Abstract
The emergence of epigenetic mechanisms as key regulators of gene expression has led to dramatic advances in understanding cancer biology. Driven by complex layers that include aberrant DNA methylation and histone modification, epigenetic aberrations have emerged as critical processes that disrupt cellular machinery and homeostasis. Recent discoveries have already translated into successful clinical trials and improved patient care, with several agents approved for hematologic disease and others undergoing study. As the field matures, substantial challenges persist that will require resolution. These include the need to decipher more fully the interplay between the epigenetic and genetic machinery, patient selection and improving treatment efficacy in solid tumors, and optimizing combination therapies to counteract chemoresistance and minimize adverse effects. Here, we review recent progress in epigenetic treatments and consider their implications for future cancer therapy.
Introduction
The field of epigenetics comprises a wide range of reversible modifications that orchestrate gene expression. The genome is organized into relaxed euchromatin and condensed heterochromatin, and DNA is interlaced among histones, which are in turn post-translationally altered to enable or disable transcription. The relative structural simplicity of DNA is therefore supported by immense modulation from epigenetic factors that are both tissue- and context-specific. It is these factors that enable a broad range of phenotypes to be manifested from a common DNA blueprint.
Advances in deciphering the fundamental machinery of the epigenome have led to significant insights into cell physiology as well as oncogenesis. This information has provided a fuller and more nuanced understanding of epigenetic abnormalities linked to genetic mutations, including the roles of methylation and acetylation. Although these marks are somatically heritable, the fact that they are also reversible suggests exciting implications for therapy. Defining (and restoring) the “normal” epigenetic landscape has consequently been the focus of active investigation, and has already generated breakthroughs in cancer detection, treatment, and prognosis. Swift approval of epigenetically targeted drugs by the US Food and Drug Administration (FDA) for hematologic conditions has cemented its role in the clinical sphereCitation1–Citation4 (), and many phase II and III clinical trials are under way for multiple conditions, including solid malignancies.Citation5–Citation7 Below, we highlight the interface between the genome and epigenome and examine the clinical impact facilitated by current and future epigenetic agents.
Table 1 Examples of approved epigenetic agents
DNA methylation and histone modification
Two of the most common types of epigenetic alterations in cancer involve aberrant changes in DNA methylation and histone modification. These alterations occur at multiple layers of regulation, directing gene expression via maintenance of restricted and permissive chromatin states. Such regulators can also be commandeered by cancer cells for oncogenic gain.Citation8 Methylation consists of the addition of a methyl group to the 5′ position of the cytosine ring in CpG dinucleotides (5 mc) and typically occurs in CpG islands within promoter regions. DNA hypermethylation in promoters can lead to the silencing of gene expression. Other areas found to harbor CpG methylation include vast areas in the genome with repetitive sequence, such as centromeres and transposon elements (involved in chromosomal stability),Citation9 CpG island shores,Citation10 noncoding regions (ie, enhancer regions and miRNAs),Citation11 and gene bodies (silencing alternative transcription start sites).Citation12 Approximately 60% of gene promoters contain CpG sites.Citation13
The central workhorse molecules that lay down DNA methylation are the DNA methyltransferases. As a maintenance enzyme, DNMT1 preserves existing methylation patterns after cell replication, and its deletion leads to apoptosisCitation13 as well as death in mice if lost during embryonic development.Citation14 In contrast, DNMT3 A and -3B are de novo methyltransferases that methylate previously unmethylated DNA. While they are in the same general class of enzymes and share similarities within their catalytic domains, their roles in tumorigenesis may differ: DNMT3A deletion may promote cancer progression,Citation15 yet DNMT3B deletion may in fact inhibit oncogenesis by liberating previously silenced tumor-suppressor genes.Citation16,Citation17 Interestingly, a substantial degree of DNA methylation in embryonic stem cells appears to occur independently of CpG sites,Citation18 and the Ten-Eleven-Translocation (TET) oxidase family has been reported to convert 5-methylcytosine to 5-hydroxymethylcytosine as a step toward demethylation, a process that does not seem to be restricted to CpG islands.Citation19 The exact roles of these phenomena in epigenetic regulation remain to be elucidated.
Besides direct manipulation of DNA, modification of the core histones (ie, two copies of H2A, H2B, H3, and H4 proteins) also play important roles in epigenetic regulation. These histones selectively bind and release DNA as nucleosomes to moderate transcription, a process that is regulated through the addition of acetyl, methyl, phosphoryl, ubiquityl, or sumoyl groups, producing a dynamic epigenetic histone code ().Citation20 These histone marks are deposited or removed by a plethora of proteins and clinical targets, including histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases (HDMs), kinases, phosphatases, and others. Here, we will summarize some salient parts of this biology, and for greater detail, the reader is directed to a couple of excellent reviews on these processes.Citation21,Citation22 Acetylation of lysines at specific sites (such as on H3K4, etc) by HATs negates the positive histone charge, permitting negatively charged DNA to take on a transcription-ready configuration. HDACs, however, remove acetyl groups from histones, causing the oppositely charged histone to bind DNA and shield it from expression. While there are exceptions to each generality, DNA methylation typically silences gene expression, while histone marks can activate or silence genes, depending on the target residues, the targeted histone and extent of the alteration.Citation23
Figure 1 (A and B) Epigenetic mechanisms of transcriptional regulation. (A) Transcriptional activation, and (B) transcriptional repression with associated histone marks.
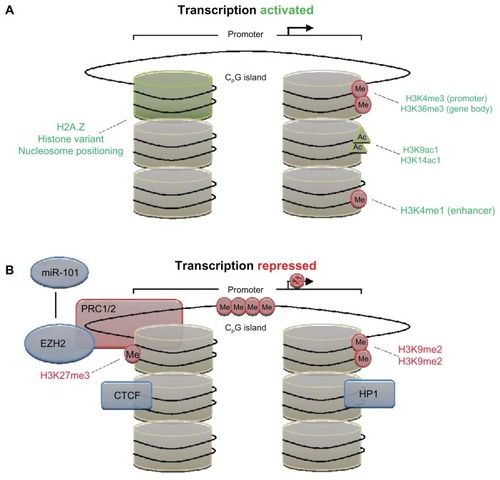
Histone marks work together. Numerous histone modifications are now known to synergize in a combinatorial way, establishing specific states to activate or inhibit expression. Permissive chromatin states, for instance, are established by histone H3 marks such as trimethylation of lysine (K) 4 over the promoter and K36 over the gene body (H3K4me3 and H3K36me3, respectively).Citation24,Citation25 Other enabling marks include monoacetylation of H3K9 and H3K14 (H3K9ac1 and H3K14ac1),Citation26 the presence of variant histones such as H2A.Z,Citation27 and methylation of enhancer elements downstream (H3K4me1).Citation28 Nucleosome positioning represents an additional contributory layer, and is determined by the intrinsic binding affinity of the DNA sequence, competitive binding of surrounding proteins, and translocation activity by adenosine triphosphate-dependent remodeling complexes.Citation29
Conversely, heterochromatin configurations that compact nucleosomes are classically indicated by H3K27me3, H3K9me3, and H3K9me2,Citation30 as well as the presence of inhibitory proteins such as CTCF and HP1.Citation31,Citation32 This is in addition to dense, localized DNA methylation. Repression can be initiated by the Polycomb group (Polycomb repression complex [PRC]) proteins, which methylate the histones in promoter regions.Citation33 Such PRC regulators, including enhancer of zeste 2 (EZH2),Citation34 are themselves regulated by noncoding RNAs such as miRNA-101 to inhibit expression.Citation35,Citation36
Collectively, such interplay between systems previously thought to be independent underscores the overarching importance of the epigenome in coordinating transcription, and of course downstream biological events. Its complexity gives rise to enormously flexible conditions that guide development and physiology, but can also be readily misappropriated for disease states such as tumorigenesis.
Epigenetic mechanisms of transcription regulation in cancer
Epigenetic dysregulation in malignant cells can be generalized across cancer types, and is characterized by global hypomethylation and focal hypermethylation.Citation37 These events occur early on in tumorigenesis, including in early stage and in situ lesions,Citation38 suggesting that these changes help establish an advantageous milieu for cancer formation. Such broad hypomethylation has been shown to induce genomic instability by activating normally quiet transcription start sites and transposon activity (increasing the likelihood of structural variation),Citation20,Citation37 while aberrantly methylated promoter areas silence key tumor suppressors. Similar studies have reported decreased acetylation from overexpressed HDACs and inhibited HATs (eg, H4K16ac1),Citation39 as well as dysregulated histone methylation (eg, H4K20me3).Citation40 Together with genetic mutations, epigenetic changes can engender favorable environments for tumorigenesis and resistance to therapy. Moreover, epigenetic alterations leading to cancer are not limited to just direct change of gene function, but also predispose to genetic mutation or dysregulation of entire signaling pathways and DNA integrity.Citation41 Genetic driver mutations affecting epigenetic players such as DNMT3A, UTX, etcCitation42 have been reported to be quite frequent events.Citation43
Aberrant promoter hypermethylation is a hallmark of cancer and is one of the most common alterations seen in malignancies. VHL1 (renal carcinoma), BRCA1 (breast cancer), and RB1 are examples of classic tumor suppressors whose dysregulation can result from either mutation or epigenetic silencing via hypermethylation.Citation44 Other tumor suppressors that are mutated (ie, CDKN2A, encoding INK4A) can instead be inactivated by epigenetic silencing.Citation45,Citation46 This was first seen years ago in a number of ad hoc analyses on candidate cancer genes.Citation47 These findings have been confirmed through high-throughput, global analyses of cancer genomes and epigenomes, which for any given cancer gene may show an absence of genetic mutations, yet reveal epigenetic-based deregulation instead.Citation48–Citation51
Other epigenetic abnormalities involve regulatory mechanisms only recently discovered. As described above, TET proteins add hydroxyl groups to 5 mC and not only initiate demethylation but also protect against unsolicited methylation. TET proteins have been found to compete with IDH1 for a common cofactor (α–ketoglutarate), which mutant IDH1 uses to produce the 2-hydroxyglutarate metabolite.Citation52 This byproduct inhibits TET2, thereby incapacitating the TET-dependent hydroxylating system and allowing methylation to accumulate in IDH1-mutated cancers. This process may contribute to the glioma CpG island methylator phenotype,Citation53 which characterizes GBM subtypes with distinct clinical and molecular features.Citation54 Interestingly, CpG island methylator phenotype-positive phenotypes appear to have better prognosis in brain and breast cancers.Citation54–Citation56 TET alterations observed in cancer include fusion transcripts (ie, MLL-TET1) as well as inactivating mutations and deletions at the TET2 locus.Citation57
Similarly, dysregulation or reprogramming of histone modifications has been observed in cancer. Viral protein binding of transcriptional regulators such as CREBBP/EP300 has been shown to induce hypoacetylation (H3 K18ac1) and subsequent transformation,Citation58,Citation59 while global overexpression of the HDAC family has been demonstrated across multiple tumor types.Citation39 Moreover, EZH2, a major HMT of the PcG system, is often overexpressed in cancer.Citation60,Citation61 The ensuing aberrant silencing of tumor-suppressor genes strongly implicates a causative oncogenic role for PcG complexes. It appears that as more mechanisms of epigenetic regulation are uncovered, so too are additional means for dysregulation and oncogenesis.
Epigenetic alterations may serve as useful biomarkers for detecting disease and predicting therapeutic efficacy. For example, hypermethylation of GSTP1 (a glutathione S-transferase) is found in approximately 85% of prostate cancer, including prostate intraepithelial neoplasia.Citation62 This is not the case in benign hyperplastic prostate tissue, and therefore its detection in blood or biopsied tissue can be used to detect the presence of prostate cancer cells.Citation63,Citation64 Similarly, MGMT and MLH1 (DNA mismatch-repair genes) have been demonstrated to undergo epigenetic inactivation in gliomasCitation65 and ovarian cancers,Citation66 respectively. Though this may facilitate oncogenesis, it can also prevent such malignancies from recovering from DNA damage caused by chemotherapeutic agents. Patients with hypermethylated MGMT or MLH1 phenotypes have accordingly been shown to have superior clinical responses following treatment with certain cytotoxic agents.Citation67 Normal cells may employ alternative repair schemes to recover in ways that cancer cells cannot.
The clinical utility of epigenetic agents in cancer management
The malleable and reversible nature of the cancer epigenome presents intriguing opportunities for novel treatments. Several drugs (specifically DNMT and HDAC inhibitors) have already been approved for select indications, and research efforts have resulted in the development of new drugs designed to be more selective and less toxic. Discovery of how tumors adapt epigenetic machinery to their advantage has also improved understanding of how cancers develop chemoresistance to existing drugs. As such, sensitizing resistant tumors with epigenetic agents to restore the impact of conventional drugs has become a major area of investigation.
Targeting DNA methylation
DNA methyltransferase inhibitors have found the earliest and greatest success as prototypical epigenetic agents. Azacitidine and decitabine were FDA-approved in the mid-2000s for treatment of myelodysplastic syndrome (MDS). As nucleoside analogues, they replace cytosine during DNA replication, in the process trapping DNMTs and targeting them for degradation. Despite indiscriminately targeting DNMTs,Citation68 they were found at low dosages to selectively reactivate gene expression with relatively few side effects.Citation69
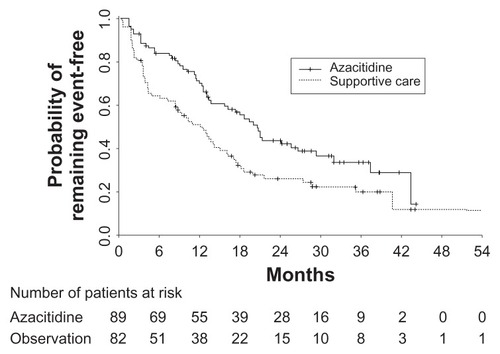
Landmark phase III trials with MDS patients comparing decitabine or azacitidine with placebo found a response rate up to 30% that was relatively durable (decitabine, median 10.3 months), including 9% demonstrating a complete response ().Citation70,Citation71 Compared to supportive-care patients, however, those treated with decitabine did not have significantly improved delayed time to acute myeloid leukemia (AML) transformation or death. More recent phase III trials analyzing azacitidine for higher-risk or elderly MDS patients, however, have demonstrated improved overall survival.Citation72 Besides MDS, DNA methyltransferase inhibitors have also shown some utility for AML. Decitabine, in a number of phase II trials, showed complete responses in 24%–52% of patients, with pronounced benefit for older patients.Citation73,Citation74 The main documented toxicities were myelosuppression (including neutropenia and thrombocytopenia), as well as nausea and vomiting.
Figure 2 Kaplan–Meier curves from a trial of azacitidine in patients with myelodysplastic syndrome.
Adapted with permission. © 2002. American Society of Clinical Oncology. All rights reserved. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20(10):2429–2440.Citation71
That a substantial portion of patients benefited to some degree from such demethylating agents, which are quite nonspecific, is somewhat surprising. There is clearly room to improve the efficacy of this class of drug, which will require intensive investigation. Potential avenues for improvement include determining the optimal duration of treatment needed and deducing azacitidine’s and decitabine’s most important molecular effects (key effector genes).Citation69 Other priorities include minimizing side effects. Two newer DNMT inhibitors include S110, which releases decitabine intracellularly,Citation75 and CP-4200, a derivative of azacitidine with an elaidic acid modification,Citation76 have both shown relatively more potent tumoricidal activity in cell lines and mouse models and await clinical investigation. Nanaomycin A, one of the first DNMT3B-specific inhibitors, has been shown to reactivate silenced tumor-suppressor genes.Citation77 Results from clinical trials will be needed to determine what incremental benefit, if any, these novel compounds will provide.
Targeting histone acetylation/deacetylation
By restoring acetylation to lysine residues on histone tails, HDAC inhibitors counteract the global overexpression of HDACs in cancer and reinstate a more permissive nucleosome structure for transcription. Inhibitors typically target the zinc-dependent active sites of HDACs, with the exception of class III HDACs (sirtuins), which are NAD+-dependent and do not primarily act on histones.Citation20,Citation78 The effects of HDAC inhibitors appear to be to promote G1 or G2/M cell-cycle arrest, as well as apoptosis and cell differentiation. Intriguingly, the basis of their efficacy (and toxicity) may not only be limited to histone modification: other reported HDAC targets include p53, signal transducer and activator of transcription 3, heat shock protein 90, and other important proteins and changing the acetylation status of these may contribute to the biological effects observed.Citation79
Although there are numerous structural classes of HDAC inhibitors, they can be stratified into broad HDAC inhibitors and class-specific agents. Vorinostat (a pan-HDAC inhibitor) and romidepsin (a class I HDAC inhibitor) have each shown > 30% response rates against cutaneous T-cell lymphoma (CTCL) in phase II trialsCitation1–Citation4 (), and accordingly were approved for use by the FDA in the late 2000s. Significant adverse events noted were diarrhea, hypercholesterolemia, and anemia for vorinostat, and nausea, cardiotoxicity, and fatigue for romidepsin.Citation79,Citation80 Though generally more tolerable than conventional chemotherapeutic agents, fatigue has been severe enough to cause discontinuation of HDAC inhibitors in up to 30% of patients.
Figure 3 Kaplan–Meier curves from a phase II trial of romidepsin in patients with cutaneous T-cell lymphoma showing progression-free survival of patients with complete response (CR) or partial response (PR), patients with stable disease (SD), and patients with progressive disease (PD) or nonevaluable (NE) patients.
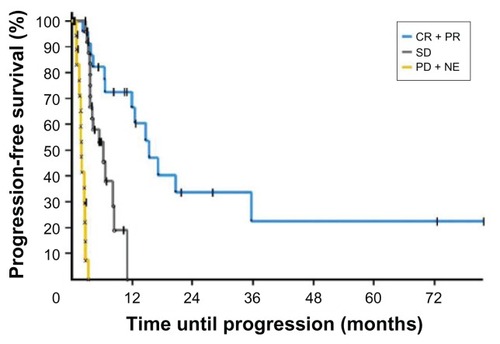
Nonetheless, enthusiasm continues to build for this class of drug, with more agents in development and undergoing trial than any other class of epigenetic drug. Panobinostat, another pan-HDAC inhibitor, is currently under investigation for numerous hematologic malignancies. A recently reported phase II trial for refractory Hodgkin’s lymphoma in 129 patients showed tumor reductions in 74%, with a median duration of response of 6.9 months.Citation81 A phase III trial with panobinostat and bortezomib is underway for multiple myeloma patients.Citation69 Similarly, the multiclass specific HDAC inhibitor mocetinostat (class I and IV) has been studied in phase II trials for refractory Hodgkin’s lymphoma, refractory diffuse large B-cell lymphoma, and follicular lymphoma.Citation79,Citation82–Citation84 Encouraging results have been seen on preliminary analysis that appear to indicate promise, including a 38% response rate for Hodgkin’s lymphoma patients.Citation82 Finally, preclinical studies show potential for novel compounds such as JQ1 and I-BET, which inhibit bromodomain (BRD)-containing proteins. BRD4, for instance, can bind acetylated histones, leading to proliferation and overexpression by recruitment of the P-TEFb complex or myc.Citation85 While known HDAC inhibitors can restore this acetylation, novel compounds such as JQ1 are potentially more specific, as they block contact between BRDs and acetylated histones.Citation86 Another synthetic drug called I-BET has also been shown to bind and neutralize the active BRD pocket, competitively reducing BRD4’s sequestering activity.Citation87 These two drugs demonstrate that targeting transcription factors and epigenetic readers can be a potentially promising avenue to explore.
Paradoxically, HAT inhibitors have also been shown to have some antitumor activity. This contrasts with the global hypoacetylation already seen in many cancers. Naturally occurring drugs such as curcumin, garcinol, and anacardic acid appear to selectively inhibit EP300, CREBBP, or KAT2B, leading to apoptosis or sensitization to therapies such as radiation.Citation88–Citation90 As no clinical trials have yet been completed, this class of agents remains largely investigational.
Histone methylation/demethylation
In contrast to the more global activity of histone acetylation enzymes, HMTs and HDMs can be somewhat more discriminating in the specific amino acid residues they target.Citation84,Citation91 This has exciting therapeutic implications, given that elimination of select histone marks could enable more tailored treatment while potentially minimizing side effects. Yet despite the promising nature of these targets, only a few clinically viable compounds have been identified so far, and all currently remain in preclinical levels of study.
Among the first histone demethylases identified was lysine-specific demethylase 1 (LSD1, or KDM1), a flavin adenine dinucleotide-dependent amino oxidase.Citation92 By targeting H3K4me1 and H3K4me2, LSD1 selectively mediates repression and has been found to be overexpressed in a significant number of cancers, including tumors of the brain, breast, and prostate, thereby making it an attractive target for drug therapy.Citation92–Citation94 Small molecules such as SL11144 and tranylcypromine appear to be potent inhibitors of LSD1,Citation95,Citation96 and have been shown in cancer cell lines to restore expression of multiple aberrantly silenced tumor suppressors, including secreted frizzled-related protein and GATA transcription factors. Research with neuroblastoma xenografts also demonstrates antitumor activity.Citation94 However, similar to HDACs, off-target effects on H3K9me2 and DNMT1 limit its immediate usefulness,Citation97 and further study is needed.
The previously mentioned Polycomb group genes also are undergoing active investigation as potential targets. The PRC1 and PRC2 complexes are responsible for H3K27me3-mediated repression of transcription.Citation98 Such histone marks can lead to DNA methylation and sustained gene silencing. EZH2, a member of the PRC2 complex, is another attractive drug target, given its overexpression in head and neck, breast, and prostate cancers,Citation99 and is targeted by a hydrolase inhibitor called 3-deazaneplanocin A (DZNep). By countering EZH2 and inhibiting H3K27 trimethylation, DZNep has been shown to induce differentiation as well as apoptosis in cancer cell lines and xenografts,Citation100,Citation101 while sparing normal cells. This apparently selective reversal of PRC2-mediated gene repression constitutes a novel treatment approach, and results from clinical trials are forthcoming. However, cancer-mutation surveys have shown that EZH2 can act as an oncogene in some cancers, but as a tumor suppressor in others. Therefore, use of EZH2 inhibitors in the clinic can only be implemented after judicious study.
Future developments in epigenetic therapy
Epigenetically targeted drugs have already become an important part of the clinical armamentarium, and in some refractory conditions are the only drugs with any activity available to patients. Yet initial successes notwithstanding, challenges remain that must be addressed by future studies. These issues involve expanding the therapeutic reach beyond hematologic cancers, identifying the synergistic potential of combination therapy for better patient outcomes, and understanding the mechanistic basis of these agents to narrow drug activity and more precisely target aberrant marks.
One of the main criticisms of epigenetic therapy has been its relatively lower efficacies when tested on solid tumors.Citation79,Citation84 This is likely due to the heterogeneity of solid tumor cells, which may make it more difficult for agents to penetrate and less difficult for tumors to develop resistance. This is especially telling with the epigenetic agents already approved for hematologic malignancies like MDS and CTCL. Vorinostat has been tested as a single agent in head and neck, breast, colorectal, and prostate cancer trials,Citation102–Citation104 and has not shown efficacy. A phase II trial with 94 non-small-cell lung cancer patients did not show significantly improved progression-free or overall survival, with 41% of patients unable to complete the six-cycle regimen.Citation105 Similarly, phase I and II trials testing romidepsin (approved for CTCL) showed very marginal clinical improvement when treating refractory solid malignancies, including kidney, prostate, and lung.Citation5,Citation6,Citation106 Unfortunately, testing epigenetic agents against refractory tumors that have become resistant to other first-line therapies increases the likelihood of failure. Furthermore, it will be important to properly select patients using molecular criteria in future trials, as these features are likely to influence heavily the likelihood of response.
Future trials will likely test combination therapies, with the hypothesis that epigenetic agents may enhance sensitivity to conventional drugs (eg, platinum or taxane chemotherapy) that have known activity. By relaxing chromatin with HDAC inhibitors, it is believed that strands are better exposed to DNA intercalation or genotoxic damage.Citation84 Such synergistic combinations are also under intense study to demonstrate reversal of chemoresistance due to epigenetic adaptation, crucial for treating disease that is recurrent or refractory to first-line treatment. Other strategies are examining the role of dual epigenetic therapies, such as DNMT inhibitors with HDAC inhibitors.Citation107,Citation108 Given previous experience with azacitidine, it will be essential to master the optimal timing (ie, sequential versus simultaneous) and dosage needed for these cocktails to be effective.
Finally, the advent of next-generation sequencing technologies has made high-throughput mutational and epigenetic analysis accessible and will ultimately allow investigators to paint a more comprehensive portrait of the determinants of response to epigenetically targeted therapies. This will greatly assist in deciphering the many nonhistone and nonpromoter effects observed in many epigenetic agents, including interactivity with miRNAs and kinases. Such large-scale analyses are already under way in mapping cancer genomes via the Cancer Genome Atlas and the International Cancer Genome Consortium. CHIP-seq, whole genome/exome sequencing, RNA-seq, and reverse-phase protein arrays, as well as the bioinformatic expertise necessary to synthesize them, have all helped make rapid strides possible.Citation109–Citation111 Epigenetic analyses of tumors have already demonstrated clinical utility, as demonstrated by the prognostic and predictive use of MGMT methylation status in GBM.Citation112 These new tools should lead to substantial improvements in finding reliable biomarkers and selecting the right drugs for the right patient.
Conclusion
With more than 100 epigenetic agents currently under investigation and a handful already approved in the last decade alone, it is clear that epigenetic therapy will continue to impact the management of cancer patients significantly. Such agents are unlikely to be a universal remedy, but their potential to prolong meaningful life and originate new avenues of treatment remains tantalizing. Much remains to be defined about the epigenome regarding its role and its dysregulation in cancer. The widespread presence of mutations in epigenetic regulators in human cancers suggests that the continued efforts in developing strategies to target epigenetic aberrations may contribute to improving efficacy of cancer management.
Disclosure
The authors report no conflicts of interest in this work.
References
- OlsenEAKimYHKuzelTMPhase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphomaJ Clin Oncol200725213109311517577020
- DuvicMTalpurRNiXPhase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL)Blood20071091313916960145
- WhittakerSJDemierreMFKimEJFinal results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphomaJ Clin Oncol201028294485449120697094
- PiekarzRLFryeRTurnerMPhase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphomaJ Clin Oncol200927325410541719826128
- SchrumpDSFischetteMRNguyenDMClinical and molecular responses in lung cancer patients receiving romidepsinClin Cancer Res200814118819818172270
- MolifeLRAttardGFongPCPhase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC)Ann Oncol201021110911319608618
- BaumanJVerschraegenCBelinskySA phase I study of 5-azacytidine and erlotinib in advanced solid tumor malignanciesCancer Chemother Pharmacol201269254755421901396
- EdenAGaudetFWaghmareAJaenischRChromosomal instability and tumors promoted by DNA hypomethylationScience2003300561845512702868
- WangYLeungFCAn evaluation of new criteria for CpG islands in the human genome as gene markersBioinformatics20042071170117714764558
- IrizarryRALadd-AcostaCWenBThe human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shoresNat Genet200941217818619151715
- VaramballySCaoQManiRSGenomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancerScience200832259081695169919008416
- GuentherMGLevineSSBoyerLAJaenischRYoungRAA chromatin landmark and transcription initiation at most promoters in human cellsCell20071301778817632057
- BirdADNA methylation patterns and epigenetic memoryGenes Dev200216162111782440
- EndresMFanGMeiselADirnaglUJaenischREffects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neuronsNeuroreport200112173763376611726790
- GaoQSteineEJBarrasaMIDeletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progressionProc Natl Acad Sci U S A201110844180611806622011581
- NoshoKShimaKIraharaNDNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancerClin Cancer Res200915113663367119470733
- LinhartHGLinHYamadaYDnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencingGenes Dev200721233110312218056424
- ListerRPelizzolaMDowenRHHuman DNA methylomes at base resolution show widespread epigenomic differencesNature2009462727131532219829295
- ItoSShenLDaiQTet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosineScience201133360471300130321778364
- Rodriguez-ParedesMEstellerMCancer epigenetics reaches mainstream oncologyNat Med201117333033921386836
- CamposEIReinbergDHistones: annotating chromatinAnnu Rev Genet20094355959919886812
- BeckDBBonasioRKanekoSChromatin in the nuclear landscapeCold Spring Harb Symp Quant Biol201175112221502408
- SawanCHercegZHistone modifications and cancerAdv Genet201070578520920745
- BarskiACuddapahSCuiKHigh-resolution profiling of histone methylations in the human genomeCell2007129482383717512414
- RosenfeldJAWangZSchonesDEZhaoKDeSalleRZhangMQDetermination of enriched histone modifications in non-genic portions of the human genomeBMC Genomics20091014319335899
- LiBCareyMWorkmanJLThe role of chromatin during transcriptionCell2007128470771917320508
- HuGCuiKNorthrupDH2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiationCell Stem Cell201312218019223260488
- HeintzmanNDHonGCHawkinsRDHistone modifications at human enhancers reflect global cell-type-specific gene expressionNature2009459724310811219295514
- GaffneyDJMcVickerGPaiAAControls of nucleosome positioning in the human genomePLoS Genet2012811e100303623166509
- BaylinSBJonesPAA decade of exploring the cancer epigenome – biological and translational implicationsNat Rev Cancer2011111072673421941284
- WenBWuHLohYHBriemEDaleyGQFeinbergAPEuchromatin islands in large heterochromatin domains are enriched for CTCF binding and differentially DNA-methylated regionsBMC Genomics20121356623102236
- KellerCAdaixoRStunnenbergRWoolcockKJHillerSBuhlerMHP1(Swi6) mediates the recognition and destruction of heterochromatic RNA transcriptsMol Cell201247221522722683269
- FujimuraYIsonoKVidalMDistinct roles of Polycomb group gene products in transcriptionally repressed and active domains of Hoxb8Development2006133122371238116687444
- AsanganiIAAteeqBCaoQCharacterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancerMol Cell2013491809323159737
- BanerjeeRManiRSRussoNThe tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2 overexpression in invasive squamous cell carcinomaOncogene201130424339434921532618
- SakuraiTBilimVNUgolkovAVThe enhancer of zeste homolog 2 (EZH2), a potential therapeutic target, is regulated by miR-101 in renal cancer cellsBiochem Biophys Res Commun2012422460761422609199
- EstellerMEpigenetics in cancerN Engl J Med2008358111148115918337604
- FeinbergAPCancer epigenetics is no Mickey MouseCancer Cell20058426726816226700
- FragaMFBallestarEVillar-GareaALoss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancerNat Genet200537439140015765097
- SeligsonDBHorvathSShiTGlobal histone modification patterns predict risk of prostate cancer recurrenceNature200543570461262126615988529
- YouJSJonesPACancer genetics and epigenetics: two sides of the same coin?Cancer Cell201222192022789535
- DawsonMAKouzaridesTHuntlyBJTargeting epigenetic readers in cancerN Engl J Med2012367764765722894577
- LeyTJDingLWalterMJDNMT3A mutations in acute myeloid leukemiaN Engl J Med2010363252424243321067377
- JonesPABaylinSBThe epigenomics of cancerCell2007128468369217320506
- IwamaAOguroHNegishiMEnhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1Immunity200421684385115589172
- JacobsJJKieboomKMarinoSDePinhoRAvan LohuizenMThe oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locusNature199939767151641689923679
- JonesPABaylinSBThe fundamental role of epigenetic events in cancerNat Rev Genet20023641542812042769
- Cancer Genome Atlas NetworkComprehensive molecular portraits of human breast tumoursNature20124907418617023000897
- Cancer Genome Atlas NetworkComprehensive molecular characterization of human colon and rectal cancerNature2012487740733033722810696
- Cancer Genome Atlas Research NetworkIntegrated genomic analyses of ovarian carcinomaNature2011474735360961521720365
- Cancer Genome Atlas Research NComprehensive genomic characterization of squamous cell lung cancersNature2012489741751952522960745
- FigueroaMEAbdel-WahabOLuCLeukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiationCancer Cell201018655356721130701
- IssaJPCpG island methylator phenotype in cancerNat Rev Cancer200441298899315573120
- TurcanSRohleDGoenkaAIDH1 mutation is sufficient to establish the glioma hypermethylator phenotypeNature2012483739047948322343889
- FangFTurcanSRimnerABreast cancer methylomes establish an epigenomic foundation for metastasisSci Transl Med201137575ra25
- van den BentMJGravendeelLAGorliaTA hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951Clin Cancer Res201117227148715521914791
- DelhommeauFDupontSDella ValleVMutation in TET2 in myeloid cancersN Engl J Med2009360222289230119474426
- FerrariRPellegriniMHorwitzGAXieWBerkAJKurdistaniSKEpigenetic reprogramming by adenovirus e1aScience200832158921086108818719284
- RastiMGrandRJMymrykJSGallimorePHTurnellASRecruitment of CBP/p300, TATA-binding protein, and S8 to distinct regions at the N terminus of adenovirus E1AJ Virol20057995594560515827174
- CaoPDengZWanMMicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1alpha/HIF-1betaMol Cancer2010910820478051
- SmitsMNilssonJMirSEmiR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesisOncotarget20101871072021321380
- JeronimoCHenriqueRHoqueMOA quantitative promoter methylation profile of prostate cancerClin Cancer Res200410248472847815623627
- BrooksJDWeinsteinMLinXCG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasiaCancer Epidemiol Biomarkers Prev1998765315369641498
- HenriqueRJeronimoCMolecular detection of prostate cancer: a role for GSTP1 hypermethylationEur Urol2004465660669 discussion 66915474280
- EstellerMGarcia-FoncillasJAndionEInactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agentsN Engl J Med2000343191350135411070098
- PlumbJAStrathdeeGSluddenJKayeSBBrownRReversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoterCancer Res200060216039604411085525
- BaylinSBOhmJEEpigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?Nat Rev Cancer20066210711616491070
- Schneider-StockRDiab-AssefMRohrbeckA5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3a and induces apoptosis in HCT-116 colon cancer cells via Gadd45- and p53-dependent mechanismsJ Pharmacol Exp Ther2005312252553615547111
- KellyTKDe CarvalhoDDJonesPAEpigenetic modifications as therapeutic targetsNat Biotechnol201028101069107820944599
- KantarjianHIssaJPRosenfeldCSDecitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized studyCancer200610681794180316532500
- SilvermanLRDemakosEPPetersonBLRandomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group BJ Clin Oncol200220102429244012011120
- FenauxPMuftiGJHellstrom-LindbergEEfficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III studyLancet Oncol200910322323219230772
- BlumWGarzonRKlisovicRBClinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabineProc Natl Acad Sci U S A2010107167473747820368434
- CashenAFSchillerGJO’DonnellMRDiPersioJFMulticenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemiaJ Clin Oncol201028455656120026803
- YooCBJeongSEggerGDelivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotidesCancer Res200767136400640817616700
- BruecknerBRiusMMarkelovaMRDelivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacyMol Cancer Ther2010951256126420442313
- KuckDCaulfieldTLykoFMedina-FrancoJLNanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cellsMol Cancer Ther20109113015302320833755
- SaundersLRVerdinESirtuins: critical regulators at the crossroads between cancer and agingOncogene200726375489550417694089
- FedericoMBagellaLHistone deacetylase inhibitors in the treatment of hematological malignancies and solid tumorsJ Biomed Biotechnol2011201147564121188171
- LaneAAChabnerBAHistone deacetylase inhibitors in cancer therapyJ Clin Oncol200927325459546819826124
- YounesASuredaABen-YehudaDPanobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II studyJ Clin Oncol201230182197220322547596
- YounesAOkiYBociekRGMocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trialLancet Oncol201112131222122822033282
- Garcia-ManeroGAssoulineSCortesJPhase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemiaBlood2008112498198918495956
- MackGSTo selectivity and beyondNat Biotechnol201028121259126621139608
- YangZHeNZhouQBrd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progressionMol Cell Biol200828396797618039861
- FilippakopoulosPQiJPicaudSSelective inhibition of BET bromodomainsNature201046873271067107320871596
- NicodemeEJeffreyKLSchaeferUSuppression of inflammation by a synthetic histone mimicNature201046873271119112321068722
- BalasubramanyamKVarierRAAltafMCurcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcriptionJ Biol Chem200427949511635117115383533
- BalasubramanyamKAltafMVarierRAPolyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expressionJ Biol Chem200427932337163372615155757
- SunYJiangXChenSPriceBDInhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiationFEBS Lett2006580184353435616844118
- LallSPrimers on chromatinNat Struct Mol Biol200714111110111517984971
- MetzgerEWissmannMYinNLSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcriptionNature2005437705743643916079795
- LimSJanzerABeckerALysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biologyCarcinogenesis201031351252020042638
- SchulteJHLimSSchrammALysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapyCancer Res20096952065207119223552
- HuangYStewartTMWuYNovel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genesClin Cancer Res200915237217722819934284
- WuYSteinbergsNMurray-StewartTMartonLJCaseroRAOligoamine analogues in combination with 2-difluoromethylornithine synergistically induce re-expression of aberrantly silenced tumour-suppressor genesBiochem J2012442369370122132744
- WangJHeviSKurashJKThe lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylationNat Genet200941112512919098913
- BrackenAPDietrichNPasiniDHansenKHHelinKGenome-wide mapping of Polycomb target genes unravels their roles in cell fate transitionsGenes Dev20062091123113616618801
- AlbertMHelinKHistone methyltransferases in cancerSemin Cell Dev Biol201021220922019892027
- GannonOEndo-MunozLBMerida de LongLHazar-RethinamMSaundersNADysregulation of the repressive H3K27 trimethylation mark in head and neck squamous cell carcinoma contributes to dysregulated squamous differentiationClin Cancer Res201319242844123186778
- TanJYangXZhuangLPharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cellsGenes Dev20072191050106317437993
- BradleyDRathkopfDDunnRVorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 ConsortiumCancer2009115235541554919711464
- VansteenkisteJVan CutsemEDumezHEarly phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancerInvest New Drugs200826548348818425418
- BlumenscheinGRJrKiesMSPapadimitrakopoulouVAPhase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancerInvest New Drugs2008261818717960324
- RamalingamSSMaitlandMLFrankelPCarboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancerJ Clin Oncol2010281566219933908
- StadlerWMMargolinKFerberSMcCullochWThompsonJAA phase II study of depsipeptide in refractory metastatic renal cell cancerClin Genitourin Cancer200651576016859580
- GorgunGCalabreseEHideshimaTA novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myelomaBlood2010115255202521320382844
- den HollanderJRimpiSDohertyJRAurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant stateBlood201011691498150520519624
- MartinJAWangZNext-generation transcriptome assemblyNat Rev Genet2011121067168221897427
- FureyTSChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactionsNat Rev Genet2012131284085223090257
- WongKMHudsonTJMcPhersonJDUnraveling the genetics of cancer: genome sequencing and beyondAnnu Rev Genomics Hum Genet20111240743021639794
- HegiMEDiserensACGorliaTMGMT gene silencing and benefit from temozolomide in glioblastomaN Engl J Med200535210997100315758010