Abstract
The importance of epigenetic regulation has been increasingly recognized in the development of cancer. In this study, we investigated the impact of smoking, a major risk factor of lung cancer, on DNA methylation by comparing the genome-wide DNA methylation patterns between lung adenocarcinoma samples from six smokers and six nonsmokers. We identified that smoking-induced DNA methylations were enriched in the calcium signaling and neuroactive ligand receptor signaling pathways, which are closely related to smoking-induced lung cancers. Interestingly, we discovered that two genes in the mitogen-activated protein kinase signaling pathway (RPS6KA3 and ARAF) were hypomethylated in smokers but not in nonsmokers. In addition, we found that the smoking-induced lung cancer-specific DNA methylations were mostly enriched in nuclear activities, including regulation of gene expression and chromatin remodeling. Moreover, the smoking-induced hypermethylation could only be seen in lung adenocarcinoma tissue but not in adjacent normal lung tissue. We also used differentially methylated DNA loci to construct a diagnostic model to distinguish smoking-associated lung cancer from nonsmoking lung cancer with a sensitivity of 88.9% and specificity of 83.2%. Our results provided novel evidence to support that smoking can cause dramatic changes in the DNA methylation landscape of lung cancer, suggesting that epigenetic regulation of specific oncogenic signaling pathways plays an important role in the development of lung cancer.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide. The estimated 5-year survival rate ranges from 6% to 16%, depending on the subtype.Citation1 The tumorigenesis of lung cancer not only involves various genetic alterations, such as point mutations, deletions, and gene amplifications, but also alterations in epigenetics, including DNA methylation and histone modifications. One of the most studied epigenetic regulations is cytosine methylation, which does not affect the primary structure of chromatin, but does affect the secondary structure, which is critical to regulation of gene expression.
In lung cancer, upregulation of DNA methyltransferase 1 (DNMT1) protein has been shown to correlate with increased hypermethylation of tumor suppressor genes and downregulation of tumor suppressor proteins.Citation2–Citation4 Recently, Belinsky evaluated the methylation of genes isolated from blood samples in a cohort of women with different levels of risk for lung cancer.Citation5 They identified that p16Ink4, MGMT, and RASSF1A were hypermethylated in the high-risk cohort. The methylation pattern of specific genes may be useful in early detection of lung cancer. In another study, Feng et al studied the DNA methylation status of 27 genes from 49 patients with non-small cell lung cancer and found that some cancer-specific changes in DNA methylation pattern can only be seen in tumor tissues but not in preneoplastic tissues.Citation6
Smoking, as a well known risk factor for lung cancer, has been widely recognized as a carcinogen with a significant role in tumorigenesis of the lung. Many studies have demonstrated that smoking can induce genomic instability by producing genetic mutations and altering epigenetic modifications. Hypermethylation of promoter regions have frequently been observed in smokers with and without lung cancer, which is consistent with the higher level of DNMT1 in lung samples from smokers when compared with that from nonsmokers.Citation3,Citation7,Citation8 The cumulative smoking dose (pack-years) has been shown to correlate well with the frequency of methylation in cancer-free heavy smokers.Citation9CCND2 and APC were frequently hypermethylated in both cancerous and noncancerous lung tissues of smokers with non-small cell lung cancer, suggesting that hypermethylation of these genes may be associated with environmental factors, such as chronic smoking. Other recent research showed that methylation of RASSF1A is associated with exposure to smoke in lung cancer.Citation10,Citation11 Hypermethylation of FHIT has been shown to be an early event in smoking-associated squamous cell lung cancer.Citation12
On the other side, some studies have shown that smoking is not clearly associated with an altered DNA methylation pattern in lung cancer patients. Tommasi et al investigated the effects of chronic exposure to a prototype smoke-derived carcinogen, benzo[a]pyrene diol epoxide (B[a]PDE), on DNA methylation in genomic regions that have been previously demonstrated to be important in lung cancer.Citation13 They demonstrated that chronic treatment of normal human cells in vitro with B[a]PDE did not alter the DNA methylation pattern in the genomic regions relevant to lung cancer within a time frame that preceded cellular transformation.Citation13 Another study by Scesnaite et al did not identify significant changes in the pattern of DNA methylation associated with smoking.Citation14 Hillemacher et al investigated the effect of smoking on the global DNA methylation pattern in 298 genomic DNA samples, but did not find a direct effect of smoking on global DNA methylation.Citation15 However, these epigenetic studies were mostly focused on special gene groups or genome loci rather than systematic epigenome-wide analysis of DNA methylation pattern. Moreover, these studies relied on a qualitative methylation-specific polymerase chain reaction method that is subjective, relying on detection of a band from electrophoresis, which does not distinguish low-level from high-level methylation.Citation16–Citation18
In this study, we used an epigenome-wide screening method based on Illumina 27K DNA methylation microarray technology to study how smoking contributes to the development of lung cancer from a DNA methylation point of view. We identified differentially methylated genes by comparing the global DNA methylation patterns between lung adenocarcinoma samples from smokers and nonsmokers. Our study provides an insightful perspective on smoking-associated DNA methylation and its role in tumorigenesis of the lung.
Materials and methods
Sample preparations
We obtained lung adenocarcinoma cancer tissues and corresponding adjacent normal lung tissues from 12 patients diagnosed with stage I–IIIa lung adenocarcinoma (six smokers and six nonsmokers) at the Shanghai Chest Hospital. All 12 patients enrolled in this study were untreated before they underwent complete tumor resection at the Shanghai Chest Hospital in 2010. Tumor staging was based on biopsy and conducted by pathologists in the Shanghai Chest Hospital. Baseline characteristics and tumor stages for all patients are listed in Supplementary Table 1. This study was approved by the ethics committee of the Shanghai Chest Hospital and the School of Medicine, Shanghai Jiao Tong University. All patients provided their written informed consent.
Illumina 27K DNA methylation microarray
The experiment was performed according to Illumina’s protocol. Briefly, the procedures included:
Bisulfite treatment, whereby 1 μg of genomic DNA was used in bisulfite conversion to convert the unmethylated cytosine into uracil.
Genomic DNA amplification, where the bisulfite-treated DNA was subjected to whole genome analysis by random hexamer primer and Phi29 DNA polymerase. The products were then enzymatically fragmented, purified from dNTPs, primers, and enzymes, before being applied to microarray chips.
Hybridization and single-base extension. There were two bead types for each CpG site per locus in the chip, which were differentiated by different bead types. A total of 200,000 beads were available in the chip. Hybridization was followed by single-base extension with hapten-labeled dideoxynucleotides.
Fluorescence staining and chip scanning. Multilayered immunohistochemical assays were performed by repeated rounds of staining with a combination of antibodies to differentiate the two types. After staining, the chip was scanned to show the intensities of the unmethylated and methylated bead types. The fluorescence intensity ratios between the two bead types were then calculated.
Analysis of methylation data. The scanned microarray images were analyzed using BeadStudio software (Illumina, San Diego, USA), which performed statistical tests on the results and normalized the raw data to reduce the effects of experimental variation and background.
Analysis of DNA methylation data
We used the MethyLumiSet Bioconductor package to handle the methylation microarray data as a data container in Methyllumi R. We studied the pattern of missing values and performed data imputation. Probes were discarded if they lay on the sex chromosomes. We then performed the Wilcoxon test to find significant differential DNA methylation regions between: normal lung tissues from smokers and normal lung tissues from nonsmokers to identify smoking-specific DNA methylation; and between lung cancer tissues from smokers and lung cancer tissues from nonsmokers to identify smoking-associated methylation changes that are specific to lung cancer.
Gene ontology and pathway analysis
We used hypergeometric analysis to identify the differentially methylated region (DMR) and obtained the DMR gene-enriched functional pathways. Gene function annotation was performed using resources from Gene Ontology. Pathway resources were obtained from KEGG and BioCarta. Gene functional cluster analysis was performed using the DAVID system.
Construction of a diagnostic model for classifying smoking-associated and nonsmoking lung cancer
We used the proper signature selection method, which combines the nearest centroid and genetic algorithm fitting tuning approach, to search for the best DNA methylation loci as diagnostic markers. Genetic algorithms are variable search procedures that are based on the principle of evolution by natural selection. The procedure works by evolving sets of variables (chromosomes) that fit certain criteria from an initial random population via cycles of differential replication, recombination, and mutation of the fittest chromosomes.Citation19 The concept of using in silico evolution for resolution of optimization problems was introduced by John Holland in 1975.Citation19
Results
Identification of smoking-specific DNA methylation in lung adenocarcinoma
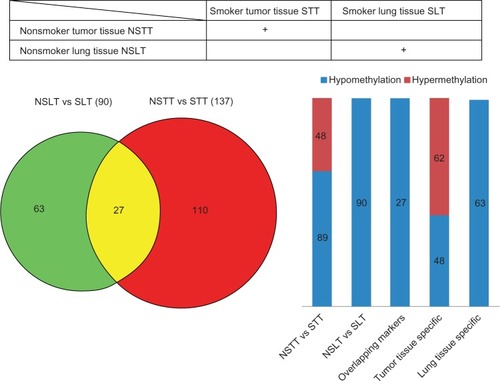
To investigate how smoking affects DNA methylation in lung adenocarcinoma, we used a DNA methylation microarray to compare the global DNA methylation pattern between lung adenocarcinoma samples from smokers and nonsmokers (six samples for each group) to identify smoking-induced DNA methylations that are specific to lung adenocarcinoma (). We identified 137 DMRs, including a number of genes having been previously shown to function in smoking and cancer, such as ADRB3, GALR1, and TRPC1. Of the 137 DMRs, 48 were hypermethylated in smokers with lung adenocarcinoma compared with nonsmokers with lung adenocarcinoma, while 89 of the 137 DMRs were hypomethylated. To filter out the smoking-induced DNA methylations that are not specific to lung adenocarcinoma, we also compared the global DNA methylation pattern for adjacent normal lung tissues from the same six smokers and six nonsmokers. We discovered 90 DMRs that were specifically induced by smoking, including two pro-oncogenic genes from the Ras/Raf/MEK/ERK (mitogen-activated protein kinase, MAPK) signaling pathway (RPS6KA3 and ARAF). Interestingly, all 90 DMRs were hypomethylated loci ( and ). By comparing the two sets of results, we found that 27 DMRs (all hypomethylated) can be found in both sets of results, indicating that these 27 DMRs are smoking-specific changes in DNA methylation that present in both lung cancer tissues and normal lung tissues of smokers. After subtracting the 27 common DMRs from the 137 DMRs that we identified in the comparison between lung cancer tissues from smokers and nonsmokers, we obtained 110 smoking-induced DNA methylations specific to lung adenocarcinoma, including 48 hypomethylated DMRs and 62 hypermethylated DMRs ( and ). The full list of the differentially methylated genes can be found in Supplementary Tables 2–4.
Figure 1 Differences in global DNA methylation pattern in lung cancer between smokers and nonsmokers. The global DNA methylation pattern in normal lung tissues from smokers (SLT) was compared with that from nonsmokers (NSLT), producing 90 differentially methylated regions. The global DNA methylation pattern of lung tumor tissues from smokers (STT) was compared with that from nonsmokers (NSTT), resulting in 137 differentially methylated regions. Twenty-seven loci can be found in both result sets. The numbers of hypermethylation and hypomethylation loci are also shown. Abbreviation: vs, versus.
To gain a functional insight from our data, we performed a Gene Ontology analysis of all candidate genes. As a result, we found that the 90 smoking-induced DMRs in the normal lung tissues of smokers were enriched in pathways including energy metabolism, phosphorylase kinase activity, and ubiquitin thiolesterase activity (). The 110 smoking-induced DMRs specific to lung cancer were mostly enriched in biological processes associated with transcription factor activity, chromatin remodeling, and neural signal processing (). Using the data we obtained from this study, we proposed a model to depict the developmental progress of smoking-induced lung cancer from an epigenetic point of view ().
Figure 2 Hypothesized model of smoking-induced lung cancer based on our data.
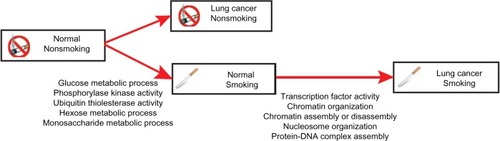
Table 1 Pathways enriched with differentially methylated genes from the comparison between normal lung tissues from smokers and nonsmokers
Table 2 Pathway enrichment analysis for smoking-induced lung cancer-specific differentially methylated genes
Diagnosis model for distinguishing smoking-associated from nonsmoking lung cancer
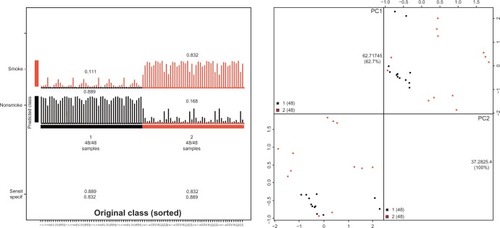
To translate our results into clinical practice, we aimed to use the smoking-specific DNA methylation pattern as a diagnosis tool to classify lung cancer samples associated with smoking and those that are not. We thus used a machine learning algorithm known as the nearest centroid approach, which is a combined model using a tuning strategy of genetic algorithms to select robust DNA methylation markers to classify smoking and nonsmoking lung cancers. We found that a set of differentially methylated DNA methylation loci can predict smoking-associated lung cancer and nonsmoking lung cancer with a sensitivity of 83.2% and a specificity of 88.9%. The overall accuracy and principal component analysis (PCA) of the classification model is shown in .
Figure 3 Diagnostic model for classifying smoking-associated lung cancer and nonsmoking lung cancer.
Abbreviations: PC, principal component; Sensit, sensitivity; specif, specificity.
Discussion
Epigenetic regulation has emerged to become an important field in studying the development of cancer. In this study, we conducted an epigenome-wide screening of DNA methylation patterns in lung adenocarcinoma samples from smokers and nonsmokers. By comparing normal lung tissue samples between smokers and nonsmokers, we identified 90 differentially methylated regions comprising genes functioning in the energy metabolism process and mitogenic kinase activity, suggesting that smoking alters cellular energy supply and cell cycle regulation in normal lung cells. Since both aberrant energy metabolism and a dysregulated cell cycle are the hallmarks of cancer, smoking makes lung cells prone to the development of neoplastic disease. Interestingly, all 90 DMRs were hypomethylated in normal lung tissues from smokers compared with those from nonsmokers. Global DNA hypomethylation has been well demonstrated in many types of cancers.Citation20 However, unlike the close link between DNA hypermethylation and suppression of tumor suppressors, the role of DNA hypomethylation in the development of cancer is still elusive.Citation21 Several lines of evidence support that DNA hypomethylation contributes to tumorigenesis by inducing genomic instability.Citation21 Our results indicate that smoking preferentially induces hypomethylation in normal lung tissues, which may predispose smokers to the development of lung cancer by inducing genomic instability.
By comparing smoking-specific DNA methylation with lung cancer-specific DNA methylation, we discovered 110 differentially methylated genes that were specifically associated with smoking-induced lung cancer. These genes are enriched in genetic and epigenetic regulatory pathways such as transcription factor activity and chromatin remodeling activity. This result indicates that smoking-induced tumorigenesis requires extensive nuclear reorganization and reflects the fundamental characteristic differences between smoking-induced lung cancer and lung cancer not related to smoking, and also provides a potential diagnostic method to distinguish smoking-induced lung cancer from lung cancer not related to smoking.
Many interesting genes that may function in smoking and cancer were identified in this study. We found that both RPS6KA3 and ARAF genes are hypomethylated in smokers but not in nonsmokers. Both genes take part in the MAPK kinase signaling pathway. The Ras/Raf/MEK/ERK (MAPK) signal transduction cascade is an important regulator of cell proliferation. RAS gene products activate proteins in the RAF family, which consists of ARAF, BRAF and RAF-1. In a lung cancer study, Lee et al identified the mutation spectrum revealed by paired genome sequences from a lung cancer patient, and suggested a model for how the multiplicity of mutations within the MAPK signaling cascade acted together to drive constitutive progrowth signals.Citation22 They found that MAPK signaling genes (SHC1, GRB2, SOS, ARAF, AP3K3, and ELK1) were upregulated in lung cancer.Citation22,Citation23 In addition, hypomethylation of MAPK signaling members in smokers is an indicator of oncogenic changes that may be valuable targets for early cancer detection.Citation22,Citation23
ADRB3, a beta-adrenergic receptor, was hypermethylated in smokers with lung cancer but not in nonsmokers with lung cancer in this study. Recent studies have revealed that nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) can directly bind and stimulate the beta-adrenergic receptor signaling pathway. NNK is derived from nicotine, and has been demonstrated to cause mutations in genes that affect cell regulation and proliferation. ADRB3 also functions in activation of the neuroactive ligand receptor interaction pathway. In an earlier study, the researchers identified other common genetic variants associated with the risk of lung cancer in the NNK disposition pathways, such as CYP2A13, the most active P450 for phase metabolic activation of NNK and the receptor (ADRB2) in its nongenotoxic pathway.Citation24 Masi et al verified the mutagenic effects of NNK in an animal model and provided a way of identifying mutations not only in ADRB2 but also in other genes that may play an essential role in the development of lung adenocarcinoma.Citation25 Therefore, it will be interesting to know if ADRB3 also has a role in smoking-induced lung cancer.
GALR1 is another interesting gene we identified in this study that is specific to smoking-associated lung cancer. GALR1 is one of the three G protein-coupled receptors that function as the receptor for galanin, which is a multifunctional neuropeptide widely expressed in the mammalian central and peripheral nervous systems. Expression of galanin and its three receptors (GALR1–3) are silenced in several types of tumor.Citation26 Hypermethylation of GALR1 has been correlated with reduced gene expression. Reported by Misawa et al, GALR1 is silenced in head and neck squamous cell carcinoma because of its hypermethylated promoter. The methylation status of the GALR1 promoter and the level of GALR1 gene expression have been well correlated in a large panel of head and neck squamous cancer cell lines and primary tumor specimens.Citation26 Ectopic expression of GALR1 suppresses tumor cell proliferation through Erk1/2-mediated regulations on cyclin-dependent kinase inhibitors and cyclin D1.Citation27 Interestingly, Jackson et al utilized a mouse model to show that galanin has a protective role against neuropeptide dependence, and these effects may be mediated by GALR1. In addition, several lines of evidence support that galanin acts through GALR1 to modulate the physiologic effects of nicotine.Citation28 However, the role of GALR1 in smoking-induced lung cancer is still unclear and deserves further investigation.
Smoking-associated lung cancer often demonstrates clinical and pathologic characteristics different from lung cancers that are not associated with smoking. Hence, it has clinical value for classifying smoking-associated lung cancer and nonsmoking lung cancer. In this study, we used smoking-specific DNA methylation loci to construct a diagnostic model to predict whether development of lung cancer is associated with smoking. In this study, our results were obtained from lung adenocarcinoma, which is the most common subtype of lung cancer. Because different types of lung cancer have distinct clinicopathologic characteristics, it should be noted that the value of our diagnostic model for other types of lung cancer is still unknown. In the next stage of study, we will need to validate this model further in a larger patient cohort and in other types of lung cancer.
In summary, we examined differences in global DNA methylation patterns between smoker and nonsmoker groups of lung cancer patients to identify differentially methylated genes associated with smoking-induced lung cancer. Our results provide novel evidence that smoking plays an important role in the development of lung cancer through DNA methylation.
Acknowledgments
This research is supported by the Chinese Ministry of Science and Technology and the Sino-Swiss International Collaboration project (2012DFG31320). This study is project (2012DFG31320). This study is supported by the Chinese Ministry of Science and Technology international collaboration foundation, project number 2010DFA32580. The authors thank all the patients who participated in this study.
Supplementary tables
Supplementary tables 1–4 http://www.dovepress.com/cr_data/supplementary_file_51041.pdf.
Disclosure
The authors report no conflicts of interest in this work.
References
- TsouJAHagenJACarpenterCLLaird-OffringaIADNA methylation analysis: a powerful new tool for lung cancer diagnosisOncogene2002215450546112154407
- DamianiLAYinglingCMLengSRomoPENakamuraJBelinskySACarcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cellsCancer Res2008689005901418974146
- LinRKHsiehYSLinPThe tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patientsJ Clin Invest201012052153220093774
- LiuFKillianJKYangMEpigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensateOncogene2010293650366420440268
- BelinskySAGene-promoter hypermethylation as a biomarker in lung cancerNat Rev Cancer2004470771715343277
- FengQHawesSESternJEDNA methylation in tumor and matched normal tissues from non-small cell lung cancer patientsCancer Epidemiol Biomarkers Prev20081764565418349282
- BaryshnikovaEDestroAInfanteMVMolecular alterations in spontaneous sputum of cancer-free heavy smokers: results from a large screening programClin Cancer Res2008141913191918347195
- MonickMMBeachSRPlumeJCoordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokersAm J Med Genet B Neuropsychiatr Genet2008159141151
- OstrowKLHoqueMOLoyoMMolecular analysis of plasma DNA for the early detection of lung cancer by quantitative methylation-specific PCRClin Cancer Res2010163463347220592015
- ToyookaSMaruyamaRToyookaKOSmoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancerInt J Cancer200310315316012455028
- KimDHJiYIHypermethylation of RASSF1A promoter is associated with the age at starting smoking and a poor prognosis in primary non-small cell lung cancerCancer Res2003633743374612839968
- LiWDengJJiangPTangJAssociation of 5′-CpG island hypermethylation of the FHIT gene with lung cancer in southern-central Chinese populationCancer Biol Ther201010997100020814237
- TommasiSKimSZhongXYWuXWPfeiferGPBesaratiniaAInvestigating the epigenetic effects of a prototype smoke-derived carcinogen in human cellsPLoS One20105e1059420485678
- ScesnaiteAJarmalaiteSMutanenPSimilar DNA methylation pattern in lung tumours from smokers and never-smokers with secondhand tobacco smoke exposureMutagenesis20112742342922217548
- HillemacherTFrielingHMoskauSMuschlerMASemmlerAKornhuberJGlobal DNA methylation is influenced by smoking behaviourEur Neuropsychopharmacol20081829529818242065
- ShivapurkarSVSuzukiMApplication of a methylation gene panel by quantitative PCR for lung cancersCancer Lett2007247567116644104
- VallbohmerBJYangDDNA methyltransferases messenger RNA expression and aberrant methylation of CpG islands in non-small-cell lung cancer: association and prognostic valueClin Lung Cancer20068394416870044
- KimYTSungSWKimJHCan aberrant promoter hypermethylation of CpG islands predict the clinical outcome of non-small cell lung cancer after curative resection?Ann Thorac Surg2005791180118815797047
- LevnerIFeature selection and nearest centroid classification for protein mass spectrometryBMC Bioinformatics200566815788095
- JonesPAFunctions of DNA methylation: islands, start sites, gene bodies and beyondNat Rev Genet20121348449222641018
- De SmetCLoriotADNA hypomethylation in cancer: epigenetic scars of a neoplastic journeyEpigenetics20105206213
- LeeWJiangZSLiuJFThe mutation spectrum revealed by paired genome sequences from a lung cancer patientNature201046547347720505728
- DhillonASHaganSRathOKolchWMAP kinase signalling pathways in cancerOncogene2007263279329017496922
- Al-WadeiHASchullerHMNicotinic receptor-associated modulation of stimulatory and inhibitory neurotransmitters in NNK-induced adenocarcinoma of the lungs and pancreasJ Pathol200821843744519274673
- MasiTCekanovaMWalkerKNitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced pulmonary adenocarcinomas in Syrian golden hamsters contain beta 2-adrenergic receptor single-nucleotide polymorphismsGenes Chromosomes Cancer20054421221715942941
- MisawaKUedaYKanazawaTEpigenetic inactivation of galanin receptor 1 in head and neck cancerClin Cancer Res2008147604761319047085
- KanazawaTKommareddiPKIwashitaTGalanin receptor subtype 2 suppresses cell proliferation and induces apoptosis in p53 mutant head and neck cancer cellsClin Cancer Res2009152222223019276245
- JacksonKJChenXMilesMFHarenzaJThe neuropeptide galanin and variants in the GalR1 gene are associated with nicotine dependenceNeuropsychopharmacology2011362339234821796100