molecules, approximately 22 nucleotides in length.Citation6 It has been reported that miRNAs can regulate gene expression by directly binding to the 3′-UTR region of their target mRNAs, resulting in translational silencing.Citation7 In the neural system, more and more miRNAs have been found to play important roles in the progression and pathogenesis of the nervous system. MiR-145 regulates the differentiation of neural stem cells by modulating the Sox2-Lin28/let-7 signaling pathway.Citation8 MiR-455-3p functions as a potential peripheral indicator for Alzheimer’s disease.Citation9 In the ischemic brain, a series of abnormally expressed miRNAs have been identified. For example, miR-298 exacerbates I/R injury following ischemic stroke by directly targeting Act1.Citation10 MiR-130a has been reported to have neuroprotective effects against ischemic stroke by regulating the PTEN/PI3K/AKT pathway.Citation11 Previous studies reported that miR-9-3p is significantly elevated in the cerebrospinal fluid following subarachnoid hemorrhage and this elevation is associated with a poor functional outcome of delayed cerebral ischemia.Citation12 MiR-9-3p has been identified as being significantly enriched in the brain relative to other tissues in patients with traumatic brain injury, and as exhibiting dramatically greater brain specificity.Citation13 The expression of miR-9-3p was also significantly higher in the cerebrospinal fluid of patients with acute ischemic stroke compared to controls.Citation14 These reports indicated that miR-9-3p might play a potential role in cerebral I/R injury. However, the function and underlying molecular mechanisms of miR-9-3p remain unclear.
Previous studies have revealed a rapid increase in ROS production immediately after acute ischemic stroke which overwhelms antioxidant capacity, resulting in further brain damage.Citation15 Recently, targeting ROS production to reduce ROS in brain tissues has becoming a potential neuroprotective treatment for ischemic stroke.Citation5 Fibroblast growth factor 19 (FGF19) is an important cytoprotective regulator that can antagonize oxidative stress and cell apoptosis under adverse conditions, including cerebral I/R injury.Citation16 The serine/threonine protein kinase, glycogen synthase kinase-3β (GSK-3β), is abundantly expressed in neurons.Citation17 Nuclear factor-E2-related factor 2 (Nrf2), a redox-sensitive transcription factor, is a direct downstream target of GSK-3β and can bind to the antioxidant response elements (ARE) in the nucleus and then activate downstream antioxidant genes such as heme oxygenase-1 (HO-1) and NADPH-quinone oxidoreductase 1 (NQO1).Citation18 GSK-3β prevents the nuclear translocation of Nrf2 and suppresses antioxidant response in cells.Citation19 Previous studies have revealed that downregulation of GSK-3β can efficiently attenuate neuronal death and protect neurons from cerebral I/R injury by activating Nrf2/ARE signaling.Citation20,Citation21 Fang et al revealed that FGF19 overexpression alleviated hypoxia/re-oxygenation-induced injury in cardiomyocyte by modulating the GSK-3β/Nrf2/ARE signaling pathway.Citation16 Increasing evidence reveals that a series of miRNAs participate in cerebral I/R injury through modulating GSK-3β/Nrf2/ARE signaling, including miR-99a,Citation22 miR-335,Citation23 and miR-322.Citation24 Hence, the identification of miRNAs targeting GSK-3β may be a potential therapeutic target for cerebral I/R injury.
Here, we demonstrated that miR-9-3p was significantly upregulated during I/R injury both in vivo and in vitro. Further, FGF19 was identified as a direct target of miR-9-3p and miR-9-3p negatively regulated FGF19 expression by binding to its 3′-UTR. Moreover, our study revealed that inhibition of miR-9-3p protected against cerebral I/R injury by targeting FGF19 to activate GSK-3β/Nrf2/ARE signaling-mediated antioxidant responses.
Materials and Methods
Animals and the Construction of an In Vivo Ischemic/Reperfusion (I/R) Injury Model
Adult C57BL/6J mice (6–13 weeks, approximately 20 ± 5 g) were purchased from the Experimental Animal Center of Jinan Central Hospital. This study was approved by the Institutional Animal Care and Use Committee of Jinan Central Hospital. All animal procedures were performed according to Guidelines for Care and Use of Laboratory Animals of Jinan Central Hospital. All mice were randomly divided into two groups: sham group and model group (n = 6 in each group). The mouse model with transient cerebral ischemia was induced through middle cerebral artery occlusion (MCAO), as previously described.Citation25 Operated mice were subjected to 2 h of ischemia before reperfusion. In the sham group, mice underwent the same operations except for the MCAO treatment. After different times (6, 12 and 24 h) for reperfusion, the neurological deficits of mice were evaluated. Afterwards, mice were sacrificed by cervical dislocation and the cortex of the ischemia side in mice brains were rapidly removed for the subsequent experiments.
Cortical Injection of microRNAs
MiR-9-3p mimics (#MC13072), miR-NC (negative control, #4,464,058), miR-9-3p inhibitor (#MH13072), and inhibitor NC (negative control, #4,464,078) were purchased from Thermo Fisher Scientific. MiR-9-3p mimics (100 μM), miR-9-3p inhibitor (100 μM), or the corresponding negative controls (100 μM) were mixed with the siRNA-Mate (GenePharma) and incubated for 20 min at room temperature. Mice were deeply anesthetized by the intraperitoneal injection of pentobarbital sodium and these miRNAs were stereotaxically injected (0.2 μL/min) into cerebral cortex of mice (bregma: −0.2 mm posterior, 3 mm dorsoventral, 4.5 mm lateral), as previously described.Citation26 After 10 min of injection, mice were exposed to MCAO for 2 h and then reperfusion for 24 h. Mice in the sham group were injected with equal amounts of physiological saline (n = 6 in each group).
Evaluation of Neurological Deficits and Brain Water Content
After 6, 12 and 24 h of reperfusion, the neurological deficits in mice were evaluated using the scales as described by Longa et al:Citation27 0 score, mice behave normally; 1 score, mice cannot fully stretch their left front legs; 2 score, mice turn around in a circle; 3 score, mice fall down to the left side; 4 score, mice cannot move by themselves and lose consciousness.
Brain water content was determined 24 h after reperfusion, as previously reported.Citation28 Briefly, infarct brain hemispheres were weighed and recorded as the wet weight using an electronic scale, and then dried overnight at 100 °C in an oven to obtain the dry weight. The brain water content was calculated according to the following formula: brain water content = [(wet weight) – (dry weight)]/(wet weight) × 100%.
Evaluation of Cerebral Infarct Volume
Cerebral infarct volume of mice was determined by TTC staining assay, as previously described.Citation29 After reperfusion, the mice were anesthetized and their brains dissected and cut into 1.5-mm slices. The slices were then soaked in a 1% TTC phosphate buffer at 37 °C for 30 min in the dark and fixed with 4% paraformaldehyde for 30 min. The images of ordered brain slices were captured and analyzed using an electronic scanner (Tsinghua Unisplendour A688, Xi’an, China). The percentage of infarct volume was calculated according to the following equation: Infarct volume (%) = Infarct volume/Total volume of slice × 100. Normal brain tissues stained red and infarct tissues were white.
Immunohistochemistry
The mice brain tissues were fixed and cut coronally into 5 mm-thick serial sections, and then immunohistochemistry was performed with an immunohistochemical kit (Boster Biological Technology, Wuhan, China) using FGF19 (1:500, ab225942, Abcam) antibody and cleaved caspase-3 (1:300; ab2302, Abcam) antibody according to the manufacturer’s instructions. A blinded investigator used a microscope to take images and chose images randomly for each section. Analysis of FGF19 expression was conducted using Image-Pro Plus software.
Cell Culture and the Construction of an in vitro I/R Injury Model
Mouse neuronal cell line HT22 was purchased from BeNa Culture Collection (BNCC, Kunshan, China) and cultured in Dulbecco modified Eagle medium (DMEM Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37 °C with additional 5% CO2. To mimic cerebral I/R injury in vitro, HT22 cells were subjected to oxygen-glucose deprivation/reperfusion (OGD/R) treatment, as previously described.Citation30 In brief, approximately 2×104 HT22 cells were cultured in glucose-free DMEM in a hypoxic chamber with 3% O2, 5% CO2 and 92% N2 for 2 h, and then cells were transferred into glucose-containing DMEM for re-oxygenation under normal conditions (95% air and 5% CO2) for 6 h, 12 h or 24 h. Cells still cultured in glucose-containing DMEM under normoxic conditions were considered as the control group.
Cell Transfection
Small interfering RNA targeting FGF19 (si-FGF19) (#sc-39,480), si-Nrf2 (#sc-37,030) and negative controls (si-NC) (#sc-37,007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). MicroRNAs (MiR-9-3p mimics, miR-NC, miR-9-3p inhibitor and inhibitor NC) and siRNAs (si-FGF19, si-Nrf2 and si-NC) were transfected into HT22 cells with a final concentration at 20nM using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. After transfection for 48 h, HT22 cells were subjected to OGD for 2 h and subsequent re-oxygenation for 24 h. Cells were then harvested for the subsequent experiments.
RT-qPCR
Total RNA of the cortex of the ischemia side in mice brains and cultured cells was extracted by using TRIzol Regent (Invitrogen), according to the manufacturer’s instructions. Approximately 2 μg of total RNA was reverse-transcribed into complementary DNA (cDNA) using a PrimeScriptTM RT reagent kit (Takara). qPCR assay was performed using SYBR Green PCR Master Mix (Takara) on an ABI PRISM 7500 Fast Real-time PCR instrument (Applied Biosystems). GAPDH and U6 were used as the internal reference for mRNA and miRNAs, respectively. The relative expression fold of targets was calculated using the 2−ΔΔCt method. The primers used were as follows: miR-9-3p forward: 5ʹ-GAGGCCCGTTTCTCTCTTTG-3ʹ, reverse: 5ʹ-AGCTTTATGACGGCTCTGTG-3ʹ; U6 forward: 5ʹ-CTCGCTTCGGCAGCACA-3ʹ, reverse: 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ; FGF19 forward: 5ʹ-CGCTGTCGGTAGCCAGAG-3ʹ, reverse: 5ʹ-CTCTGCACGCCCTTGATG-3ʹ; HO-1 forward: 5ʹ-GGAACTTTCAGAAGGGCCAG-3ʹ, reverse: 5ʹ-GTCCTTGGTGTCATGGGTCA-3ʹ; NQO-1 forward: 5ʹ-GTATCCTGCCGAGTCTGTT-3ʹ, reverse: 5ʹ-GATCCCTTGCAGAGAGTACA-3ʹ; GAPDH forward: 5ʹ-CCACCCATGGCAAATTCCATGGCA-3ʹ, reverse: 5ʹ-TCTAGACGGCAGGTCAGGTCCACC-3ʹ.
Western Blot
Total protein of the cortex of the ischemia side in mice brains or cultured cells was extracted using a RIPA lysis buffer (Beyotime Institute of Biotechnology, China). Approximately equal amounts of protein were separated by 12% SDS-PAGE and transferred onto PVDF membranes. After blocking in 5% skim milk, the membranes were incubated with primary antibodies, including FGF19 (1:500, ab225942, Abcam), GSK-3β (1:500, ab93926, Abcam), phosphorylated-GSK-3β (1:500, ab75814, Abcam), Nrf2 (1:500, ab62352, Abcam), Lamin B2 (1:1000, ab8983, Abcam) and GAPDH (1:2000, ab8425, Abcam), overnight at 4 °C. Afterwards, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3000, ab205718, Abcam) at room temperature for 1 h. The protein bands were visualized using an enhanced chemiluminescent (ECL) kit.
MTT Assay
Cell viability was evaluated using an MTT Cell Viability Assay Kit (Invitrogen; Thermo Fisher Scientific, Inc.), according to the standard protocols. In brief, transfected or treated HT22 cells cultured in 96-well plates were added to 10 μL of MTT stock solution for 4 h at 37 °C. Then, 100 μL of dissolution reagent was added to each well and incubated for another 4 h. The absorbance was detected using an optical density measurement at 570 nm under a microplate absorbance reader (Bio-Rad, Sunnyvale, CA, USA). Cell viability of the control group was regarded as 100% and cell viabilities in other groups were calculated as percentages of the control.
Detection of Intracellular ROS
Intracellular ROS levels were measured using a Reactive Oxygen Species Assay Kit (Beyotime Biotechnology, Shanghai, China) and MitoSOX Red Mitochondrial Superoxide Indicator (YEASEN, Shanghai, China), respectively, according to the manufacturers' instructions. ROS levels were detected by fluorescence intensity at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. MitoSOX fluorescence intensity was detected at an excitation wavelength of 510 nm and an emission wavelength of 580 nm.
Luciferase Reporter Assay
Fragments of wild type (WT) and mutant type (MUT) 3′-UTR of FGF19 containing the putative binding site with miR-9-3p were inserted into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega). The recombinant luciferase reporter vectors were co-transfected with miR-9-3p mimics or miR-NC into HT22 cells using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, cells were lysed and the relative luciferase activity of FGF19 3′-UTR WT or FGF19 3′-UTR MUT was detected by the dual luciferase reporter system (Promega). For detection of Nrf2/ARE transcriptional activity, HT22 cells were co-transfected with Nrf2/ARE reporter vector (Promega), phRL-TK Renilla luciferase vector (Promega), and miR-9-3p mimics/inhibitor or corresponding negative controls (miR-NC and inhibitor NC). Forty-eight hours after culturing, the luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). Data are presented as the ratio of Renilla luciferase activity in cell lysates to firefly luciferase.
Statistical Analysis
All data were presented as means ± standard deviation (SD) and each experiment was separately repeated three times. Data analysis was performed using SPSS 21.0 software. A single comparison between the two groups was determined by Student’s t-test, and comparisons between multiple groups were analyzed by one-way analysis of variance (ANOVA). P <0.05 was considered to be statistically significant.
Results
Downregulation of miR-9-3p Attenuated Ischemic Brain Infarction In Vivo
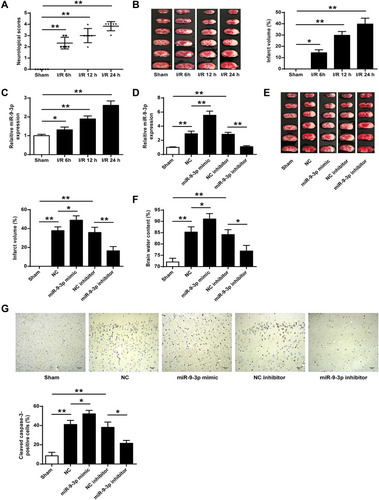
To explore the role of miR-9-3p in ischemic brain damage, the experimental model with cerebral ischemia was used. We found that both neurological scores and infarct volume of mice subjected to MCAO followed by reperfusion for 6, 12 and 24 h were increased compared with those in sham mice (p <0.01, ; p <0.05, ), suggesting that the mouse model with cerebral I/R injury was successfully established. Then the expression of miR-9-3p in the cortex of the ischemic regions of I/R mice was detected by RT-qPCR, and the results showed that the level of miR-9-3p was significantly increased in brain tissues of mice subjected to I/R injury compared with that in sham mice (p <0.05, ). To further determine the role of mIR-9-3p in ischemic brain damage, mice were injected with miR-9-3p mimics/inhibitor or corresponding negative controls and then exposed to MCAO/R treatment. RT-qPCR results indicated that miR-9-3p mimics increased miR-9-3p level and miR-9-3p inhibitor decreased its expression (p <0.05, ). Moreover, the infarct volume and brain water content of mice in the miR-NC and inhibitor NC group was still increased compared with that in sham mice (p <0.01), overexpression of miR-9-3p (miR-9-3p mimics) further enhanced infarct volume and brain water content of mice compared with that in the miR-NC group (p <0.05), while downregulation of miR-9-3p (inhibitor) obviously decreased infarct volume and brain water content of mice compared with that in the inhibitor NC group (p <0.01, p <0.05, and ). By performing immunohistochemistry assay, we found that MCAO treatment significantly upregulated the number of cleaved caspase-3 positive cells in cerebral cortex compared with the sham group (p <0.01), and miR-9-3p mimics further increased the expression of cleaved caspase-3 compared with miR-NC (p <0.05), while miR-9-3p inhibitor obviously reduced the number of cleaved caspase-3 positive cells compared with the NC inhibitor (p <0.05, ). These results indicated that miR-9-3p might promote ischemic brain damage.
Figure 1 MiR-9-3p was upregulated in brain tissues of I/R mice and downregulated in attenuated ischemic brain infarction in vivo. (A–C) Mice were subjected to MCAO/R for 6 h, 12 h or 24 h. (A) Neurobehavioral outcomes. (B) Infarct volumes. (C) The expression of miR-9-3p in brain tissues was detected by RT-qPCR. (D–F) Mice were injected with miR-9-3p mimics/inhibitor or corresponding negative controls and then exposed to MCAO/R for 24 h. (D) The expression of miR-9-3p in brain tissues was detected by ER-qPCR. (E) Infarct volumes. (F) Brain water content. (G) Mice were injected with miR-9-3p mimics/inhibitor or corresponding negative controls and then exposed to MCAO/R for 24 h. The expression of cleaved caspase-3 in cerebral cortex was evaluated by immunohistochemistry. (×200, scale bar = 100μm). n = 6 in each group. Data were expressed as mean ± SD. *p <0.05, **p <0.01.
Downregulation of miR-9-3p Attenuated OGD/R-Induced Neuronal Injury in vitro
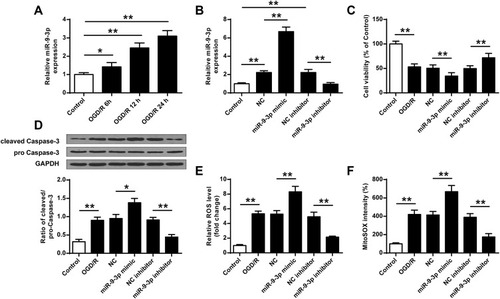
To further determine the effects of miR-9-3p in ischemic brain damage, mouse neuronal cell line HT22 were treated by OGD/R to mimic ischemic conditions in vitro. RT-qPCR showed that the expression of miR-9-3p increased in HT22 cells exposed to OGD/R for 6, 12 and 24 h compared with that in the control group (p <0.05, p <0.01, ). Subsequently, HT22 cells were transfected with miR-9-3p mimics/inhibitor or corresponding negative controls and then treated by OGD/R for 24 h. Transfection efficiency was detected by RT-qPCR, and the results indicated that miR-9-3p level was significantly upregulated by transfection of miR-9-3p mimics and downregulated by transfection of miR-9-3p inhibitor (p <0.01, ). MTT assay showed that OGD/R treatment significantly decreased cell viability compared with the control group (p <0.01), overexpression of miR-9-3p further decreased cell viability in OGD/R-treated HT22 cells compared with miR-NC (p <0.01), while downregulation of miR-9-3p increased cell viability in OGD/R-treated HT22 cells compared with inhibitor NC (p <0.01) (). Western blot indicated that OGD/R treatment significantly promoted the expression of cleaved caspase-3 compared with the control group (p <0.01) and upregulation of miR-9-3p further enhanced cleaved caspase-3 expression in OGD/R-treated HT22 cells compared with miR-NC (p <0.05), while downregulation of miR-9-3p markedly decreased its expression in OGD/R-treated HT22 cells compared with the inhibitor NC group (p <0.01) (). In addition, OGD/R treatment significantly promoted the production of intracellular ROS and mitochondrial ROS (p <0.01), and overexpression of miR-9-3p further increased the production of ROS in OGD/R-exposed HT22 cells (p <0.01), while downregulation of miR-9-3p obviously decreased ROS production in OGD/R-treated HT22 cells (p <0.01) ( and ). These results suggested that downregulation of miR-9-3p protected HT22 cells from OGD/R-induced injury in vitro.
Figure 2 MiR-9-3p was upregulated by OGD/R treatment in HT22 cells and downregulation of miR-9-3p attenuated OGD/R-induced neuronal injury in vitro. (A) HT22 cells were treated by OGD/R for 6, 12 and 24 h, and the relative expression of miR-9-3p was detected by RT-qPCR. (B–F) HT22 cells were transfected with miR-9-3p mimics/inhibitor or corresponding negative controls and then treated by OGD/R for 24 h. (B) The relative expression of miR-9-3p was detected by RT-qPCR. (C) Cell viability was detected by MTT assay. (D) The protein expression of cleaved caspase-3 was detected by Western blot. (E and F) Intracellular (E) and mitochondrial (F) ROS level were measured using respective detection kits. Data were expressed as mean ± SD. *p <0.05, **p <0.01.
FGF19 Was a Target of miR-9-3p
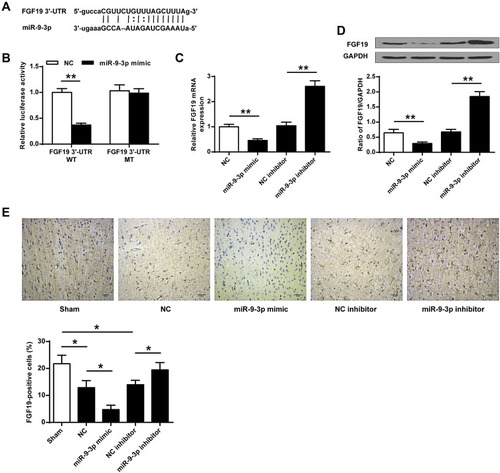
To explore the underlying mechanisms of miR-9-3p, TargetScan was used to predict the potential targets of miR-9-3p, and the prediction suggested that there was a putative binding site between miR-9-3p and 3′-UTR of FGF19 (), indicating that FGF19 might be a target of miR-9-3p. Then, luciferase reporter assay was performed, and the results showed that miR-9-3p mimics significantly decreased the relative luciferase activity of the FGF19 3′-UTR WT vector compared with miR-NC (p <0.01) but exhibited no change on the luciferase activity of the FGF19 3′-UTR MUT vector (). Meanwhile, overexpression of miR-9-3p significantly decreased the expression of FGF19 both at the mRNA and protein levels compared with miR-NC (p <0.01), and downregulation of miR-9-3p increased FGF19 expression compared with inhibitor NC (p <0.01, and ). In addition, the results of immunohistochemistry assay showed that MCAO treatment significantly reduced the number of FGF19-positive cells in cerebral cortex compared with the sham group (p <0.05), miR-9-3p mimics further reduced the expression of FGF19 in cerebral cortex compared with miR-NC (p <0.05), while miR-9-3p inhibitor obviously increased the number of FGF19-positive cells in cerebral cortex compared with NC inhibitor (p <0.05, ). These results indicated that FGF19 was a target of miR-9-3p.
Figure 3 FGF19 was a target of miR-9-3p. (A) The putative binding site between miR-9-3p and 3′-UTR of FGF19 was predicted by TargetScan. (B) The relative luciferase activity of FGF19 3′-UTR WT or MUT vector was detected by dual luciferase reporter system. (C and D) HT22 cells were transfected with miR-9-3p mimics/inhibitor or corresponding negative controls. The expression of FGF19 was detected by RT-qPCR (C) and Western blot (D). (E) Mice were injected with miR-9-3p mimics/inhibitor or corresponding negative controls and then exposed to MCAO/R for 24 h. The expression of FGF19 in cerebral cortex was evaluated by immunohistochemistry. (×200, scale bar=100μm). n = 6 in each group. Data were expressed as mean ± SD. *p <0.05, **p <0.01.
miR-9-3p Regulated the GSK-3β/Nrf2/ARE Antioxidant Signaling
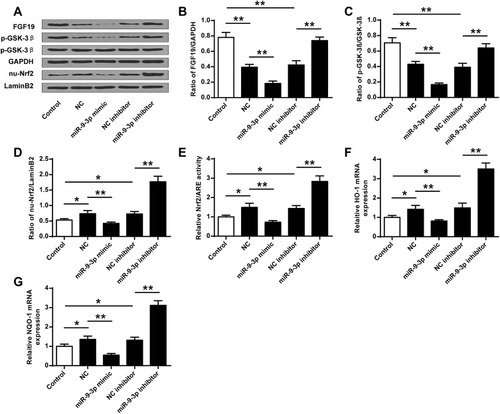
Due to the ROS production in OGD/R-treated HT22 cells and the effects of miR-9-3p, we explored the effects of miR-9-3p on GSK-3β/Nrf2/ARE antioxidant signaling. We found that OGD/R treatment significantly decreased the protein expression of FGF19 and p-GSK-3β, while it increased the expression of nuclear Nrf2 (nu-Nrf2) in HT22 cells compared with the control group; upregulation of miR-9-3p decreased the expression of FGF19, p-GSK-3β and nu-Nrf2 in OGD/R-treated HT22 cells compared with miR-NC; and downregulation of miR-9-3p increased the expression of FGF19, p-GSK-3β and nu-Nrf2 in OGD/R-treated HT22 cells compared with inhibitor NC (–). Meanwhile, overexpression of miR-9-3p decreased the Nrf2/ARE activity while downregulation of miR-9-3p increased Nrf2/ARE activity (). In addition, the expression of Nrf2/ARE downstream target genes including HO-1 and NQO1 was increased by OGD/R treatment, significantly downregulated by miR-9-3p mimics transfection, and upregulated by miR-9-3p inhibitor transfection ( and ). These results suggested that miR-9-3p was involved in the regulation of GSK-3β/Nrf2/ARE antioxidant signaling to affect the OGD/R injury in vitro.
Figure 4 MiR-9-3p regulated the GSK-3β/Nrf2/ARE antioxidant signaling. HT22 cells were transfected with miR-9-3p mimics/inhibitor or corresponding negative controls and then treated by OGD/R for 24 h. (A) The protein expression of FGF19, p-GSK-3β and nuclear Nrf2 (nu-Nrf2) was detected by Western blot. Quantitative analysis of FGF19 (B), p-GSK-3β (C) and nu-Nrf2 (D). (E) Relative Nrf2/ARE transcriptional activity was determined by luciferase reporter assay. (F and G) The expression of HO1 (F) and NQO1 (G) was detected by RT-qPCR. Data were expressed as mean ± SD. *p <0.05, ** < 0.01.
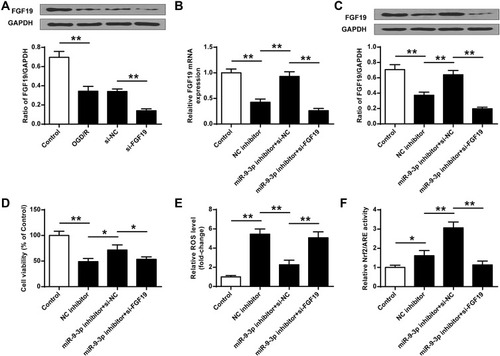
Silencing of FGF19 Reversed the Protective Effects of miR-9-3p Inhibition on OGD/R Injury
To determine whether miR-9-3p functions by targeting FGF19, HT22 cells were transfected with siRNA targeting FGF19 (si-FGF19) or si-NC and then exposed to OGD/R treatment. Western blot assay showed that OGD/R treatment significantly reduced the expression of FGF19 compared with the control group (p <0.01), and silencing of FGF19 further decreased FGF19 expression in OGD/R-treated HT22 cells compared with si-NC (p <0.01) (). Next, HT22 cells were co-transfected with miR-9-3p inhibitor and si-NC, or co-transfected with miR-9-3p inhibitor and si-FGF19. The results of RT-qPCR () and Western blot () revealed that miR-9-3p inhibitor increased FGF19 expression compared with inhibitor NC in OGD/R-treated HT22 cells (p <0.01), while co-transfection of miR-9-3p inhibitor and si-FGF19 significantly decreased FGF19 expression compared with the miR-9-3p inhibitor/si-NC group. Meanwhile, miR-9-3p inhibition mediated neuroprotective effects including cell viability, and ROS production in OGD/R-treated HT22 cells was partially reversed by FGF19 silencing (p <0.05) ( and ). In addition, the enhancement of miR-9-3p inhibition on the Nrf2/ARE signaling pathway was also partially attenuated by FGF19 silencing (p <0.05, ). These results suggested that silencing of FGF19 attenuated the protective role of miR-9-3p inhibition on OGD/R injury.
Figure 5 Silencing of FGF19 reversed the protective effects of miR-9-3p inhibition on OGD/R injury. (A) HT22 cells were transfected with si-FGF19 or si-NC, and then exposed to OGD/R for 24 h. The expression of FGF19 was detected by Western blot. (B–F) HT22 cells were transfected with inhibitor NC, or co-transfected with miR-9-3p inhibitor and si-NC, or co-transfected with miR-9-3p inhibitor and si-FGF19. Next, cells were exposed to OGD/R for 24 h. (B and C) The expression of FGF19 was detected by RT-qPCR (B) and Western blot (C). (D) Cell viability was measured by MTT assay. (E) Intracellular ROS level was measured using a corresponding detection kit. (F) Relative Nrf2/ARE transcriptional activity was determined by luciferase reporter assay. Data were expressed as mean ± SD. *p <0.05, **p <0.01.
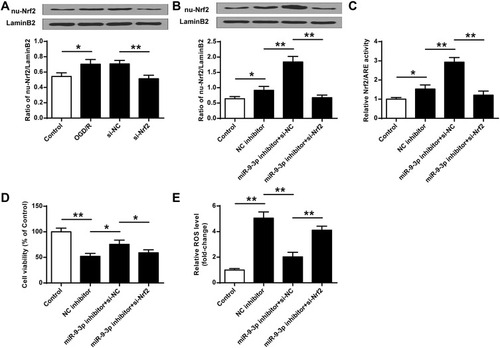
Blocking of Nrf2 Signaling Abrogated miR-9-3p Inhibition Mediated Neuroprotective Effects in vitro
To confirm the neuroprotective effects of miR-9-3p inhibition by acting on Nrf2 antioxidant signaling, HT22 cells were transfected with si-Nrf2 or si-NC, and then exposed to OGD/R for 24 h. RT-qPCR results indicated that OGD/R treatment increased nu-Nrf2 expression compared with the control group (p <0.05), while silencing of Nrf2 markedly decreased nu-Nrf2 expression in OGD/R-treated HT22 cells compared with si-NC (p <0.01, ). Then, HT22 cells were transfected with inhibitor NC, or co-transfected with miR-9-3p inhibitor and si-NC, or co-transfected with miR-9-3p inhibitor and so-Nrf2, and then cells were exposed to OGD/R for 24 h. We found that si-Nrf2 transfection significantly restricted the expression of nuclear Nrf2 (nu-Nrf2) and decreased Nrf2/ARE transcriptional activity cells in OGD/R-treated HT22 cells which were elevated by miR-9-3p inhibitor (p <0.05, and ). In addition, the neuroprotective effects of miR-9-3p inhibitor including cell viability and ROS production were obviously reversed by Nrf2 silencing (p <0.05, and ). These results indicated that blocking of Nrf2 signaling significantly attenuated miR-9-3p inhibition mediated neuroprotective effect in vitro.
Figure 6 Blocking of Nrf2 signaling abrogated miR-9-3p inhibition mediated neuroprotective effects in vitro. (A) HT22 cells were transfected with si-Nrf2 or si-NC, and then exposed to OGD/R for 24 h. The expression of nu-Nrf2 was detected by Western blot. (B–E) HT22 cells were transfected with inhibitor NC, or co-transfected with miR-9-3p inhibitor and si-NC, or co-transfected with miR-9-3p inhibitor and so-Nrf2, then cells were exposed to OGD/R for 24 h. (B) The expression of nu-Nrf2 was detected by Western blot. (C) Relative Nrf2/ARE transcriptional activity was determined by luciferase reporter assay. (D) Cell viability was measured by MTT assay. (E) Intracellular ROS level was measured using a corresponding detection kit. Data were expressed as mean ± SD. *p <0.05, **p <0.01.
Discussion
Previous studies have demonstrated that the abnormal expressions of miRNAs always occurred in the brain in response to I/R.Citation31 Thorough understanding of mechanisms in specific miRNAs may contribute to the development of potential targets for protecting against I/R injury. In this study, we found that miR-9-3p level was significantly upregulated in mice brains with I/R in vivo, as well as in OGD/R-treated HT22 cells in vitro. Moreover, downregulation of miR-9-3p effectively alleviated infarct volume and neurological outcomes of I/R mice and also attenuated OGD/R-induced HT22 cell injury and ROS production. Mechanistically, our findings showed that miR-9-3p bound to the 3′-UTR region of FGF19 mRNA to inhibit its expression, subsequently resulting in the inactivation of the GSK-3β/Nrf2/ARE signaling pathway as well as the suppression of the following antioxidant response.
MiR-9-3p, a newly-identified miRNA, has been revealed to play important roles in the pathogenic processes of malignant tumors, including proliferation, invasion, migration, apoptosis and epithelial-mesenchymal transition (EMT) including hepatocellular carcinoma,Citation32 gastric cancer,Citation33 bladder cancer,Citation34 nasopharyngeal carcinoma,Citation35 and medullary thyroid carcinoma,Citation36 and so on. For example, overexpression of miR-9-3p augments cell apoptosis induced by H2O2 through targeting and downregulating Herpud1 in glioma.Citation37 MiR-9-3p has a tumor-suppressor role through targeting TAZ (WWTR1) in hepatocellular carcinoma cells.Citation38 MiR-9-3p regulates the multidrug resistance of chronic myelogenous leukemia by targeting ABCB1.Citation39 Recent studies have revealed that miR-9-3p was significantly upregulated in cerebrospinal fluid of stroke patients compared with that of normal subjects.Citation12,Citation14,Citation40 However, the role and specific mechanisms of miR-375 in I/R-induced injury in the brain remains unclear. In agreement with a previous study finding that miR-9-3p was significantly elevated in the cerebrospinal fluid of stroke patients,Citation40 our study found that miR-9-3p was significantly upregulated in the I/R model, both in vitro and in vivo. Studies on embryonic stem cells imply that miR-9 plays a key role in neural fate determination in mammalian brain development.Citation41 It suggests that cerebral I/R injury induced miR-9-3p expression, and then affected the neural fate determination in mammalian brain development. Moreover, we demonstrated for the first time that miR-9-3p had a stimulative role in the progression of cerebral I/R injury, which was shown by overexpression of miR-9-3p, and inhibition of miR-9-3p alleviated cerebral I/R injury, suggesting that miR-9-3p might be a potential therapeutic target for ischemic stroke.
Increasing evidence has revealed that miRNAs can function as competing endogenous RNAs (ceRNAs) to regulate gene expression at the post-transcription level.Citation42 Target mRNAs of miR-375 have been reported to include Herpud1,Citation37 fibroblast growth factor 5 (HBGF-5),Citation32 endothelial cell-specific molecule 1 (ESM1),Citation34 fibronectin 1 (FN1), β1 integrin (ITGB1) and α5 integrin (ITGAV).Citation35 Herein, we identified a new target of miR-9-3p, FGF19. A dual-luciferase reporter assay was performed and confirmed that miR-9-3p could bound to 3′-UTR of FGF19. FGF19 was significantly upregulated in response to cerebral I/R injury in cardiomyocyte, and FGF19 promoted the activation of Nrf2-mediated antioxidant response element (ARE) antioxidant signaling by inhibiting GSK-3β activity.Citation16 Here, FGF19 was markedly downregulated in OGD/R-treated HT22 cells and silencing of FGF19 decreased the expression of nu-Nrf2, and relative Nrf2/ARE activity and HT22 cell viability, which was in agreement with previous studies. Interestingly, our study showed a new target of miR-9-3p in cerebral I/R injury and extended the functions of miRNAs in human diseases.
During the I/R process, the reestablishment of blood flow will cause further damage to the ischemic tissue via ROS accumulation, interference with cellular ion homeostasis, and the inflammatory responses to cell death.Citation43 In the last decades, several antioxidant mechanisms against I/R injury have been identified and studied, including catalase (CAT), superoxide dismutase (SOD), Glutathione S-transferase (GST), NADPH quinone oxidoreductase-1 (NQO1) and heme oxygenase-1 (HO-1),Citation44,Citation45 of which, NQO1 and HO-1 are downstream genes of the Nrf2/ARE response regulatory system.Citation46 In addition, GSK-3β can prevent the nuclear translocation of Nrf2 and then inhibit the gene expression of Nrf2/ARE downstream genes associated with the cell antioxidant response.Citation47 Hence, targeting GSK-3β may be a potential therapeutic goal for preventing ROS production causing cell injury. In this study, we demonstrated that miR-9-3p was a directly negative regulator of FGF19, and miR-9-3p promoted cerebral I/R injury by regulating the FGF19/GSK-3β/Nrf2/ARE2-mediated antioxidant response. Our study provided a new regulatory mechanism of miRNAs involved in the development of I/R injury.
In addition, an increasing number of studies have demonstrated that FGF19 has several other downstream targets in various pathogenic processes, such as FGFR4 in breast cancer,Citation48 IL-6/STAT3 signaling in hepatocarcinogenesis,Citation49 AMPK/PGC-1α signaling involved in palmitic acid-induced mitochondrial dysfunction, oxidative stress in skeletal muscle,Citation50 and so on. Whether one or more targets, except for GSK-3β/Nrf2/ARE2, mediated the role of miR-9-3p in cerebral I/R injury should be determined in the future.
Conclusion
In summary, our study revealed that inhibition of miR-9-3p protected against cerebral I/R injury both in vivo and in vitro by targeting FGF19 to activate GSK-3β/Nrf2/ARE signaling pathway-mediated antioxidant responses, suggesting that miR-9-3p might be a potential therapeutic target for cerebral I/R injury.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- Kim T, Mehta SL, Morris-Blanco KC, et al. The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing α-synuclein. Sci Signal. 2018;11. doi:10.1126/scisignal.aat4285
- Zuo G, Zhang D, Mu R, et al. Resolvin D2 protects against cerebral ischemia/reperfusion injury in rats. Mol Brain. 2018;11(1):9. doi:10.1186/s13041-018-0351-129439730
- Galkin A. Brain ischemia/reperfusion injury and mitochondrial complex I damage. Biochemistry. 2019;84(11):1411–1423. doi:10.1134/s000629791911015431760927
- Stegner D, Klaus V, Nieswandt B. Platelets as modulators of cerebral ischemia/reperfusion injury. Front Immunol. 2019;10:2505. doi:10.3389/fimmu.2019.0250531736950
- Yang Q, Huang Q, Hu Z, Tang X. Potential neuroprotective treatment of stroke: targeting excitotoxicity, oxidative stress, and inflammation. Front Neurosci. 2019;13:1036. doi:10.3389/fnins.2019.0103631611768
- Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–1207. doi:10.1016/j.jaci.2017.08.03429074454
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi:10.1016/j.cell.2009.01.00219167326
- Morgado AL, Rodrigues CMP, Solá S. MicroRNA-145 regulates neural stem cell differentiation through the Sox2-Lin28/let-7 signaling pathway. Stem Cells. 2016;34(5):1386–1395. doi:10.1002/stem.230926849971
- Kumar S, Vijayan M, Reddy PH. MicroRNA-455-3p as a potential peripheral biomarker for alzheimer’s disease. Hum Mol Genet. 2017;26(19):3808–3822. doi:10.1093/hmg/ddx26728934394
- Sun H, Zhong D, Wang C, et al. MiR-298 exacerbates ischemia/reperfusion injury following ischemic stroke by targeting act1. Cell Physiol Biochem. 2018;48(2):528–539. doi:10.1159/00049181030021197
- Zheng T, Shi Y, Zhang J, et al. MiR-130a exerts neuroprotective effects against ischemic stroke through PTEN/PI3K/AKT pathway. Biomed Pharmacother. 2019;117:109117. doi:10.1016/j.biopha.2019.10911731226635
- Bache S, Rasmussen R, Wolcott Z, et al. Elevated miR-9 in cerebrospinal fluid is associated with poor functional outcome after subarachnoid hemorrhage. Transl Stroke Res. 2020;11(6):1243–1252. doi:10.1007/s12975-020-00793-132248435
- O’Connell GC, Smothers CG, Winkelman C. Bioinformatic analysis of brain-specific miRNAs for identification of candidate traumatic brain injury blood biomarkers. Brain Injury. 2020;34(7):965–974. doi:10.1080/02699052.2020.176410232497449
- Sørensen SS, Nygaard A-B, Carlsen AL, et al. Elevation of brain-enriched miRNAs in cerebrospinal fluid of patients with acute ischemic stroke. Biomarker Res. 2017;5(1):24. doi:10.1186/s40364-017-0104-9
- Rodrigo R, Fernandez-Gajardo R, Gutierrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS & neurological disorders. Drug Targets. 2013;12(5):698–714. doi:10.2174/1871527311312050015
- Fang Y, Zhao Y, He S, et al. Overexpression of FGF19 alleviates hypoxia/reoxygenation-induced injury of cardiomyocytes by regulating GSK-3β/Nrf2/ARE signaling. Biochem Biophys Res Commun. 2018;503(4):2355–2362. doi:10.1016/j.bbrc.2018.06.16129964017
- Leroy K, Brion JP. Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J Chem Neuroanat. 1999;16:279–293. doi:10.1016/s0891-0618(99)00012-510450875
- Satoh T, Okamoto S-I, Cui J, et al. Activation of the keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] Phase II inducers. Proc Natl Acad Sci U S A. 2006;103(3):768–773. doi:10.1073/pnas.050572310216407140
- Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi:10.1074/jbc.M51373720016551619
- Leng Y, Liang M-H, Ren M, et al. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28(10):2576–2588. doi:10.1523/jneurosci.5467-07.200818322101
- Pang T, Wang Y-J, Gao Y-X, et al. A novel GSK-3β inhibitor YQ138 prevents neuronal injury induced by glutamate and brain ischemia through activation of the Nrf2 signaling pathway. Acta Pharmacol Sin. 2016;37(6):741–752. doi:10.1038/aps.2016.327108601
- Yang X, Ji H, Yao Y, et al. Downregulation of circ_008018 protects against cerebral ischemia–reperfusion injury by targeting miR-99a. Biochem Biophys Res Commun. 2018;499(4):758–764. doi:10.1016/j.bbrc.2018.03.21829605297
- Wu N, Zhang X, Bao Y, et al. Down-regulation of GAS5 ameliorates myocardial ischaemia/reperfusion injury via the miR-335/ROCK1/AKT/GSK-3β axis. J Cell Mol Med. 2019;23(12):8420–8431. doi:10.1111/jcmm.1472431625671
- Dong W, Xie F, Chen X-Y, et al. Inhibition of Smurf2 translation by miR-322/503 protects from ischemia-reperfusion injury by modulating EZH2/Akt/GSK3β signaling. Am J Physiol Cell Physiol. 2019;317(2):C253–C261. doi:10.1152/ajpcell.00375.201830649914
- Zeng J, Zhu L, Liu J, Zhu T, Xie Z. Metformin protects against oxidative stress injury induced by ischemia/reperfusion via regulation of the lncRNA-H19/miR-148a-3p/Rock2 axis. Oxid Med Cell Longev. 2019;2019:8768327. doi:10.1155/2019/876832731934270
- Papadakis M, Hadley G, Xilouri M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19(3):351–357. doi:10.1038/nm.309723435171
- Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res. 1997;19(6):641–648. doi:10.1080/01616412.1997.117408749427967
- Ma C, Wang X, Xu T, et al. Qingkailing injection ameliorates cerebral ischemia-reperfusion injury and modulates the AMPK/NLRP3 inflammasome signalling pathway. BMC Complement Altern Med. 2019;19(1):320. doi:10.1186/s12906-019-2703-531747940
- Cheng F, Zhong X, Lu Y, et al. Refined qingkailing protects MCAO mice from endoplasmic reticulum stress-induced apoptosis with a broad time window. Evid Based Complement Alternat Med. 2012;2012:567872. doi:10.1155/2012/56787222536287
- Sun X, Wang D, Zhang T, et al. Eugenol attenuates cerebral ischemia-reperfusion injury by enhancing autophagy via AMPK-mTOR-P70S6K pathway. Front Pharmacol. 2020;11:84. doi:10.3389/fphar.2020.0008432153404
- Wang C, Pan Y, Cheng B, Chen J, Bai B. Identification of conserved and novel microRNAs in cerebral ischemia-reperfusion injury of rat using deep sequencing. J Mol Neurosci. 2014;54(4):671–683. doi:10.1007/s12031-014-0383-725063377
- Tang J, Li Y, Liu K, et al. Exosomal miR-9-3p suppresses HBGF-5 expression and is a functional biomarker in hepatocellular carcinoma. Minerva Med. 2018;109(1):15–23. doi:10.23736/s0026-4806.17.05167-928750499
- Meng Q, Xiang L, Fu J, et al. Transcriptome profiling reveals miR-9-3p as a novel tumor suppressor in gastric cancer. Oncotarget. 2017;8(23):37321–37331. doi:10.18632/oncotarget.1631028418879
- Cai H, Yang X, Gao Y, et al. Exosomal MicroRNA-9-3p secreted from BMSCs downregulates ESM1 to suppress the development of bladder cancer. Mol Ther Nucleic Acids. 2019;18:787–800. doi:10.1016/j.omtn.2019.09.02331734559
- Ding Y, Pan Y, Liu S, Jiang F, Jiao J. Elevation of MiR-9–3p suppresses the epithelial-mesenchymal transition of nasopharyngeal carcinoma cells via down-regulating FN1, ITGB1 and ITGAV. Cancer Biol Ther. 2017;18(6):414–424. doi:10.1080/15384047.2017.132358528613134
- Chen Y, Zhang S, Zhao R, Zhao Q, Zhang T. Upregulated miR-9-3p promotes cell growth and inhibits apoptosis in medullary thyroid carcinoma by targeting BLCAP. Oncol Res. 2017;25:1215–1222. doi:10.3727/096504016x1479171535595727938505
- Yang L, Mu Y, Cui H, Liang Y, Su X, Roemer K. MiR-9-3p augments apoptosis induced by H2O2 through down regulation of herpud1 in glioma. PLoS One. 2017;12(4):e0174839. doi:10.1371/journal.pone.017483928430789
- Higashi T, Hayashi H, Ishimoto T, et al. miR-9-3p plays a tumour-suppressor role by targeting TAZ (WWTR1) in hepatocellular carcinoma cells. Br J Cancer. 2015;113(2):252–258. doi:10.1038/bjc.2015.17026125451
- Li Y, Zhao L, Li N, et al. miR-9 regulates the multidrug resistance of chronic myelogenous leukemia by targeting ABCB1. Oncol Rep. 2017;37(4):2193–2200. doi:10.3892/or.2017.546428260112
- Yan Q, Sun SY, Yuan S, Wang XQ, Zhang ZC. Inhibition of microRNA-9-5p and microRNA-128-3p can inhibit ischemic stroke-related cell death in vitro and in vivo. IUBMB Life. 2020;72(11):2382–2390. doi:10.1002/iub.235732797712
- Krichevsky AM, Sonntag K-C, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006;24(4):857–864. doi:10.1634/stemcells.2005-044116357340
- Qi X, Zhang D-H, Wu N, et al. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52(10):710–718. doi:10.1136/jmedgenet-2015-10333426358722
- Minutoli L, Puzzolo D, Rinaldi M, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016;2016:2183026. doi:10.1155/2016/218302627127546
- Bougioukas I, Didilis V, Emmert A, et al. Apigenin reduces NF-κB and subsequent cytokine production as protective effect in a rodent animal model of lung ischemia-reperfusion injury. J Invest Surg. 2018;31(2):96–106. doi:10.1080/08941939.2017.129651228340319
- Shimizu S, Tsounapi P, Dimitriadis F, et al. Testicular torsion-detorsion and potential therapeutic treatments: a possible role for ischemic postconditioning. Int J Urol. 2016;23(6):454–463. doi:10.1111/iju.1311027217335
- Hu Q, Ren J, Li G, et al. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9(3):403. doi:10.1038/s41419-018-0436-x29540694
- Shen Y, Chen S, Zhao Y. Sulfiredoxin-1 alleviates high glucose-induced podocyte injury though promoting Nrf2/ARE signaling via inactivation of GSK-3β. Biochem Biophys Res Commun. 2019;516(4):1137–1144. doi:10.1016/j.bbrc.2019.06.15731284950
- Liu Y, Cao M, Cai Y, et al. Dissecting the role of the FGF19-FGFR4 signaling pathway in cancer development and progression. Front Cell Dev Biol. 2020;8:95. doi:10.3389/fcell.2020.0009532154250
- Zhou M, Yang H, Learned RM, Tian H, Ling L. Non-cell-autonomous activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat Commun. 2017;8:15433. doi:10.1038/ncomms1543328508871
- Guo A, Li K, Xiao Q. Fibroblast growth factor 19 alleviates palmitic acid-induced mitochondrial dysfunction and oxidative stress via the AMPK/PGC-1α pathway in skeletal muscle. Biochem Biophys Res Commun. 2020;526:1069–1076. doi:10.1016/j.bbrc.2020.04.00232305136