
Abstract
Muscular dystrophies are a heterogeneous group of genetically inherited degenerative disorders defined by dystrophic features on pathological assessment of muscle biopsy specimens. Muscular dystrophies and lymphoma are not common concomitant diseases. Chimeric antigen receptor (CAR) T-cell immunotherapy for lymphoma patients with inherited degenerative diseases, such as muscular dystrophies, has not been previously reported. We report a relapsed/refractory diffuse large B-cell lymphoma (DLBCL) patient with progressive muscular dystrophy (PMD) characterized by progressive muscle weakness that affected the limb, axial and facial muscles. He was identified to be a germline DYSF p.R204* homozygous mutation carrier. The patient received a murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” and suffered from a mild case of grade 1 cytokine release syndrome (CRS). One month after the CAR T-cell infusion, he achieved complete remission of his lymphoma without minimal residual disease (MRD), as assessed by radiography. One year after the infusion, the Deauville score was stable at 1. Currently, patient has been in remission for over three years after receiving anti-CD19 and anti-CD22 CAR T-cell therapy. This case provides evidence for the use of CAR T-cell therapy in lymphoma patients with inherited degenerative disorders. Achieving remission of the lymphoma and subsequent administration of γ-globulin as well as zoledronic acid reduced the muscular dystrophy symptoms.
Introduction
Muscular dystrophies are a heterogeneous group of disorders characterized by clinical features of progressive muscle weakness and dystrophic features on pathological assessments of muscle biopsy specimens.Citation1–Citation5 The heterogeneity of muscular dystrophies is mainly reflected in the distribution of the affected musculature, degree of respiratory and cardiac compromise, and involvement of other organ systems, such as the eyes and central nervous system. The genetic basis is an important factor in the pathogenesis underlying these disorders. Mutations of more than 40 genes, including LAMA2, ITGA7, POMT1, SEPN1, and SYNE1, have been reported to be related to muscular dystrophies.Citation2,Citation3,Citation6–Citation10 These genes encode laminin, collagen, integrin and other skeletal muscle-related proteins. Duchenne muscular dystrophy represents the most common muscular dystrophy in children, with an annual incidence of approximately one in 5000 males and an estimated point prevalence of 8.29 per 100,000 males.Citation3 Becker muscular dystrophy has a prevalence of 7.29 per 100,000 males.Citation3,Citation11 Therapeutic approaches focus on improving the structural integrity of muscle fibers by restoring dystrophin production or on the secondary consequences of dystrophin deficiency, such as inflammatory processes, fibrosis, muscle degeneration, or decreased muscle mass.
Non-Hodgkin’s lymphoma accompanied by muscular dystrophy was reported in a previous study twenty years ago. In that case, the tumor caused intussusception as well as intestinal bleeding. Emergency surgery was thus performed, and the patient was doing well without any signs of recurrence 13 months after surgery.Citation12 Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoma worldwide. Standard second-line treatment for relapsed or refractory large B-cell lymphoma is high-dose chemotherapy with autologous stem-cell transplantation (ASCT). In recent years, chimeric antigen receptor (CAR) T cell therapy has emerged as a promising therapeutic strategy for relapsed/refractory DLBCL patients.Citation13,Citation14 Two large CAR-T clinical trials compared the safety and efficacy of CAR T-cell therapy with ASCT as second-line treatment for large B-cell lymphoma and reach seemingly discordant conclusions. The ZUMA-7 trial supported that CAR T-cell product axicabtagene ciloleucel was superior to ASCT.Citation15 While in the BELINDA trial, CAR T-cell product tisagenlecleucel did not show a benefit over ASCT.Citation16 Thus, CAR T-cell therapy would be an alternative treatment for patients who were considered high risk for ASCT. However, DLBCL patients with other congenital diseases treated with CAR T-cell therapy have not been widely studied.
Here, we report a DLBCL patient with congenital muscular dystrophy who received a murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” and achieved complete remission without minimal residual disease (MRD).Citation17 To date, this patient has been in complete remission for 38 months. Furthermore, his muscular dystrophy has greatly improved.
Case Description
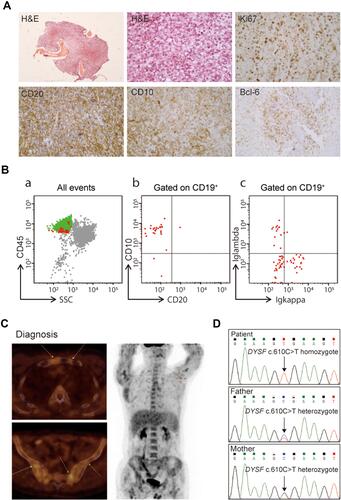
In March 2009, a 24-year-old man was diagnosed with progressive muscular dystrophy, which was confirmed by muscle and neural biopsies. Eight years later, this patient presented to a local hospital in May 2017 with pain in his left hip joint. Magnetic resonance imaging (MRI) showed infection in the left hip bone marrow. Immunohistochemical (IHC) staining of the bone biopsy samples revealed stage IV DLBCL with an international prognostic index (IPI) of 2 () and the following results: CD30 (+), CD21 (+), CD20 (+), CD10 (+), CK (-), Bcl-6 (+), CD43 (+), MPO (+), Syn (-) and MUM-1 (-). The fraction of Ki-67-positive tumor cells was over 70%. However, no obvious abnormalities were found during further cytology and flow cytometry analyses using bone marrow aspirates (). Moreover, positron emission tomography-computed tomography (PET/CT) revealed varying degrees of bone damage accompanied by abnormal radioactive uptake in the bilateral tibia, left scapula, chest bone, ribs, multiple vertebrae, pelvis and humerus (). The maximum standardized uptake value (SUVmax) was between 6.9 and 20.6.
Figure 1 Hematopathology results of a progressive muscular dystrophy patient with diffuse large B-cell lymphoma. (A) Hematoxylin and eosin (H&E) and immunohistochemistry staining of bone biopsy samples. (B) Flow cytometry analysis of bone marrow aspirates at diagnosis. (C) Computed tomography and positron emission tomography images at diagnosis. (D) Confirmation of the peripheral blood DYSF c.610 C>T mutation in this patient and his parents using Sanger sequencing.
The patient was started on induction chemotherapy with six courses of rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin (R-EPOCH) from August to December 2017. Then, he underwent four courses of high-dose methotrexate (HDMTX), each at a dose of 3.5 g/m2. Unfortunately, PET/CT revealed radioactive uptake at the primary tumor location, indicating that the patient did not achieve complete remission.
The patient was referred to our hospital in March 2018 for CAR T-cell therapy. Whole-exome sequencing of the bone marrow cells was performed, and a DYSF c.610 C>T p.R204* homozygous mutation was found with a mutant allele fraction of nearly 100%. The DYSF-coded protein dysferlin is a skeletal muscle protein that is involved in muscle contraction and the process of the sarcolemma.Citation18–Citation21 DYSF dysfunction has been reported to be associated with type 2B limb girdle muscular dystrophy.Citation22–Citation24 The nonsense substitution in R204 led to a depletion of almost the whole protein, whose full-length is 2081 amino acids. Family pedigree studies showed that both the patient’s father and mother were DYSF p.R204* heterozygosis mutation carriers (). Moreover, family history studies revealed that the patient’s parents were cousins.
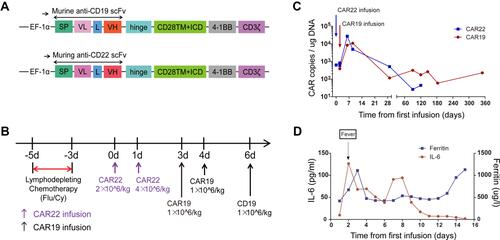
To stop tumor progression, the patient was given decitabine (25 mg/m2) for four days, etoposide (100 mg per day, one day) and liposomal doxorubicin (40 mg per day, one day) before CAR T-cell therapy. Then, he received lymphodepleting chemotherapy with fludarabine (30 mg/m2) and cyclophosphamide (20 mg/kg) for 3 days (days −5 to −3). Subsequently, he received a murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” (). Preparation of CAR T-cells was described in Supplementary Methods. Autologous CD22-targeted CAR T-cells (CAR22) (2*10^6 cells/kg) were infused on day 0, and 4*10^6 CAR22 cells/kg were infused on day +1, followed by CD19-targeted CAR T-cells (CAR19) (1*10^6 cells/kg) on days +3, +4 and +6 (). From day +1 to day +3, the patient suffered from a mild fever of 38.5°C, without respiratory or gastrointestinal symptoms. After receiving 30 mg oral rifampicin, the patient’s body temperature dropped to normal. This cytokine release syndrome (CRS) symptom was determined to be grade 1 according to the grading system published by Lee et al.Citation25 No immune effector cell-associated neurotoxicity (ICAN) symptoms were observed after the infusion. Both murine CAR19 and CAR22 transgene copy numbers as well as the levels of sIL6 and ferritin were tracked (). Calculation of CAR copies number was described in Supplementary Methods. The probe-primer sets for CAR19/CAR22 detection were listed in Supplementary Table S1.
Figure 2 The protocol for the murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” and therapeutic response. (A) Schematic diagram of murine CAR19 and CAR22 CAR vectors. SP, signal peptide; VH, variable H chain; L, linker; VL, variable L chain. (B) The protocol for CAR T infusion in combination with chemotherapy including fludarabine and cyclophosphamide. (C) Timeline of murine CAR22 and CAR19 transgene copy numbers. (D) Dynamic changes in sIL-6 and ferritin after CAR T-cell infusion.
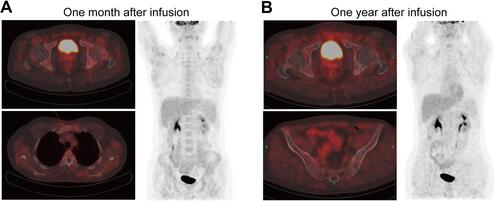
One month after the infusion, the PET-CT results suggested an obvious improvement in this patient compared to the PET-CT results from March 2018. The patient achieved complete remission (CR) with a Deauville score of 3. Only a limited portion of the manubrium sternum and ischial tuberosity showed mildly increased radioactive uptake. The SUV max was between 2.9 and 3.7 (). One year after the CAR T-cell infusion, the patient’s Deauville score was stable at 1 according to the PET-CT results ().
Figure 3 Computed tomography and positron emission tomography images one month (A) and one year after murine monoclonal anti-CD19 and anti-CD22 CAR T-cell “cocktail” infusion (B).
After the CAR T-cell infusion, the patient received 10 g γ-globulin injections every month for 12 months and 4 mg zoledronic acid every month for 24 months. To date, this patient has been in CR for 38 months. Moreover, his muscular dystrophy has greatly improved.
Discussion
This study reported a DLBCL patient with congenital muscular dystrophy who received CAR T-cell therapy and achieved long-term CR. The patient’s parents were cousins and were both DYSF p.R204* heterozygosis mutation carriers. DYSF dysfunction has been reported to be associated with type 2B limb girdle muscular dystrophy. The DYSF p.R204* homozygous mutation could be the genetic basis of the patient’s muscular dystrophy. There was no evidence of whether lymphoma was associated with the patient’s genetic factors. However, the incidence of tumors is elevated in people whose parents are close relatives.
The clinical onset of muscular dystrophies varies, ranging from birth to childhood or adulthood. Generally, obvious clinical signs manifest at birth or in the first few months of life for congenital muscular dystrophy patients. However, many other forms, such as Duchenne muscular dystrophy or some limb girdle muscular dystrophies, manifest in childhood or adolescence; milder, later-onset limb girdle muscular dystrophies and most cases of myotonic dystrophy and facioscapulohumeral muscular dystrophy do not manifest until adulthood. In this case, the patient’s muscular dystrophy manifested in adulthood.
After receiving rescue chemotherapy, this relapsed/refractory DLBCL patient should have undergone ASCT according to traditional standards. This relapsed/refractory DLBCL patient combined with myopathy, showed decreased mobility and a high Eastern Cooperative Oncology Group (ECOG) score greater than 2 points. Autologous hematopoietic stem cell transplantation, pretreated with high-dose chemotherapy may increase mortality at this condition. Therefore, CAR T-cell therapy was chosen as a better alternative strategy. Based on the occurrence of mild CRS during CAR T-cell therapy and the absence of exacerbated muscular dystrophy symptoms, this approach is considered to be safe in patients with B-cell lymphomas combined with genetic diseases. Antigen escape-related relapse is a major challenge for long-term disease control in CAR T- cell therapy. For this reason, we administered sequential CAR T- cell infusions targeting CD19 and CD22 to prevent antigen escape.Citation17,Citation26
In this patient, recovery from relapsed/refractory DLBCL after CAR T-cell therapy may contribute to the improvement of his basic physical condition, which has led to increases in both his physical strength and muscle strength. This case provides evidence for the use of CAR T-cell therapy in lymphoma patients with inherited degenerative disorders.
Data Sharing Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Approval and Consent to Participate
This study was approved by the Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20160310). Informed consent was obtained from the patient in strict accordance with the principles of the Declaration of Helsinki. This study is registered at www.chictr.org.cn as ChiCTR-OPN-16008526.
Consent for Publication
Patient consent for publication was obtained.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Acknowledgments
The authors would like to thank all members of the study team, the patient, and his family. We would also like to thank the Bio-Raid Company for the preparation of the CAR T-cells.
Disclosure
The authors declare that they have no competing interests in this work.
Additional information
Funding
References
- Carter JC, Sheehan DW, Prochoroff A, Birnkrant DJ. Muscular Dystrophies. Clin Chest Med. 2018;39(2):377–389. doi:10.1016/j.ccm.2018.01.004
- Mercuri E, Bonnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019;394(10213):2025–2038. doi:10.1016/S0140-6736(19)32910-1
- Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381(9869):845–860. doi:10.1016/S0140-6736(12)61897-2
- Emery AE. The muscular dystrophies. Lancet. 2002;359(9307):687–695. doi:10.1016/S0140-6736(02)07815-7
- Emery AE. Fortnightly review: the muscular dystrophies. BMJ. 1998;317(7164):991–995. doi:10.1136/bmj.317.7164.991
- Sarkozy A, Foley AR, Zambon AA, Bonnemann CG, Muntoni F. LAMA2-related dystrophies: clinical phenotypes, disease biomarkers, and clinical trial readiness. Front Mol Neurosci. 2020;13:123. doi:10.3389/fnmol.2020.00123
- Burkin DJ, Kaufman SJ. The α7β1 integrin in muscle development and disease. Cell Tissue Res. 1999;296(1):183–190. doi:10.1007/s004410051279
- Hu P, Yuan L, Deng H. Molecular genetics of the POMT1-related muscular dystrophy-dystroglycanopathies. Mutat Res Rev Mutat Res. 2018;778:45–50. doi:10.1016/j.mrrev.2018.09.002
- Caggiano S, Khirani S, Dabaj I, et al. Diaphragmatic dysfunction in SEPN1-related myopathy. Neuromuscul Disord. 2017;27(8):747–755. doi:10.1016/j.nmd.2017.04.010
- Puckelwartz M, McNally EM. Emery-Dreifuss muscular dystrophy. Handb Clin Neurol. 2011;101:155–166.
- Tawil R, van der Maarel S, Padberg GW, van Engelen BG. 171st ENMC international workshop: standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2010;20(7):471–475. doi:10.1016/j.nmd.2010.04.007
- Uotani H, Hirokawa S, Saito F, et al. Non-Hodgkin’s lymphoma of the ascending colon in a patient with becker muscular dystrophy: report of a case. Surg Today. 2001;31(11):1016–1019. doi:10.1007/s005950170015
- Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980
- Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. doi:10.1016/S1470-2045(18)30864-7
- Locke FL, Miklos DB, Jacobson CA, et al. Investigators and contributing kite members. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386(7):640–654.
- Bishop MR, Dickinson M, Purtill D, et al. Second-line tisagenlecleucel or standard care in aggressive B-cell lymphoma. N Engl J Med. 2022;386(7):629–639. doi:10.1056/NEJMoa2116596
- Wang N, Hu X, Cao W, et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood. 2020;135(1):17–27. doi:10.1182/blood.2019000017
- Cardenas AM, Gonzalez-Jamett AM, Cea LA, Bevilacqua JA, Caviedes P. Dysferlin function in skeletal muscle: possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp Neurol. 2016;283:246–254. doi:10.1016/j.expneurol.2016.06.026
- Han R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle. 2011;1(1):10. doi:10.1186/2044-5040-1-10
- Sharma A, Yu C, Leung C, et al. A new role for the muscle repair protein dysferlin in endothelial cell adhesion and angiogenesis. Arterioscler Thromb Vasc Biol. 2010;30(11):2196–2204. doi:10.1161/ATVBAHA.110.208108
- Han R, Campbell KP. Dysferlin and muscle membrane repair. Curr Opin Cell Biol. 2007;19(4):409–416. doi:10.1016/j.ceb.2007.07.001
- Aoki M, Takahashi T. [Mutational and clinical features of Japanese patients with dysferlinopathy (Miyoshi myopathy and limb girdle muscular dystrophy type 2B)]. Rinsho Shinkeigaku. 2005;45(11):938–942. Japanese.
- Cao XQ, Joypaul K, Cao F, Gui LL, Hu JT, Mei W. Anesthetic management of a patient with limb-girdle muscular dystrophy 2B:CARE-compliant case report and literature review. BMC Anesthesiol. 2019;19(1):155. doi:10.1186/s12871-019-0813-8
- Aldosari KH, Al-Ghamdi S, Alkhathlan KM, Alkhalidi HM. Phenotypic and genotypic analysis of limb-Girdle muscular dystrophy type 2B. Neurosciences. 2020;25(3):214–217. doi:10.17712/nsj.2020.3.20200002
- Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
- Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–1226. doi:10.1158/2159-8290.CD-18-0442