
Abstract
Lapatinib is an oral-form dual tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR or ErbB/Her) superfamily members with anticancer activity. In this study, we examined the effects and mechanism of action of lapatinib on several human leukemia cells lines, including acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and acute lymphoblastic leukemia (ALL) cells. We found that lapatinib inhibited the growth of human AML U937, HL-60, NB4, CML KU812, MEG-01, and ALL Jurkat T cells. Among these leukemia cell lines, lapatinib induced apoptosis in HL-60, NB4, and Jurkat cells, but induced nonapoptotic cell death in U937, K562, and MEG-01 cells. Moreover, lapatinib treatment caused autophagic cell death as shown by positive acridine orange staining, the massive formation of vacuoles as seen by electronic microscopy, and the upregulation of LC3-II, ATG5, and ATG7 in AML U937 cells. Furthermore, autophagy inhibitor 3-methyladenine and knockdown of ATG5, ATG7, and Beclin-1 using short hairpin RNA (shRNA) partially rescued lapatinib-induced cell death. In addition, the induction of phagocytosis and ROS production as well as the upregulation of surface markers CD14 and CD68 was detected in lapatinib-treated U937 cells, suggesting the induction of macrophagic differentiation in AML U937 cells by lapatinib. We also noted the synergistic effects of the use of lapatinib and cytotoxic drugs in U937 leukemia cells. These results indicate that lapatinib may have potential for development as a novel antileukemia agent.
Introduction
Acute myeloid leukemia (AML) is a group of heterogeneous diseases with abnormally active proliferation of hematopoietic precursors, which block patients’ normal hematopoiesis, causing neutropenia and anemia.Citation1 Although more than 50% of adult AML patients show complete remission with conventional chemotherapeutic drugs, only 20%–30% of patients show long-term disease-free survival.Citation2 Despite being associated with complicated genetic mutations, ~50% of AML cases correlate with mutations caused by the aberrant activation of receptor tyrosine kinases (eg, FLT3 and c-KIT) or downstream effectors (eg, Ras).Citation1–Citation3 Thus, in recent years, some small-molecule inhibitors, for example, sorafenib, gefitinib, erlotinib, and quizartinib (AC220) have been tested in preclinical studies as targeted therapies for AML.Citation1,Citation4,Citation5
Lapatinib (GlaxoSmithKline plc, London, UK) is a small-molecule inhibitor against the receptor tyrosine kinase of epidermal growth factor receptor (EGFR) or ErbB/Her family members.Citation6–Citation8 EGFR/ErbB/Her superfamily members, including EGFR/ErbB1/Her1, ErbB2/Her2/Neu, ErbB3/Her3, and ErbB4/Her4, are common targets for anticancer drugs because their functions are commonly dysregulated in many cancers.Citation9–Citation11 Lapatinib, which is taken orally, was originally approved by the US Food and Drug Administration in combination with capecitabine for advanced breast cancers that have developed resistance to the anti-ErbB2 monoclonal antibody trastuzumab (Herceptin; Genentech, Inc., South San Francisco, CA, USA) or other drugs.Citation6,Citation10,Citation12,Citation13
Differentiation therapies play important roles in leukemia treatment. Differentiation therapies in oncology include changing malignant tumors to curable tumors or terminally differentiated cells that undergo no further proliferation.Citation14 This includes the induction of cancer cell differentiation and tumor eradication by either stopping tumor cell proliferation or increasing drug sensitivity after differentiation. One successful method of differentiation therapy is treating acute promyelocytic leukemia (APL) with all-trans retinoic acid. All-trans retinoic acid changes malignant leukemia to treatable cell forms by inducing differentiation of these leukemia cells into drug-sensitive cells. Other drugs that induce differentiation include guanosine triphosphate, herbimycin A, and butyrates for erythroid differentiation of chronic myelogenous leukemia (CML),Citation14–Citation16 and 12-O-tetradecanoylphorbol-13-acetate (TPA), HIV-tat protein, thrombopoietin, and dimethyl sulfoxide (DMSO) for the megakaryocytic differentiation of CML or APL.Citation17–Citation20
In our previous studies, we found that lapatinib induced autophagic cell death in CML and hepatoma.Citation21,Citation22 In this study, we focused on lapatinib-induced cytotoxicity mechanisms in AML cells. We found that lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner via either autophagic or apoptotic cell death depending on the different cell types. The induction of autophagic cell death and macrophagic differentiation by lapatinib has been demonstrated in U937 cells. We also found a synergistic effect of lapatinib and other cytotoxic drugs in AML U937 cells. The results of our study suggest that lapatinib may be a potentially useful therapy for leukemia.
Materials and methods
Cell culture
The following human leukemia cell lines were used in our study: AML-derived HL-60 and U937 cells,Citation23 CML-derived MEG-01 and KU812 cells, APL-derived NB4 cells,Citation22 and Jurkat T lymphoma. These cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA) and penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C under 5% CO2. Genetic recombination experiments in our study were approved by the Biosafety Committee of Chang Jung Christian University.
Cell treatments
Lapatinib (GlaxoSmithKline) or erlotinib (Hoffman-La Roche Ltd., Basel, Switzerland) tablets were ground and dissolved in DMSO (Sigma-Aldrich Co., St Louis, MO, USA) to prepare a 1,000-fold stock solution. Autophagy inhibitor 3-methyladenine (3-MA) was dissolved in an RPMI 1640 medium. The pan caspase inhibitor Z-VAD-fmk (R&D Systems, Inc., Minneapolis, MN, USA), etoposide (Sigma-Aldrich Co.), paclitaxel (Sigma-Aldrich Co.), and autophagy inducer thapsigargin (Sigma-Aldrich Co.) at concentrations of 20 mM, 1 µM, 25 µM, and 150 µM, respectively, were dissolved in DMSO to prepare a 1,000-fold stock solution. Treatment with protein kinase C activator TPA (Sigma-Aldrich Co.) was used as the positive control for the differentiation of leukemia cells.Citation24 The 10,000-fold TPA stock solution (1 mM) was prepared in DMSO.
Viability analysis
After treatments, cell viability was analyzed using the trypan blue assay (Thermo Fisher Scientific) or the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. Cell numbers or optical density (OD) values of the control cells (DMSO vehicle-treated) were calculated as 100% viability. Data from triplicate (or more) experiments were presented as mean ± standard deviation (SD). P-values (P<0.05, P<0.01, P<0.001) were calculated by Student’s t-test between lapatinib-treated and DMSO control cells or as indicated.
Observation of morphology and LC3 aggregation by microscopy
After treatments with drugs for 3 days, the cells were centrifuged on slides using cytospin apparatus (Thermo Fisher Scientific), and were fixed and stained with Liu’s stain (Muto Pure Chemicals, Tokyo, Japan) according to the manufacturer’s instructions. Cell morphology was observed under a microscope. For the observation of LC3 aggregation, the stable expression of GFP-LC3 in U937 leukemia cells was induced by the transduction of a GFP-LC3-containing lentivirus and blasticidin selections as described previously.Citation25
Flow cytometry
For the detection of apoptotic cells with DNA laddering (hypodiploid or sub-G1 phase) by flow cytometry (FACS Calibur; Becton Dickinson, Franklin Lakes, NJ, USA), treated cells were spun and resuspended in a hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100) with 5 µg/mL propidium iodide (Sigma-Aldrich Co.). Total dead cells (propidium iodide-positive cells) and apoptotic cells were simultaneously detected by flow cytometry.Citation21,Citation22 The percentage of acridine orange-positive cells was detected after cells were collected and stained with acridine orange (10 µg/mL, Sigma-Aldrich Co.) for 15 minutes. For determining the percentage of CD14- or CD68-positive cells, the cells were collected and stained with 0.5 µg/mL antibodies against CD14 (BD Pharmingen, San Diego, CA, USA) or CD68 (Dako, Hamburg, Germany) at 4°C for 20 minutes and stained with Alexa Fluor 488-conjugated anti-Mouse IgG secondary antibody (Thermo Fisher Scientific) at 4°C for another 20 minutes. After washing with phosphate-buffered saline (PBS), CD14- or CD68-positive cells were measured and analyzed by flow cytometry. For phagocytosis analysis, cells were treated, collected, washed with PBS, and mixed with green fluorescent latex beads (Sigma-Aldrich Co.), and then were split into two tubes. One tube was incubated on ice as a control, while the other tube was incubated at 37°C for 2 hours. After washing with PBS, fluorescent beads containing cells (with phagocytotic ability) were analyzed by flow cytometry. For the detection of intracellular reactive oxygen species (ROS) production, cells were treated with DMSO or different concentrations of lapatinib for 24 hours, and then incubated at 37°C for 30 minutes with a medium containing 2 µM 2′,7′-dichlorofluorescein diacetate (Molecular Probes, Eugene, OR, USA) for cytosolic hydrogen peroxide detection. ROS production was then measured by flow cytometry. All the flow cytometric data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Western blot analysis
After the drug treatment, U937 cells were spun and lysed in lysis buffer (Sigma-Aldrich Co.) with protease inhibitors (Hoffman-La Roche Ltd.). Cell lysates with 50 µg of protein were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transferring to polyvinylidene fluoride membranes for immunoblotting. The following antibodies were used for immunoblotting: actin (Sigma-Aldrich Co.), ATG5 (Cell Signaling, Beverly, MA, USA), Beclin-1 (or ATG6, Cell Signaling), ATG7 (Santa Cruz Biotechnology Inc., Dallas, TX, USA), LC3 (or ATG8; Abgent, San Diego, CA, USA), and BNIP (Abcam, Cambridge, UK).
Transmission electron microscopy
After treatment, U937 cells were spun and then fixed with a 2.5% glutaraldehyde-containing cacodylate buffer (pH 7.4). Then the cells were stained with 1% osmium tetroxide, dehydrated, and embedded in Spurr’s resin according to the manufacturer’s instructions (Electron Microscopy Science, Fort Washington, PA, USA). The cell morphology was observed by transmission electron microscopy (JEM-1200EXII; JEOL, Tokyo, Japan).
Knockdown of autophagy-related genes by shRNA expression system
A lentiviral short hairpin RNA (shRNA) expression system from the RNAi Consortium (TRC) was applied to knockdown the expression of the autophagy-related genes: ATG5, ATG7 (clone #1 or #2, TRCN0000007584 or TRCN0000007587, respectively), Beclin-1 (or ATG6, clone #1 or #2, TRCN0000033549 or TRCN0000033552, respectively), and RFP (as nontargeting shRNA control) as described previously.Citation21 We obtained these pLKO.1-shRNA constructions from the National RNAi Core Facility in Taiwan. The shRNA construct preparation and lentiviral production and infection were performed according to the TRC and the Core Facility protocol as described previously.Citation21,Citation22 After puromycin selection for at least 1 week, cells were treated with lapatinib, and cell viability was validated by trypan blue and MTS assays as described earlier.
Results
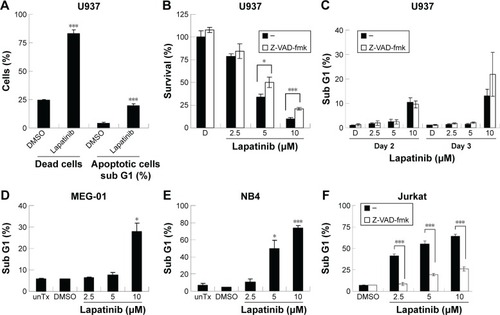
Lapatinib-induced apoptotic and nonapoptotic cell death in several kinds of leukemia cells
In this study, we first performed a comparison of the effect of lapatinib on several leukemia cell lines. We used both MTS and trypan blue assays for viability validation. As shown in , lapatinib reduced cell viability in a dose- and time-dependent manner in AML-derived U937, ALL Jurkat T, and CML-derived KU812 leukemia cells. Compared to other leukemia cell lines, KU812 is less sensitive to lapatinib-induced cytotoxicity. We also found lapatinib-induced mitochondrial collapse in U937 cells (). These findings in relation to cell viability reduction and mitochondrial collapse are consistent with the cytotoxic effects of lapatinib in CML-derived K562 and MEG-01 and AML-derived HL-60 and NB4 cells observed in our previous study.Citation22
Figure 1 Lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner.
Abbreviations: DMSO, dimethyl sulfoxide; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; OD, optical density.
We then tested the mechanisms involved in lapatinib-induced cytotoxicity. We found a dramatic induction of apoptosis by lapatinib in HL-60 cells, and also induction of apoptosis and autophagic cell death in K562 cells. Approximately, 30% of the lapatinib-treated K562 cells showed DNA fragmentation.Citation22 As shown in , in lapatinib-treated U937 cells, growth inhibition was mainly caused due to the presence of high percentages of dead cells, in contrast to the low percentages of sub-G1 cells. The low percentages of sub-G1 cells suggest nonapoptotic cell death in lapatinib-treated U937 cells. These data are similar to those obtained for CML-derived MEG-01 () and K562 cells.Citation22 However, lapatinib induced production of massive amounts of sub-G1 cells in NB4 and Jurkat T cells (), like in AML-derived HL-60 cells.Citation22 In addition, we confirmed the induction of apoptosis by lapatinib in U937 and Jurkat cells in the presence or absence of the broad-spectrum caspase inhibitor Z-VAD-fmk. As shown in , the percentages of sub-G1 cells were effectively reduced by the caspase inhibitor Z-VAD-fmk in Jurkat T cells but not in U937 cells. In agreement, we found that Z-VAD-fmk could not protect most of the U937 cells from lapatinib-induced cell death (), suggesting nonapoptotic cell death in lapatinib-treated U937 cells. Thus, according to our results (, and our previous study),Citation22 lapatinib induced apoptosis in AML HL-60, APL NB4, Jurkat T lymphoma, and small portions of CML K562 cells, but induced nonapoptotic cell death in the majority of K562, U937, and CML MEG-01 cells.
Figure 2 The induction of different death mechanisms by lapatinib in several kinds of leukemia lines.
Abbreviations: DMSO, dimethyl sulfoxide; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; unTx, untreated.
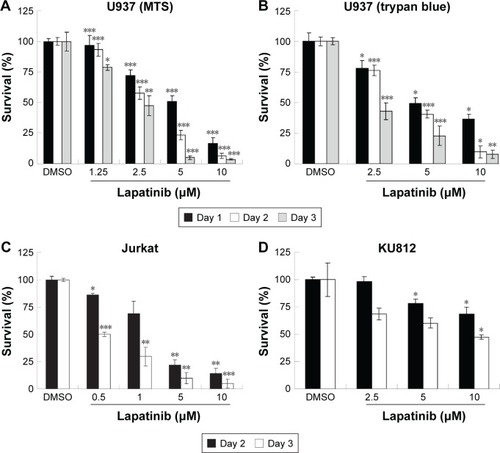
Autophagic cell death in lapatinib-treated AMLU937 leukemia cells
Since lapatinib effectively induces nonapoptotic cell death in AML U937 cells, we wondered whether lapatinib induced autophagic cell death, the second type of programmed cell death in U937 cells. As shown in , after treatment with lapatinib, massive cells with vacuoles (autophagic morphology) were observed. These morphological data were similar to those obtained for cells treated with the differentiation inducer TPA.Citation24 To further confirm lapatinib-induced autophagic cell death in U937 cells, we used the GFP-LC3 overexpression system and fluorescence microscopy.Citation26,Citation27 In U937 cells, we detected cells with punctuated GFP-LC3 (LC3 aggregates) after lapatinib or autophagy inducer thapsigargin treatment ().
Figure 3 The induction of autophagic cell death by lapatinib in U937 cells.
Abbreviations: DMSO, dimethyl sulfoxide; TPA, 12-O-tetradecanoylphorbol-13-acetate; TG, thapsigargin; 3-MA, 3-methyladenine; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
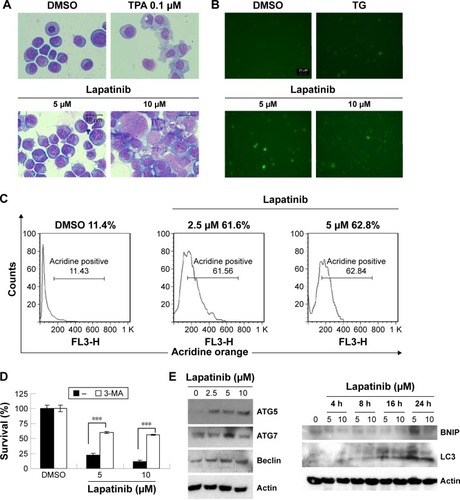
To further confirm the autophagic cell death of U937 cells, we used acridine orange staining. As shown in , high numbers of acridine orange-positive cells were detected in lapatinib-treated cells. We also determined whether lapatinib-induced cytotoxicity was blocked in the presence of the autophagy inhibitor 3-MA. As shown in , 3-MA effectively blocked lapatinib-induced cytotoxicity in U937 cells. In addition, we checked the expression levels of these autophagy-related proteins. As shown in , we found a clear increase in Beclin-1 (ATG6), ATG5, ATG7, LC3, and BNIP in lapatinib-treated leukemia by immunoblotting.
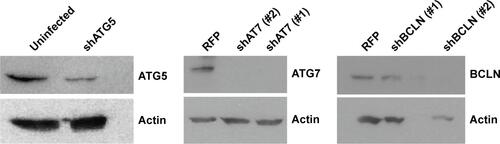
Upon further examination of lapatinib-induced autophagic cell death by transmission electronic microscope (TEM), we found massive cells with vacuoles enclosing a lot of unhealthy mitochondria (). Furthermore, we performed autophagy inhibition by using U937 cells with shRNA expression corresponding to autophagy regulatory proteins ATG7, ATG5, and Beclin-1 (). U937 leukemia cells were transducted with shRNAs containing plasmids from TRC as described in the “Materials and methods” section and previous reports.Citation21,Citation22 As shown in , the expression of shRNA against ATG7, ATG5, and Beclin-1 (but not nonspecific shRNA against RFP) inside the cells dramatically decreased lapatinib sensitivity according to examination by the MTS () and trypan blue assays (). These data, showing the expression of shRNA, further confirmed lapatinib-induced autophagic cell death in AML U937 cells.
Figure 4 The induction of massive vacuoles inside cells by lapatinib in U937 cells.
Abbreviations: DMSO, dimethyl sulfoxide; TEM, transmission electron microscope.
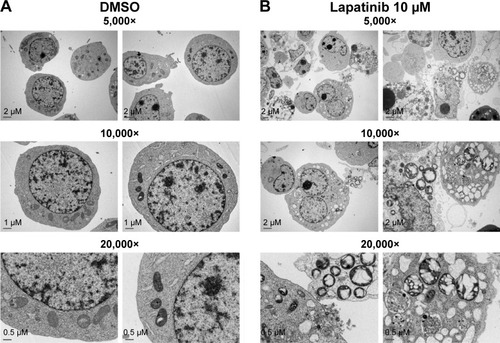
Figure 5 The RNAi-mediated downregulation of autophagy-related genes hinders lapatinib-induced autophagic cell death in U937 cells.
Abbreviations: RNAi, RNA interference; shRNA, short hairpin RNA; DMSO, dimethyl sulfoxide; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; Unif, uninfected cells.
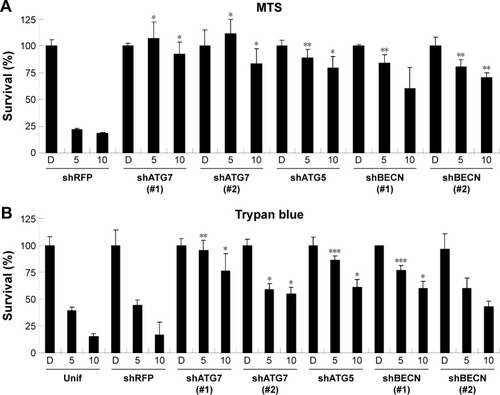
Differentiation induction by lapatinib in AML leukemia cells
Since we have previously demonstrated the induction of megakaryocytic differentiation marker CD61 by lapatinib in K562 cells,Citation22 we determined whether lapatinib could also cause differentiation in U937 cells. As shown in , we found lapatinib induced the upregulation of CD14 and CD68 surface markers in U937 cells. This is similar to the effects caused by treatment with the differentiation inducer TPA.Citation28–Citation30 TPA induces the differentiation of AML cells into monocyte/macrophage-like cells or polymorphonuclear neutrophil cells.Citation31 For further confirmation of differentiation in lapatinib-treated cells, phagocytic activity and ROS production assays were performed.Citation24 As shown in , phagocytosis ability was dramatically increased with lapatinib treatment, similar to the positive-control TPA treatment. The cellular ROS level was also increased in lapatinib-treated U937 cells (). The upregulation of CD14 and CD68 surface markers and cellular ROS level suggests the induction of monocyte/macrophage differentiation by lapatinib treatment in AML U937 cells, similar to the effects of TPA treatment.
Figure 6 The upregulation of CD14 and CD68 macrophagic differentiation markers and the phagocytosis ability of lapatinib in AML U937 leukemia cells.
Abbreviations: AML, acute myeloid leukemia; DMSO, dimethyl sulfoxide; TPA, 12-O-tetradecanoylphorbol-13-acetate; DCFH-DA, 2′,7′-dichlorofluorescein diacetate; ROS, reactive oxygen species; MFI, mean fluorescence intensity.
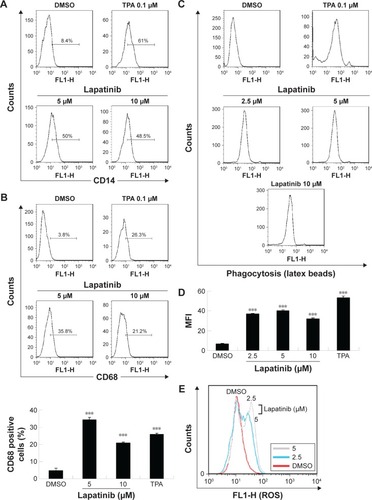
Synergistic effects of lapatinib and other anticancer drugs in U937 leukemia cells
To reduce the side effects and increase the therapeutic effects, cotreatment is a useful strategy for clinical therapies. We, therefore, wondered whether a combination of lapatinib and other anticancer drugs would show enhanced cytotoxic effects in leukemia. We first choose erlotinib, a targeted therapy drug and EGFR inhibitor that shows off-target effects in AML leukemia cells and patients.Citation4,Citation5,Citation32–Citation38 As shown in and consistent with previous reports, erlotinib alone failed to effectively inhibit U937 cell growth. However, in combination with low doses of lapatinib, both drugs showed dramatic growth inhibition of U937 cells. Again, a combination of low doses of lapatinib and etoposide or paclitaxel effectively increased the cytotoxic effects as compared to a single drug treatment. These results suggest the synergistic effects of lapatinib and other anticancer drugs in U937 leukemia cells. These synergistic effects of lapatinib and other anticancer drugs suggest the possible benefits of the development of combination therapies in leukemia treatment.
Figure 7 The synergistic effects of lapatinib and other cytotoxic drugs in U937 leukemia cells.
Abbreviations: DMSO, dimethyl sulfoxide; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
Discussion
In this study, we found lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner () either via autophagic or apoptotic cell death depending on the type of cell lines ().Citation22 This includes apoptosis induction in AML HL-60, APL NB4, Jurkat T lymphoma, and a minority of CML K562 cells, and the induction of nonapoptotic cell death in AML U937, CML MEG-01, and the majority of K562 cells by lapatinib. In U937 cells, autophagic cell death induced by lapatinib was demonstrated by massive vacuole formation observed by TEM () and microscopy (); LC3 aggregations (); acridine orange-positive cells (); cell death inhibition by 3-MA () (but not Z-VAD-fmk []); upregulation of the autophagy-related proteins ATG5, ATG7, Beclin, BNIP, and LC3 (); and cell death inhibition by knockdown of shRNA against ATG5, ATG7, and Beclin ().Citation26 Moreover, we also found induction of differentiation by lapatinib in U937 cells in correlation with the upregulation of monocyte/macrophage markers CD14 and CD68 ().Citation23,Citation36 Lapatinib also increased the phagocytosis ability of U937 cells, as shown by massive incorporation of fluorescent beads and higher ROS production ().Citation23 We further found that the use of lapatinib and other cytotoxic drugs, including elortinib, etoposide, and paclitaxel, in U937 cells has a synergistic effect that increased tumor cytotoxicity (). These findings suggest that a combination therapy using lapatinib and other anticancer drugs would further benefit anti-AML applications.
A few studies have focused on lapatinib-induced autophagic cell death. This study is consistent with our previous findings regarding the induction of autophagic cell death in hepatoma cells, induction of autophagic cell death, apoptosis, and differentiation by lapatinib in CML K562 cells.Citation21,Citation22 In addition to our findings, some studies have also discussed the induction of autophagy by lapatinib, such as in HCT116 colon cancer cellsCitation39 and in lapatinib and obatoclax cotreated breast cancer cells.Citation40,Citation41
Consistent with the findings of our study, some EGFR inhibitors, for example, sorafenib, gefitinib, erlotinib, and quizartinib (AC220), have been reported as targeted therapies for AML treatment in preclinical studies.Citation1,Citation4,Citation5 Irwin et al also found the antiproliferation effect of lapatinib in human ALL Z119 and Z181 cell lines with the Philadelphia chromosome and ErbB2 expression.Citation42 Also, similar to the findings of our study, cytotoxic activity and differentiation induction were found for gefitinib and erlotinib and other targeted therapy drugs against EGFR, in leukemia cells and clinical responses.Citation4,Citation5,Citation32–Citation38,Citation43 According to the previous findings and our preliminary results (data not shown), there are no clear expression patterns for EGFR or ErbB2 in AML or in some of the tested cell lines, such as HL-60,Citation33,Citation36,Citation44,Citation45 despite the demonstration of a EGF-EGFR autocrine loop in U937 cells.Citation46 Thus, the real targets of lapatinib, as well as gefitinib and erlotinib EGFR inhibitors need to be further investigated.Citation43,Citation47
Conclusion
We found that lapatinib induced cytotoxicity in several leukemia cell lines via either autophagic or apoptotic cell death. Lapatinib-induced autophagic cell death was observed in AML U937 cells. We also found differential induction by lapatinib and that treatment with lapatinib and other cytotoxic drugs (in AML U937 cells) had a synergistic effect. Our study suggests that a combination of lapatinib and other anticancer drugs may potentially be used as an AML therapy for clinical application.
Acknowledgments
The authors thank Miranda Loney and Ronald Lovel for English editing, Dr King-Song Jeng for experimental assistance, and the National RNAi Core Facility, Academia Sinica, Taiwan, for the pLKO.1-shRNA constructions. This study was supported by grants MMH-E-102-13 and MMH-E-103-13 from Mackay Memorial Hospital, grants NSC 100-2314-B-309-004- and NSC 100-2314-B-195-007-MY3 from the National Science Council of Taiwan, and grants MOST 103-2313-B-309-001- and MOST 104-2320-B-309-001- from the Ministry of Science and Technology, Republic of China.
Supplementary materials
Figure S1 Lapatinib-induced mitochondria collapse in U937 cells.
Notes: U937 cells were treated with DMSO or 2.5 µM lapatinib for 2 days. Cells were stained with DiOC6 (molecular probes) and the MTP inside the cells was analyzed by flow cytometry.
Abbreviations: DMSO, dimethyl sulfoxide; MTP, mitochondria transmembrane potential.
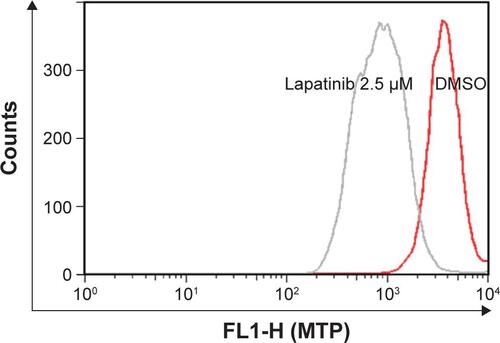
Figure S2 Expression of ATG7, ATG5, and BCLN in shRNA knockdown cells.
Notes: U937 cells with shRNA expression as indicated were prepared. Cell lysates from shRNA expressing U937 cells were collected and subjected to Western blot analysis for determining the expression of ATG7, ATG5, BCLN, and actin (loading control). Two shATG7 or shBECN clones (#1 or #2) were used.
Abbreviations: BCLN, Beclin-1; shRNA, short hairpin RNA.
Disclosure
The authors report no conflicts of interest in this work.
References
- HatzimichaelEGeorgiouGBenetatosLBriasoulisEGene mutations and molecularly targeted therapies in acute myeloid leukemiaAm J Blood Res201331295123358589
- TallmanMSGillilandDGRoweJMDrug therapy for acute myeloid leukemiaBlood200510641154116315870183
- KellyLMGillilandDGGenetics of myeloid leukemiasAnnu Rev Genomics Hum Genet2002317919812194988
- BoehrerSAdesLGalluzziLErlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparisonBiochem Pharmacol200876111417142518617157
- BoehrerSAdesLBraunTErlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical studyBlood200811142170218017925489
- MacFarlaneRJGelmonKALapatinib for breast cancer: a review of the current literatureExpert Opin Drug Saf201110110912121091041
- RusnakDWAlligoodKJMullinRJAssessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell linesCell Prolif200740458059417635524
- RusnakDWLackeyKAffleckKThe effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivoMol Cancer Ther200112859412467226
- HolbroTHynesNEErbB receptors: directing key signaling networks throughout lifeAnnu Rev Pharmacol Toxicol20044419521714744244
- KopperLLapatinib: a sword with two edgesPathol Oncol Res20081411818409020
- ItoYTokudomeNSugiharaTTakahashiSHatakeKDoes lapatinib, a small-molecule tyrosine kinase inhibitor, constitute a breakthrough in the treatment of breast cancer?Breast Cancer200714215616217485900
- TevaarwerkAJKolesarJMLapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancerClin Ther200931Pt 22332234820110044
- RyanQIbrahimACohenMHFDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2Oncologist200813101114111918849320
- SpiraAICarducciMADifferentiation therapyCurr Opin Pharmacol20033433834312901941
- HonmaYMatsuoYHayashiYOmuraSTreatment of Philadelphia-chromosome-positive human leukemia in SCID mouse model with herbimycin A, bcr-abl tyrosine kinase activity inhibitorInt J Cancer19956056856887860143
- MorceauFDupontCPalissotVGTP-mediated differentiation of the human K562 cell line: transient overexpression of GATA-1 and stabilization of the gamma-globin mRNALeukemia20001491589159710995005
- MeshkiniAYazdanparastRInvolvement of ERK/MAPK pathway in megakaryocytic differentiation of K562 cells induced by 3-hydrogenkwadaphninToxicol In Vitro20082261503151018586450
- WilliamsCAMondalDAgrawalKCThe HIV-1 Tat protein enhances megakaryocytic commitment of K562 cells by facilitating CREB transcription factor coactivation by CBPExp Biol Med (Maywood)20052301187288416339753
- RouyezMCBoucheronCGisselbrechtSDusanter-FourtIPorteuFControl of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathwayMol Cell Biol1997179499150009271377
- TetterooPAMassaroFMulderASchreuder-van GelderRvon dem BorneAEMegakaryoblastic differentiation of proerythroblastic K562 cell-line cellsLeuk Res1984821972066232432
- ChenYJChiCWSuWCHuangHLLapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinomaOncotarget20145134845485424947784
- HuangHLChenYCHuangYCLapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia k562 cellsPLoS One2011612e2901422216158
- ChenYJHuangWPYangYCPlatonin induces autophagy-associated cell death in human leukemia cellsAutophagy20095217318319066447
- DattaRYoshinagaKKanekiMPandeyPKufeDPhorbol ester-induced generation of reactive oxygen species is protein kinase cbeta-dependent and required for SAPK activationJ Biol Chem200027552410004100311042219
- SuWCChaoTCHuangYLWengSCJengKSLaiMMRab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagyJ Virol20118520105611057121835792
- KlionskyDJAbdallaFCAbeliovichHGuidelines for the use and interpretation of assays for monitoring autophagyAutophagy20128444554422966490
- KroemerGGalluzziLVandenabeelePClassification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009Cell Death Differ200916131118846107
- SchmittgenTDLeeEJJiangJReal-time PCR quantification of precursor and mature microRNAMethods2008441313818158130
- MollinedoFLopez-PerezRGajateCDifferential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL-60 cellsGene20084191–2162618547747
- CollinsSJRuscettiFWGallagherREGalloRCNormal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxideJ Exp Med19791494969974219131
- ClergetMPollaBSErythrophagocytosis induces heat shock protein synthesis by human monocytes-macrophagesProc Natl Acad Sci U S A1990873108110852153967
- PitiniVArrigoCAltavillaGErlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancerJ Clin Oncol200826213645364618640945
- LindhagenEErikssonAWickstromMSignificant cytotoxic activity in vitro of the EGFR tyrosine kinase inhibitor gefitinib in acute myeloblastic leukaemiaEur J Haematol200881534435318637032
- MirandaMBDuanRThomasSMGefitinib potentiates myeloid cell differentiation by ATRALeukemia20082281624162718305561
- RavindranathanMKlementichFJJonesDVJrPotential interaction of chemotherapy and gefitinib in the induction of hematologic neoplasiaLeukemia200721122546254717657221
- StegmaierKCorselloSMRossKNWongJSDeangeloDJGolubTRGefitinib induces myeloid differentiation of acute myeloid leukemiaBlood200510682841284815998836
- TakigawaNTakeuchiMShibayamaTSuccessful treatment of a patient with synchronous advanced non-small cell lung cancer and acute myeloid leukemia by a combination of gefitinib, low-dose cytarabine and aclarubicinAnticancer Res2005253c2579258216080496
- EnnishiDSezakiNSenooTTeruiYHatakeKHinoNA case of acute promyelocytic leukemia during gefitinib treatmentInt J Hematol200684328428517020873
- GalluzziLAaronsonSAAbramsJGuidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotesCell Death Differ20091681093110719373242
- CruickshanksNTangYBoothLHamedHGrantSDentPLapatinib and obatoclax kill breast cancer cells through reactive oxygen species-dependent endoplasmic reticulum stressMol Pharmacol20128261217122922989520
- TangYHamedHACruickshanksNFisherPBGrantSDentPObatoclax and lapatinib interact to induce toxic autophagy through NOXAMol Pharmacol201281452754022219388
- IrwinMENelsonLDSantiago-O’FarrillJMSmall molecule ErbB inhibitors decrease proliferative signaling and promote apoptosis in philadelphia chromosome-positive acute lymphoblastic leukemiaPLoS One201388e7060823936456
- BoehrerSGalluzziLLaineyEErlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemiaCell Cycle201110183168317521897118
- HahnCKBerchuckJERossKNProteomic and genetic approaches identify Syk as an AML targetCancer Cell200916428129419800574
- AllenHHsuanJClarkSExpression of epidermal-growth-factor receptor in the K562 cell line by transfection. Altered receptor biochemistryBiochem J199027137857902173908
- Eales-ReynoldsLJLaverHModjtahediHEvidence for the expression of the EGF receptor on human monocytic cellsCytokine200116516917211814311
- LaineyESebertMThepotSErlotinib antagonizes ABC transporters in acute myeloid leukemiaCell Cycle201211214079409223095522