Abstract
Despite remarkable progress in cancer-drug discovery, the delivery of novel, safe, and sustainably effective products to the clinic has stalled. Using Src as a model, we examine key steps in drug development. The preclinical evidence on the relationship between Src and solid cancer is in sharp contrast with the modest anticancer effect noted in conventional clinical trials. Here, we consider Src inhibitors as an example of a promising drug class directed to invasion and metastasis and identify roadblocks in translation. We question the assumption that a drug-induced tumor shrinkage in preclinical and clinical studies predicts a successful outcome. Our analysis indicates that the key areas requiring attention are related, and include preclinical models (in vitro and mouse models), meaningful clinical trial end points, and an appreciation of the role of metastasis in morbidity and mortality. Current regulations do not reflect the natural history of the disease, and may be unrelated to the key complications: local invasion, metastasis, and the development of resistance. Alignment of preclinical and clinical studies and regulations based on mechanistic trial end points and platforms may help in overcoming these roadblocks. Viewed kaleidoscopically, most elements necessary and sufficient for a novel translational paradigm are in place.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
The mismatch between science and translation is best illustrated by advances in cancer research,Citation1,Citation2 especially tumor virology,Citation3 and the dearth of novel, safe, and sustainably effective drugs for solid cancer introduced to the clinic.Citation4–Citation6 This anomaly prompts the question: Where are we going wrong?Citation7,Citation8
Fifty years ago, Thomas Kuhn explained that science does not progress linearly, but through paradigmatic shifts; conventional and seemingly logical paradigms lose their utility when they cease to be fruitful.Citation9 In this context, “fruitful” refers to translational potential, and anomalies between preclinical expectations and clinical reality are signs that all is not well. In solid cancer, an abysmally low approval rate, ineffective drug performance, market recalls, and unaffordable prices complicate the problem.Citation8,Citation10 In order to identify whether the cause lies in discovery, development, or in the approval process, which in a manner governs development, a rethink is in order.
Provocative questionsCitation11 are the best starting point for a collective rethink, and we address three issues in solid cancer: nonpredictive preclinical models, the Response Evaluation Criteria in Solid Tumors (RECIST) framework for evaluating tumor response to intervention, and an under appreciation of the role of metastasis in morbidity and mortality. We question the operational assumption that tumor shrinkage is an index of overall survival/regression and a reduction in metastatic potential, especially since this assumption governs drug development and may direct attention away from local invasion and metastasis. Dissemination is the leading cause of mortality, and the most important improvements in morbidity and mortality will result from the prevention (or elimination) of metastasis.Citation12–Citation15 Accordingly, using the knowledge of Src inhibitors in solid cancer, we review the gaps between preclinical expectations and clinical reality in the evaluation of Src inhibitors, and indicate areas that may need emphasis.
Src and invadopodia in cancer cell invasion and metastasis
The year 2011 marks the centenary of Peyton Rous’s discovery of the chicken sarcoma virus.Citation16,Citation17 Six decades after this discovery, the agent was identified as the viral Src gene (v-Src) and it was established that a host gene (c-Src or Src) was captured by the virus.Citation18 In 1966, at the age of 85 years, and 55 years after the publication of work on the tumor-producing virus, Rous was awarded the Nobel Prize. In 1989, Harold Varmus and Michael Bishop were awarded the Nobel Prize for their discovery of the cellular origin of retroviral oncogenes as exemplified by Src.Citation18 Martin chronicles events along the winding “road to Src” and the discovery of the first human protooncogene,Citation19 while Becsei-Kilborn details the multiple reasons for the delayed recognition of this discovery.Citation20 Today, Src is considered a key consideration in cancer cell invasion and metastasis.Citation21–Citation26
Src and related signaling mechanisms influence key elements in carcinogenesis, and invadopodia may represent the proximate mechanism related to local invasion and metastasis. But under current regulations, it is likely that Src inhibitors will recapitulate the experience of the matrix metalloproteinase inhibitors – failure. Today, mechanism-based drugs that do not decrease tumor size are declared clinically ineffective.
Invasion of adjacent tissue is an early step in the metastatic cascade and the key determinant of the metastatic potential of tumor cells. The invasion process is complex, and is best understood in the context of the cancer cells’ interactions with their environment.Citation27–Citation30 This includes signaling pathways involved in epithelial–mesenchymal transition (EMT),Citation31,Citation32 chemotaxis,Citation33,Citation34 and structural and biomechanical properties of the extracellular matrix (ECM) and surrounding cells.Citation35–Citation40 About 90% of cancers originate from epithelial tissue. EMT describes the morphological change in a normal cell to an invasive and possibly metastatic one. This transition results in a migratory phenotype that is responsible for penetrating the basement membrane and invading adjacent tissue. Focal degradation of the ECM as well as invasion through the basement membrane is affected by the formation and activity of invadopodia. Invadopodia are actin-based protrusions of tumor cells that mediate proteolysis of ECM constituentsCitation41–Citation43 ().
Figure 1 (A–C) Invadopodia in invasion. (A) Steps of the invasion/metastasis process. In most carcinomas, cells from the primary tumor undergo an epithelial–mesenchymal transition and gain a migratory phenotype that allows for degradation of the ECM. These modified cells then penetrate the BM barrier, invade adjacent tissue, and supply a vasculature. (B and C) Invadopodia are dynamic cellular protrusions with an ability to invade surrounding tissue via degradation of the ECM. (B) Transmission electron microscopy image of sarcoma cell section with invadopodia penetrating a dermis-based matrix; scale bar 500 nm.Citation43 (C) Schematic depicting the organization and key signaling components of invadopodia.
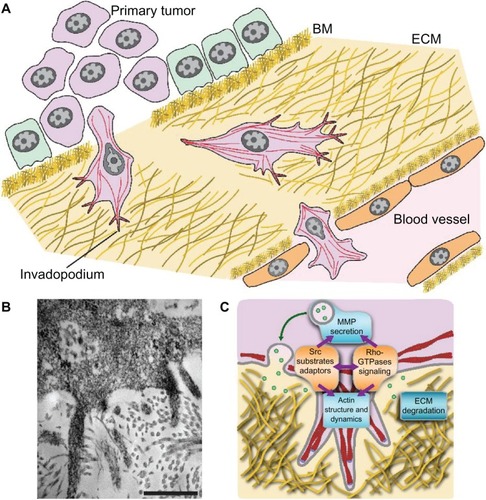
Cancer cells have been shown to generate sufficient actomyosin force to deform collagen fibers and push through the ECM. However, focal degradation of the ECM precedes invasion, and it is now established that the invasive and metastatic potential of the cancer cells is related to their ability to form invadopodia. Local invasion is driven by two invadopodial processes: EMT-facilitated motility and migration, and protease-mediated degradation of the ECM.Citation44–Citation46 The Src family kinases are critical for invadopodial formation and function.
Targeting Src/invadopodia for the development of anti-invasive drugs
Broad coherent, and consistent preclinical evidence indicates that Src plays a role in the advancement and metastasis of solid cancer, and that invadopodia are an important and proximate driver of local invasion in metastasis.Citation44–Citation48
Src inhibitors: rationale and preclinical evidence justifying development in solid cancer
Rationale
The rapidly emerging interest in invadopodia in cancer invasion and metastasis has placed the Src proto-oncogene and related signaling pathways at the focal point of anticancer drug discovery. The rationale for development of Src inhibitors in solid cancer is distinctive and differentiated since it is not directed primarily to cell proliferation but towards progression of the disease, namely invasion and metastasis. In the context of preclinical studies, Plé and colleagues at AstraZenecaCitation49 have outlined elements supporting this strategy:
Src kinase is overexpressed and upregulated in several human tumor types.
Increased Src activity in tumor cells reduces cell adhesion, facilitates motility, and thereby promotes an invasive phenotype. Src kinase plays a key role in EMT and the conversion of epithelial tumor cells to an invasive phenotype.
Increased Src kinase activity is linked with disruption E-cadherin-mediated cell-cell adhesion and the function of focal adhesions, which are critical for cell migration.
Inhibition of Src kinase limits bone metastases. Three Src inhibitors are undergoing advanced clinical development in solid cancer:
the thiazole carboxamide dasatinib (BMS- 354825, SPRYCEL®, Bristol-Myers Squibb)
the anilinoquinazoline saracatinib (AZD0530, AstraZeneca)
the quinolinecarbonitrile bosutinib (SKI-606, Wyeth/Pfizer).
All are orally active, small-molecule, adenosine triphosphate (ATP)-binding, competitive inhibitors of tyrosine phosphorylation (). Dasatinib (Sprycel BMS-354825; Bristol-Myers Squibb, Princeton, NJ, USA) is an orally active, small-molecule (molecular weight [MW] = 488) multikinase inhibitor of several Src family kinases as well as c-Kit, platelet-derived growth-factor receptor, Bcr-Abl, and ephrin-receptor kinases. It is an ATP-competitive inhibitor and inhibits Src tyrosine kinase (half-maximal inhibitory concentration [IC50] = 0.55 nM). Dasatinib was approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of adult patients with chronic myelogenous leukemia in the chronic phase resistant or intolerant to prior therapy that included imatinib (June 2006), and for the treatment of newly diagnosed adult patients with Philadelphia chromosome-positive chronic myelogenous leukemia in the chronic phase (October 2010).Citation50 Saracatinib (AZD0530; AstraZeneca, Reims, France) is an orally active, small-molecule (MW = 542), highly selective, dual-specific inhibitor of Src/Abl kinase inhibitor. It is an ATP-competitive inhibitor and inhibits Src tyrosine kinase (IC50 = 2.7 nM) and Abl kinase (IC50 = 30 nM).Citation51 Bosutinib (SKI-606; Wyeth/Pfizer, Pearl River, NJ, USA) is an orally active, small-molecule (MW = 530) dual inhibitor of Src/Abl kinase inhibitor. It is an ATP-competitive inhibitor and inhibits Src tyrosine kinase (IC50 = 1.2 nM) and Abl kinase (IC50 = 1 nM).Citation52
Table 1 Src inhibitors: specificity and clinical phase
Evidence
Overall, studies in in vitro and in vivo models of cancer have confirmed the ability of Src inhibitors to control tumor-cell motility and invasion. Cell proliferation and survival were unaffected at concentrations sufficient to block cell migration and invasion.Citation53–Citation55
Pichot and colleagues examined the effect of dasatinib in a drug-sensitive breast cancer cell line (MDA-MB-231), and demonstrated that dasatinib inhibited the formation of invadopodia and invasiveness in sensitive cells.Citation56 Furthermore, the combination of dasatinib and doxorubicin synergistically decreased proliferation and viability in the dasatinib insensitive MCF7 cell line, lowering the IC50 of doxorubicin by more than one log unit. Dong and colleagues examined the effect of saracatinib on the highly metastatic murine sarcoma cell line KHT. Saracatinib inhibited major elements in the metastatic cascade, including Src and focal adhesion kinase, and decreased cell migration and invasion. Pretreatment of KHT cells with saracatinib prior to injection markedly lowered lung colonies in mice in a dose-dependent manner, suggesting an antimetastatic effect.Citation57 Schweppe and colleagues examined the effect of saracatinib on cell lines from papillary and anaplastic cancer. In addition to noting inhibition of growth and invasion, they demonstrated the involvement and sensitivity of an Src–focal adhesion kinase complex in this cancer type.Citation58 They further examined the effect of dasatinib in an orthotopic metastasis mouse model of papillary thyroid cancer. Here, dasatinib blocked growth and metastasis.Citation59 Rabbani and colleagues examined the effect of bosutinib on the highly invasive human prostate cancer cell lines PC-3 and DU-145. Bosutinib pretreatment of PC-3 cells prior to injection markedly lowered skeletal lesions in mice.Citation60 Morton and colleagues demonstrated the effect of dasatinib in inhibiting the development of metastases in a mouse model of pancreatic ductal adenocarcinoma.Citation61 In head-and-neck squamous cell carcinoma cell lines, Ammer and colleagues demonstrated the effect of saracatinib in inhibiting cell growth, cell-cycle progression, and transwell Matrigel invasion.Citation62 Dose-dependent decreases in Src activation and phosphorylation of the invasion-associated substrates focal adhesion kinase, p130CAS, and cortactin were also observed. Further, saracatinib treatment displayed a dose-dependent inhibitory effect on invadopodia formation, ECM degradation and matrix metalloproteinase 9 activation. They concluded that inhibition of Src kinase by saracatinib impairs the proinvasive activity of head-and-neck squamous cell carcinoma by inhibiting Src substrate phosphorylation important for invadopodia formation and associated matrix metalloproteinase activity. Because metastatic bone colonization consists of an initial latent phase mediated by an Src survival response and a later regrowth phase, there are opportunities to interrupt one or both phases of colonization. The relevance of Src activation in bone-specific metastasis in prostate and breast cancer is well established.Citation63
Src inhibitors: success in the laboratory, and failure in the clinic
Despite the elegant case made for Src invadopodia in invasion and metastasis,Citation21–Citation26,Citation47–Citation64 overall results of Src inhibitors as monotherapy and in conventional clinical trials in solid cancer have shown “little or modest activity.”Citation65 We now review possible causes of this anomaly.
Preclinical models
Today, there is no ideal preclinical strategy that can predict the efficacy of agents in clinical trials. Preclinical models range from the simple, rapid, and convenient to the complex, delayed, and cumbersome (). In mouse xenograft models, size can be measured with calipers, but imaging is needed for genetically engineered mouse models (GEMMs). Although tumor cell lines and xenograft models are still in use today, the former do not address stromal interactions, while the latter are biased towards cytotoxic agents. In a superb perspective, Burchill concludes that complementary strategies are best used, and the selection of models should be based on a clear definition of the desired information.Citation66
Table 2 Preclinical cancer models – the quest for predictive utility and industrialization
Tumor xenografts, unlike conventional xenografts, use the patient’s tumor, not permanent cell lines. In an impressive treatise, Decaudin described advances in the primary human tumor xenograft model (“tumorgrafts”) that appear quite promising but await validation.Citation67 Tumor xenografts in immunodeficient mice have the advantages of convenience and visualization of tumor growth, and may have the ability to predict clinical efficacy of candidate drugs.Citation68–Citation71 GEMMs are created by allowing for overexpression of defined oncogenes, knock-in of genetic point mutations, and knockout of tumor suppressors. GEMMs address certain deficiencies of the tumor xenograft model, especially immunodeficiency, but introduce new concerns. Since predictive utility in a high-throughput system is a major roadblock in anticancer drug research, a clear demonstration of superiority over the tumor xenograft model may justify the effort and costs involved with GEMMs. The selection of models depends on the questions that need to be answered, and at this time tumorgrafts and GEMMs have the potential to provide prescriptive but limited information.Citation72–Citation75
According to Céspedes and colleagues, the ideal mouse model should show histopathologic features similar to the human tumor, progress through the same stages, and involve the same genes and biochemical pathways in its initiation and progression.Citation71 Further, the tumor response may reflect the response of the human tumor to a specific therapy, and thereby predict efficacy in clinical trials. Singh and colleagues systematically studied tumor growth and responses to treatment in two GEMMs involving non-small-cell lung cancer and a pancreatic adenocarcinoma, and compared the results to clinical trial data using erlotinib and bevacizumab.Citation72 In this retrospective analysis, they found encouraging correlations between outcomes in GEMMs and clinical trials.
Importantly, a clinically relevant animal model should be metastatic. In this context, Francia and colleagues describe various models of aggressive multiorgan spontaneous metastasis after surgical resection of orthotopically transplanted human tumor xenografts.Citation76 In solid cancer, the key differentiator is invasiveness, which depends on cell motility and the ability to cross tissue boundaries, and biomarkers and metastasis assays could direct the discovery of novel invasive agents. Although defined steps in the metastatic cascade can be studied in isolation and in vitro, a 3-D and lifelike in vitro model would be useful.Citation38,Citation77–Citation82 Griffith and Swartz have outlined “design principles” for the creation of 3-D in vitro models that can recreate the interwoven set of biochemical and mechanical cues in the cellular microenvironment that are relevant to invasion.Citation82 3-D in vitro models, by mimicking features of the in vivo environment, span the gap between 2-D cell cultures and whole-animal systems, and can thereby further anticancer drug research.Citation78
RECIST, and its limitations
In clinical trials, tumor shrinkage and prevention of new lesions is a standard measure of efficacy. According to the RECIST trial, the following definitions apply: complete response, the disappearance of all target lesions; partial response, at least a 30% decrease in the sum of the longest diameter of all target lesions; progressive disease, at least a 20% increase in the sum of the longest diameter of all target lesions or the appearance of new lesions; and stable disease, neither partial response nor progressive disease.Citation83 A key regulatory element for approval of a candidate agent in solid cancer is a RECIST-based response: tumor shrinkage is accepted as a surrogate measure for a beneficial and sustained effect on local invasion and metastasis. With RECIST, intervention in solid cancer with novel drugs targeting invasiveness of cancer cells may be declared (or even predicted to be) clinically ineffective, since they rarely reduce tumor size.Citation84–Citation86
Further, in the RECIST scheme, the categories are arbitrary and wide. It gets more complex when one realizes that terms such as “tumor size” and “tumor shrinkage” refer to volume and therefore require measurements in three dimensions. Changes in tumor size are more sensitive to volumetric rather than linear measurements, thus allowing for a much earlier detection of response and progression.Citation87
In the context of targeted agents in solid cancer, the assumption that a decrease in tumor size is a surrogate index of improvement has not been validated.Citation88 Importantly, with cytostatic-induced necrosis and cavitation, evaluation based on tumor size alone, as is done in RECIST, is no longer an adequate method.Citation85 Accordingly, attempts to validate “predictive” biomarkers within a regulatory construct (RECIST) based on tumor size, especially with targeted agents, will be difficult to interpret, and for a simple reason: a mismatch in terms between the natural history of the disease, and tumor size, and the questionable assumption that “tumor shrinkage” is a surrogate index of improvement.
With good reason, Weber has stated that tumor response is a fundamental concept in clinical oncology, but perhaps the least understood.Citation89 Mozley and colleagues at Merck list the key concerns about RECIST-based response assessments: “tumors do not always expand or contract uniformly, changes in line lengths represent only a small fraction of the available information in the images, and the stable disease category is so broad that it is not always adequately sensitive to changes in tumor mass.”Citation90 Birchard and colleagues studied 99 consecutive patients with advanced non-small-cell lung cancer using RECIST. There was no relationship between early tumor response and patient survival, and patients who had an initial reduction in tumor size did not have an improved survival compared with patients with initial disease progression. In addition, there was no particular percentage reduction in tumor size that was found to correlate with survival.Citation91 This study confirms the meta-analysis conducted by Sekine and colleagues in more than 50 trials in patients with non-small-cell lung cancer; the correlation coefficient between response rate and median patient survival was 0.5.Citation88,Citation92
In this context, imaging technologies based on signaling pathways and metabolism, not just tumor size, have the potential to extract in vivo mechanistic information in real time, generate longitudinal data sets in intact host environments, and directly translate from preclinical cancer models to the clinic.Citation93–Citation97
Src inhibitors: opportunities
Metastasis
The primary problem in drug research in solid cancer is that discovery studies, by definition, are mechanistic in nature, while clinical evaluation is empiric and is based on tumor shrinkage. Further, although the rationale and evidence for the use of Src inhibitors in metastasis is impressive, there is no clear regulatory route to demonstrate a clinical effect of a drug on tumor metastasis.Citation15,Citation98 Interpretation and decision-making is limited to what we observe and measure. Since conventional preclinical development plans focus on the primary tumor, and not on metastasis, it is likely that the specific action of a candidate drug on metastasis - braking, acceleration, or a permissive effect - may be missed. A possible differential effect of a drug on the primary tumor and metastasis could be discordant on account of direct or indirect mechanisms. Pharmacologic-induced shrinkage of the primary tumor alone may not necessarily confer overall benefit, because deficient pericyte coverage of tumor vessels may facilitate metastasis via hypoxia-associated EMT and the MET signaling pathway.Citation99,Citation100 As an example, preclinical studies suggest the beneficial effects of inhibition of tumor angiogenesis may be linked to an increase in local invasion and metastasis.Citation101–Citation103 Accordingly, proposals to integrate preclinical and clinical programs on metastasis are self-evident.Citation12–Citation15
Drug resistance and tumor heterogeneity
The continuing resistance of solid cancer to therapy, especially with selective kinase inhibition, is an important and urgent concern. This is a likely consequence of tumor heterogeneity that allows for the emergence of preexisting low-frequency cancer cells that harbor resistant mutations. The initial clinical response is not sustained.
In breast cancer cell lines, Zhang and colleagues demonstrated that resistance to trastuzumab (Herceptin) was linked to hyperactivation of Src, and that this resistance could be reversed by Src inhibition using saracatinib.Citation104 Their data supports the conclusion that Src is a critical signaling node that is hyperactivated in various trastuzumab-resistance models. This discovery, that Src is a druggable node that may prevent resistance, has an important bearing on rational combination therapy using cytostatic drugs: trastuzumab + Src inhibitors (saracatinib). Based on a review of the preclinical database on Src inhibitors, Zhang and Yu conclude that Src inhibitor- containing combinatorial regimens have potential in overcoming resistance to current anticancer therapies and in preventing metastatic recurrence.Citation22 These translational initiatives promise both resistance prevention (or reversal), a separate beneficial effect on disease progression and metastasis, and also a lower predicted toxicity profile.Citation105 In this context, the repeated pattern of an initial response followed by a relapse and resistance consequent upon tumoral heterogeneity may be mitigated and/or delayed by the initial administration of defined combination therapy. The FDA has addressed these concerns and is now encouraging the development of combination therapies in cancer,Citation106 and has announced a pathway for the accelerated identification and regulatory approval of investigational cancer drugs.Citation107
Why focus on regulations?
Regulations, not science, define and determine both the process of drug development and the specifications of the commercial product. Clinical and regulatory thinking are the key determinants of the quality and rate of the translational throughput from science to medicines. And in cancer, it is now evident that the surrogate measure of efficacy, a reduction in tumor size (also termed “response”), does not extrapolate to sustained clinical benefit. With targeted therapy, the emergence of resistance should be anticipated and candidate combinations evaluated earlier in phase II trials.
Justification for a rethink
The productivity crisis in pharmaceuticals is multifactorial, and a simple and single strategy is unlikely to be successful. The prevailing paradigm in cancer drug development - tumor shrinkage leads to improved survival - is based on the central assumption that cellular proliferation (and mutations) is mechanistically related to invasive and metastatic capability. A failure in the productivity of this paradigm is the primary reason for a rethink.
Earlier, in 1962, Kuhn envisioned a thematic and sequential process to explain scientific progress,Citation9 and the steps are well explained by Kaiser:Citation108
A mature scientific program is characterized by paradigms: guiding concepts, theories, and methods.
In experiments, anomalies sometime arise between results and expectations.
When accumulated anomalies cannot be co-opted into the existing paradigm, the field enters a state of crisis and productivity ceases.
Resolution comes only with the introduction of a new paradigm that addresses the anomalies.
Ideally, a crisis in translational productivity should encourage paradigm rethinks.
The century-old productivity stream of targeted drugs can be traced to the concepts of Paul Ehrlich (1854–1915), namely his translational strategy for the development of safe and effective “magic bullets” (Zauberkugel).Citation109 Today, we face a translational roadblock; we have more attractive new drug classes for solid cancer in the laboratory than safe and effective medicines in the clinic,Citation110 and the locus of this anomaly is clearly at the interface between rational science and the empirical and outdated assessment of clinical efficacy. Drews has explained that it is risky to identify and develop drugs on the basis of incomplete and insufficiently validated hypotheses.Citation111 Specifically in cancer, tumor size (burden) is a consequence of the accumulation of clonal cells,Citation112 and this is unrelated to the mechanisms driving distant metastasis. Accordingly, tumor shrinkage, especially in trials with Src inhibitors, may not qualify as a surrogate measure of overall clinical efficacy.Citation113
Interestingly, although science is driven by ideas and tools,Citation114 the life sciences may have been more receptive to new tools (the science-industry complex) rather than new ideas. Whether the primacy of new tools or new ideas is responsible for the advancement of science is a false choice; both are necessary, but the latter needs more emphasis. New tools have supported prevailing paradigms, but have also identified anomalies. Here, a Kuhnian mindset is essential for the advancement and reception of alternative ideas that address these anomalies.
Today, anomalies have brought us to a decision node: should the development, clinical evaluation, and regulatory criteria for the approval of anticancer drugs be modified to reflect the shift from an antiproliferative strategy based on the experience of cytotoxic agents to one based on pathophysiologic mechanisms? The reasoning, taken together, is that if the mechanisms determining cellular proliferation and local invasion and metastasis are separate and distinct, then a unitary and establishment mindset fixated on cellular proliferation and tumor size may be antithetical to clinical objectives.
Conclusion: homage to magister mundi
We have looked at Src and invadopodia, and have outlined integrative strategies to lift translational roadblocks in solid cancer. Looking back, we realize that a rethink would have been unnecessary had we followed the guidance of Ehrlich.Citation110,Citation115 An Ehrlichian realignment between medicinal chemistry, cell biology, preclinical development, and clinical trials has the potential to redirect anticancer efforts towards anti-invasion and antimetastatic objectives, and operate towards the delivery of safe medicines with meaningful efficacy. If this approach is fruitful, the increase in productivity should also lead to affordable medicines for all.Citation8,Citation10
Acknowledgments
We thank Professor Jan Svoboda (Institute of Molecular Genetics, Prague, Czech Republic) for his fundamental insights into the early studies on Src in cancer, and M Sunil Kumar, PhD, of the Godavari Pharma Group, Hyderabad, India. We express deep appreciation to our reviewers. This work was supported, in part, by the Kellner Family Foundation Principal Investigator Grant and a grant of the Ministry of Education of the Czech Republic (MSM0021620858).
Disclosure
The authors report no conflicts of interest in this work.
References
- DeVitaVTJrRosenbergSATwo hundred years of cancer researchN Engl J Med2012366232207221422646510
- HanahanDWeinbergRAHallmarks of cancer: the next generationCell2011144564667421376230
- JavierRTButelJSThe history of tumor virologyCancer Res200868197693770618829521
- ScannellJWBlanckleyABoldonHWarringtonBOpinion: diagnosing the decline in pharmaceutical R&D efficiencyNat Rev Drug Discov201211319120022378269
- PammolliFMagazziniLRiccaboniMThe productivity crisis in pharmaceutical R&DNat Rev Drug Discov201110642843821629293
- KolaILandisJCan the pharmaceutical industry reduce attrition rates?Nat Rev Drug Discov20043871171515286737
- FernandesMJudging a cancer drug: Avastin’s story [letter]New York Times201116055423section A
- BrábekJFernandesMAffordable cancer careLancet Oncol20121312322225715
- KuhnTSThe Structure of Scientific Revolutions: 50th Anniversary EditionChicagoUniversity of Chicago Press2012
- SullivanRPeppercornJSikoraKDelivering affordable cancer care in high-income countriesLancet Oncol2011121093398021958503
- BenowitzSProvocative questions initiative to fund innovative cancer researchJ Natl Cancer Inst20121041396696722745472
- WeberGFWhy does cancer therapy lack effective anti-metastasis drugs?Cancer Lett2013328220721123059758
- SleemanJPChristoforiGFoddeRConcepts of metastasis in flux: the stromal progression modelSemin Cancer Biol201222317418622374376
- EckhardtBLFrancisPAParkerBSAndersonRStrategies for the discovery and development of therapies for metastatic breast cancerNat Rev Drug Discov201211647949722653217
- SleemanJSteegPSCancer metastasis as a therapeutic targetEur J Cancer2010467177180
- RousPA sarcoma of the fowl transmissible by an agent separable from the tumor cellsJ Exp Med191113439741119867421
- WeissRAVogtPK100 years of Rous sarcoma virusJ Exp Med2011208122351235522110182
- StehelinDVarmusHEBishopJMVogtPKDNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNANature19762605547170173176594
- MartinGSThe road to SrcOncogene200423487910791715489909
- Becsei-KilbornEScientific discovery and scientific reputation: the reception of Peyton Rous’ discovery of the chicken sarcoma virusJ Hist Biol201043111115720503720
- CanelMSerrelsAFrameMCBruntonVGE-cadherin-integrin crosstalk in cancer invasion and metastasisJ Cell Sci2013126Pt 239340123525005
- ZhangSYuDTargeting Src family kinases in anti-cancer therapies: turning promise into triumphTrends Pharmacol Sci201233312212822153719
- SirventABenistantCRocheSOncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancerAm J Cancer Res20122435737122860228
- TsudaMTanakaSRoles for Crk in cancer metastasis and invasionGenes Cancer201235–633434023226571
- ElsbergerBStewartBTatarovOEdwardsJIs Src a viable target for treating solid tumors?Curr Cancer Drug Targets201010768369420578988
- AleshinAFinnRSSRC: a century of science brought to the clinicNeoplasia201012859960720689754
- LuPWeaverVMWerbZThe extracellular matrix: a dynamic niche in cancer progressionJ Cell Biol2012196439540622351925
- BissellMJHinesWCWhy don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progressionNat Med201017332032921383745
- BoxCRogersSJMendiolaMEcclesSATumor-microenvironmental interactions: paths to progression and targets for treatmentSemin Cancer Biol201020312813820599506
- BrábekJMierkeCTRöselDVeselýPFabryBThe role of the tissue microenvironment in the regulation of cancer cell motility and invasionCell Commun Signal201082220822526
- LobodaANebozhynMVWattersJWEMT is the dominant program in human colon cancerBMC Med Genomics20114921251323
- SavagnerPThe epithelial-mesenchymal transition (EMT) phenomenonAnn Oncol201021Suppl 7S87S92
- RoussosETCondeelisJSPatsialouAChemotaxis in cancerNat Rev Cancer201111857358721779009
- Bravo-CorderoJJHodgsonLCondeelisJDirected cell invasion and migration during metastasisCurr Opin Cell Biol201224227728322209238
- WirtzDKonstantopoulosKSearsonPCThe physics of cancer: the role of physical interactions and mechanical forces in metastasisNat Rev Cancer201111751252221701513
- CukiermanEBassiDEPhysico-mechanical aspects of extracellular matrix influences on tumorigenic behaviorsSemin Cancer Biol201020313914520452434
- GuckJLautenschlägerFPaschkeSBeilMCritical review: cellular mechanobiology and amoeboid migrationIntegr Biol2010211–12575583
- PankováKRöselDNovotnýMBrábekJThe molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cellsCell Mol Life Sci2010671637119707854
- KumarSWeaverVMMechanics, malignancy, and metastasis: the force journey of a tumor cellCancer Metastasis Rev2009281–211312719153673
- MierkeCTRöselDFabryBBrábekJContractile forces in tumor cell migrationEur J Cell Biol2008878–966967618295931
- YamaguchiHPathological roles of invadopodia in cancer invasion and metastasisEur J Cell Biol20129111–1290290722658792
- SaltelFDaubonTJuinAInvadosomes: intriguing structures with promiseEur J Cell Biol2011902–310010720605056
- ToldeORoselDVeselyPFolkPBrábekJThe structure of invadopodia in a complex 3D environmentEur J Cell Biol201089967468020537759
- DestaingOBlockMRPlanusEAlbiges-RizoCInvadosome regulation by adhesion signalingCurr Opin Cell Biol201123559760621550788
- WangYMcNivenMAInvasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK–p130Cas complexJ Cell Biol2012196337538522291036
- MatsuiHHaradaISawadaYSrc, p130Cas, and mechanotransduction in cancer cellsGenes Cancer201235–639440123226577
- StylliSSKayeAHLockPInvadopodia: at the cutting edge of tumour invasionJ Clin Neurosci200815772573718468901
- EckertMALwinTMChangATTwist1-induced invadopodia formation promotes tumor metastasisCancer Cell201119337238621397860
- PléPAGreenTPHennequinLFDiscovery of a new class of anilinoquinazoline inhibitors with high affinity and specificity for the tyrosine kinase domain of c-SrcJ Med Chem200447487188714761189
- LombardoLJLeeFYChenPDiscovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assaysJ Med Chem200447276658666115615512
- HennequinLFAllenJBreedJN-(5-chloro-1,3-benzodioxol-4-yl)-7-(2-(4-methylpiperazin-1-yl)ethoxy.-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitorJ Med Chem200649226465648817064066
- BoschelliDHWuBBarrios SosaACSynthesis and Src kinase inhibitory activity of 2-phenyl- and 2-thienyl-7-phenylaminothieno[3,2-b]pyridine-6-carbonitrilesJ Med Chem200548113891390215916442
- JallalHValentinoMLChenGBoschelliFAliSRabbaniSAA Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, growth, and metastasis in vitro and in vivoCancer Res20076741580158817308097
- VulturABuettnerRKowolikCSKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cellsMol Cancer Ther2008751185119418483306
- GreenTPFennellMWhittakerRPreclinical anticancer activity of the potent, oral Src inhibitor AZD0530Mol Oncol20093324826119393585
- PichotCSHartigSMXiaLDasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cellsBr J Cancer20091011384719513066
- DongMRiceLLeplerSPampoCSiemannDWImpact of the Src inhibitor Saracatinib on the metastatic phenotype of a fibrosarcoma KHT tumor modelAnticancer Res201030114405441321115886
- SchweppeREKeregeAAFrenchJDSharmaVGrzywaRLHaugenBRInhibition of Src with AZD0530 reveals the Src-focal adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancerJ Clin Endocrinol Metab20099462199220319293266
- ChanCMJingXPikeLATargeted inhibition of SRC kinase with dasatinib blocks thyroid cancer growth and metastasisClin Cancer Res201218133580359122586301
- RabbaniSAValentinoMLArakelianASBoschelliFSKI-606 (bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasisMol Cancer Ther2010951147115720423991
- MortonJPKarimSAGrahamKDasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinomaGastroenterology2010139129230320303350
- AmmerAGKelleyLCHayesKESaracatinib impairs head and neck squamous cell carcinoma invasion by disrupting invadopodia functionJ Cancer Sci Ther200912526120505783
- BoyceBXingLSrc inhibitors in the treatment of metastatic bone disease: rationale and clinical dataClin Investig (Lond)201111216951706
- CreedonHBruntonVGSrc kinase inhibitors: promising cancer therapeutics?Crit Rev Oncog201217214515922471705
- PulsLNEadensMMessersmithWCurrent status of Src inhibitors in solid tumor malignanciesOncologist201216556657821521831
- BurchillSAWhat do, can, and should we learn from models to evaluate potential anticancer agents?Future Oncol20062220121116563089
- DecaudinDPrimary human tumor xenografted models (‘tumorgrafts’) for good management of patients with cancerAnticancer Drugs201122982784121623183
- OcanaAPandiellaASiuLLTannockIFPreclinical development of molecular-targeted agents for cancerNat Rev Clin Oncol20118420020921135887
- CaponigroGSellersWRAdvances in the preclinical testing of cancer therapeutic hypothesesNat Rev Drug Discov201110317918721358737
- TeicherBAIn vivo/ex vivo and in situ assays used in cancer research: a brief reviewToxicol Pathol200937111412219098118
- CéspedesMVCasanovaIParreñoMManguesRMouse models in oncogenesis and cancer therapyClin Transl Oncol20068531832916760006
- SinghMLimaAMolinaRAssessing therapeutic responses in Kras mutant cancers using genetically engineered mouse modelsNat Biotechnol201028658559320495549
- FranciaGKerbelRSRaising the bar for cancer therapy modelsNat Biotechnol201028656156220531333
- RoblesAIVarticovskiLHarnessing genetically engineered mouse models for preclinical testingChem Biol Interact20081711215916417362899
- CarverBSPandolfiPPMouse modeling in oncologic preclinical and translational researchClin Cancer Res200612185305531117000663
- FranciaGCruz-MunozWManSXuPKerbelRSMouse models of advanced spontaneous metastasis for experimental therapeuticsNat Rev Cancer201111213514121258397
- BakerBMChenCSDeconstructing the third dimension – how 3D culture microenvironments alter cellular cuesJ Cell Sci2012125Pt 133015302422797912
- LiLLuYOptimizing a 3D culture system to study the interaction between epithelial breast cancer and its surrounding fibroblastsJ Cancer2011245846621915190
- HarunagaJSYamadaKMCell-matrix adhesions in 3DMatrix Biol2011307–836336821723391
- KrauseSMaffiniMVSotoAMSonnenscheinCThe microenvironment determines the breast cancer cells’ phenotype: organization of MCF7 cells in 3D culturesBMC Cancer20101026320529269
- YamadaKMCukiermanSModeling tissue morphogenesis and cancer in 3DCell2007130460161017719539
- GriffithLGSwartzMACapturing complex 3D tissue physiology in vitroNat Rev Mol Cell Biol20067321122416496023
- EisenhauerEATherassePBogaertsJNew response evaluation criteria in solid tumors: revised RECIST guideline version 1.1Eur J Cancer200945222824719097774
- TumaRSSometimes size doesn’t matter: reevaluating RECIST and tumor response rate endpointsJ Natl Cancer Inst200698181272127416985244
- DiederichSImaging beyond RECIST: CT and MRI in molecular therapiesCancer Imaging201212234735023023112
- BradburyPSeymourLTumor shrinkage and objective response rates: gold standard for oncology efficacy screening trials, or an outdated end point?Cancer J200915535436019826353
- GoldmacherGVConklinJThe use of tumour volumetrics to assess response to therapy in anticancer clinical trialsBr J Clin Pharmacol201273684685422242836
- SullivanDCGatsonisCResponse to treatment series: part 1 and introduction, measuring tumor response – challenges in the era of molecular medicineAJR Am J Roentgenol20111971151721701005
- WeberWAAssessing tumor response to therapyJ Nucl Med200950Suppl 1S1S10
- MozleyPMBendtsenCZhaoBMeasurement of tumor volumes improves RECIST-based response assessments in advanced lung cancerTransl Oncol201251192522348172
- BirchardKRHoangJKHerndonJEJrPatzEFJrEarly changes in tumor size in patients treated for advanced stage nonsmall cell lung cancer do not correlate with survivalCancer2009115358158619117348
- SekineIKubotaKNishiwakiYSasakiYSaijoNResponse rate as an endpoint for evaluating new cytotoxic agents in phase II trials of non-small-cell lung cancerAnn Oncol1998910107910849834819
- WeberWACzerninJPhelpsMEHerschmanHRTechnology insight: novel imaging of molecular targets is an emerging area crucial to the development of targeted drugsNat Clin Pract Oncol200851445418097456
- DesarIMvan HerpenCMvan LaarhovenHWBarentszJOOyenWJvan der GraafWTBeyond RECIST: molecular and functional imaging techniques for evaluation of response to targeted therapyCancer Treat Rev200935430932119136215
- SerkovaNJTranslational imaging endpoints to predict treatment response to novel targeted anticancer agentsDrug Resist Updat2011144–522423521640633
- Garcia FigueriasRPadhaniARGohVJNovel oncologic drugs: what they do and how they affect imagesRadiographics201131172059209122084189
- KangHLeeHYLeeKSKimJHImaging-based tumor treatment response evaluation: review of conventional, new and emerging conceptsKorean J Radiol201213437139022778559
- ElvinPGarnerAPTumor invasion and metastasis: challenges facing drug discoveryCurr Opin Pharmacol20055437438115955738
- GerhardtHSembHPericytes: gatekeepers in tumour cell metastasis?J Mol Med (Berl)200886213514417891366
- CookeVGLeBleuVSKeskinDPericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by MET signaling pathwayCancer Cell2012211668122264789
- Pàez-RibesMAllenEHudockJAntiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasisCancer Cell200915322023119249680
- EbosJMLKerbelRSAntiangiogenic therapy:impact on invasion, disease progression, and metastasisNat Rev Clin Oncol20118421022121364524
- ShojaeiFAnti-angiogenesis therapy in cancer: current challenges and future perspectivesCancer Lett2012320213013722425960
- ZhangSHuangWCLiPCombating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathwaysNat Med201117446146921399647
- MuthuswamySKTrastuzumab resistance: all roads lead to SRCNat Med201117441641821475230
- WoodcockJGriffinJPBehrmanREDevelopment of novel combination therapiesN Engl J Med20113641198598721323535
- EssermanLJWoodcockJAccelerating identification and regulatory approval of investigational cancer drugsJAMA2011306232608260922187281
- KaiserDIn retrospect: the structure of scientific revolutionsNature20124847393164166
- StrebhardtKUllrichAPaul Ehrlich’s magic bullet concept: 100 years of progressNat Rev Cancer20088647348018469827
- LordCJAshworthABiology-driven cancer drug development: back to the futureBMC Biol201083820385032
- DrewsJCase histories, magic bullets and the state of drug discoveryNat Rev Drug Discov20065863564016883301
- AndreeffMGoodrichDWPardeeABCell proliferation, differentiation, and apoptosisBastRCJrKufeDWPollockREHolland-Frei Cancer Medicine5th edHamilton (ON)BC Decker20001732
- FrameMCSrc in cancer: deregulation and consequences for cell behaviourBiochim Biophys Acta20021602211413012020799
- DysonFJIs science mostly driven by ideas or by tools?Science201233861131426142723239721
- DrewsJPaul Ehrlich: magister mundiNat Rev Drug Discov20043979780115340389