Abstract
The BRAF inhibitor (BRAFi) treatment has led to impressive responses in BRAFV600E mutation-positive melanomas, but responses are not durable in many patients. As most of the BRAFi escape mechanisms involve ERK reactivation, combinations with MEK inhibitors (MEKi) are currently tested to improve BRAFi-mediated response durations. Additionally, such a combination is expected to reduce MEKi-induced skin toxicities, as these drugs are thought to have antagonistic effects on ERK activation in keratinocytes. However, preclinical in vivo data exploring the combination of BRAFi and MEKi to achieve improved tumor control in the absence of skin toxicities are limited. Using a murine Tyr::CreERT2;PtenLoxP/LoxP;BrafCA/+ melanoma model, we have determined the effect of BRAFi and MEKi treatment and their combination on melanoma control and occurrence of adverse events. We found that the MEKi dosed beyond the maximum tolerable dose (MTD) led to stronger control of tumor growth than did the BRAFi, but mice had to be removed from treatment because of skin toxicity. The combination of BRAFi and MEKi reduced MEKi-associated skin toxicity. This allowed high and long-term dosing of the MEKi, resulting in long-term tumor control. In contrast to previous hypotheses, the addition of a BRAFi did not restore the MEKi-mediated downregulation of pERK1/2 in skin cells. Our data describe, for the first time, the alleviation of MEKi-mediated dose-limiting toxicity by addition of a BRAFi in a mouse melanoma model. Additional clinical Phase I studies should be implemented to explore MEKi dosing beyond the single drug MTD in combination with BRAFi.
Introduction
The mitogen-activated protein kinase (MAPK) pathway, consisting of the kinase cascade RAS-RAF-MEK-ERK, is the most commonly activated signaling pathway in melanoma. This MAPK pathway activation is mostly driven by the mutations in BRAF (eg, BRAFV600E mutation in 40%–50% of melanomas) or in NRAS (about 20% of melanomas).Citation1,Citation2 The NRAS mutation can lead to constitutive activity of this protein, indirectly stimulating continuous activation of the BRAF protein, whereas the BRAF mutations can directly activate BRAF. Subsequently, the activated BRAF serine threonine kinase upregulates downstream signaling to mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase (ERK), respectively, leading to an oncogenic gain-of-function in the affected cell.
As this aberrant activity of the MAPK pathway drives tumor cell proliferation and survival, many targeted therapies have been developed to inhibit key proteins in this kinase cascade, such as BRAF, MEK or ERK. BRAFV600E inhibitors (BRAFi) like vemurafenib (ZELBORAF®; Genentech, South San Francisco, CA, USA) and dabrafenib (TAFINLAR®, GlaxoSmithKline, Philadelphia, PA, USA) have shown remarkable clinical activity in clinical Phase III studies.Citation3,Citation4
MEK inhibitors (MEKi) have also been shown to induce responses, but they introduce a struggle with dose-limiting toxicities because of targeting a nonmutated protein present in a broad range of normal tissues.Citation5–Citation7 The second-generation MEK1/2 inhibitor selumetinib (AstraZeneca, Wilmington, DE, USA) failed to significantly improve progression-free survival (PFS),Citation6 possibly resulting from insufficient patient selection and low dosing because of dose-limiting (skin) toxicities. However, the third-generation MEKi trametinib (Mekinist™; GlaxoSmithKline) has been recently shown to improve PFS and overall survival (OS) in a clinical Phase III study.Citation5 However, skin toxicities remained the leading adverse events and most common reason for dose reductions.Citation5,Citation8
Treatment with either BRAFi or MEKi alone generally does not lead to long-term melanoma control because of drug resistance. Acquired resistance to BRAFi can occur in an ERK-dependent or ERK-independent manner.Citation9 The commonly occurring ERK-dependent escape mechanisms often converge on the activation of the upstream kinase MEK, making MEKi attractive drugs to combine with BRAFi in treatment.Citation10 Vice versa, treatment with BRAFi might prevent escape from treatment with MEKi, given that the amplification of oncogenic drivers of ERK signaling, such as BRAF, has recently been identified as a mechanism of acquired resistance to MEKi.Citation11–Citation14
Combining BRAFi and MEKi in treatment can also result in reduction of skin-associated adverse events.Citation15 It has been postulated that BRAFi-associated keratoacanthomas occur due to paradoxical upregulation of phosphorylated ERK (pERK) in skin keratinocytes, whereas MEKi-induced skin toxicities are thought to be driven by drug-induced down-regulation of pERK levels in these cells.Citation7,Citation14 This diametrically opposed pERK regulation by BRAFi and MEKi may be balanced out if these drugs are combined, resulting in fewer skin-associated adverse events than has been observed with either drug alone.
Recently, a clinical Phase II study testing the combination of the BRAFi dabrafenib and the MEKi trametinib indeed showed that this treatment combination was able to delay treatment escape and also able to reduce the incidence and severity of skin toxicity observed with the single treatments. In detail, combining the BRAFi with the MEKi in melanoma treatment resulted in an improved response rate (RR) of 76%; the PFS was extended to 9.4 months, as compared with the 54% RR and 5.8 months PFS observed for patients treated with BRAFi alone.Citation15 This combination therapy result also appears to be much better than the findings in the clinical Phase III study testing the MEKi trametinib as a single treatment (22% RR, 4.8 months PFS).Citation5 Although the incidence of grade 1 and 2 pyrexia and gastrointestinal adverse events was increased when patients received the combination treatment as compared with dabrafenib alone, the skin-associated adverse events such as rash, keratoacanthomas, and squamous cell carcinoma occurred less frequently.
Similar findings were observed in another clinical Phase I trial testing the combination of the BRAFi vemurafenib with the MEKi cobimetinib (Exelixis, South San Francisco, CA, USA).Citation16 Of note, the increased incidence of pyrexia was not observed in this study and therefore seems to be specific for combinations with dabrafenib.
Despite these promising data, the frequency of complete responses was rather low and the relapse rate was high.Citation15 These findings may be related to the fact that the MEKi was dosed below the MTD.Citation8 Potentially, the efficacy of the treatment combination could have been improved if the dose of the MEKi had been increased. In the study presented here, we aimed to investigate preclinically the effect of combined BRAFi and high-dose MEKi (beyond the MTD) treatment on tumor control and skin toxicity in an inducible mouse model of BrafV600E/+/Pten−/− melanoma.
Materials and methods
All described animal experiments were ethically approved by the Animal Experimentation Committee of The Netherlands Cancer Institute. Mice were treated in accordance with the Dutch law on animal experimentation.
Melanoma induction and growth analysis
Tumor induction on the skin of the Tyr::CreERT2; PtenLoxP/LoxP;BrafCA/+ mice was performed as previously described.Citation17 In detail, 2 μL of 5 mM 4-hydroxytamoxifen (Sigma-Aldrich, St Louis, MO, USA) in pure dimethyl sulfoxide (DMSO; Sigma-Aldrich) was topically applied for 5 minutes to the shaven right flank of 4- to 10-week-old mice for 3 consecutive days. As a result, functional Pten in melanocytes was lost by the deletion of exon 5 and one copy of Brafwt was replaced for BrafV600E. The combination of these two genetic events resulted in malignant transformation of melanocytes. Tumor outgrowth was followed twice weekly by digital photographs of the tumor, including a size reference. Tumor size was then analyzed in two dimensions using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
PLX4720 and trametinib treatment
PLX4720 treatment was administered by a chow diet containing 417 mg/kg PLX4720 (the precursor of vemurafenib) or the control chow containing no compound. Both chows were provided by Plexxikon Inc (Berkeley, CA, USA). PLX4720 plasma level analyses showed ranges between 350 and 400 μM, which is higher than plasma levels obtained in the clinical setting.Citation17
The MEK inhibitor trametinib was dissolved in 0.5% hydroxypropyl methylcellulose (Sigma-Aldrich) and 0.2% Tween-80 (Sigma-Aldrich) and was dosed orally at 0.75 mg/kg, once daily, 5 days per week for the indicated durations; MEKi-untreated animals received vehicle control only. For the dose-range experiments the drug was dosed at 0.01, 0.05, 0.1, 0.5, or 0.75 mg/kg, once daily, 5 days per week, for 7 weeks. Unfortunately, there was no test available at that time point to determine the plasma levels of the drugs for these mice.
Immunohistochemistry of skin samples
Immunohistochemistry of formalin-fixed paraffin-embedded skin samples for pERK1/2 was performed as previously described.Citation18 Primary antibody to the pERK1/2 (Thr202/Tyr204) clone D13.14.4E (Cell Signaling Technology, Danvers, MA, USA) was used to detect active ERK1/2.
Western blot of skin samples
Western blot of the skin samples was performed as previously described.Citation17 The primary antibodies employed included mouse anti-pERK1/2 (Thr202/Tyr204) clone 20G11 and mouse anti-β-Actin (Millipore, Billerica, MA, USA) to correct for equal loading. Proteins were detected on the Odyssey infrared imager and quantification of signals was performed using Odyssey software (LI-COR Biosciences, Lincoln, NE, USA).
Results
Selective BRAFi and MEKi treatment control growth of BrafV600E/+/Pten−/− murine melanomas
The Tyr::CreERT2;PtenLoxP/LoxP;BrafCA/+-inducible melanoma model that was used for the experiments in this work has been described recentlyCitation17–Citation19 Upon induction using tamoxifen, these mice develop rapidly growing BrafV600E/+/Pten−/− tumors resembling human spindle cell melanoma. To investigate the effect of the selective BRAFi PLX4720 and the MEKi trametinib beyond the MTD on melanoma outgrowth, we treated tumor-bearing mice with either of these drugs or their combination ().Citation20,Citation21 As expected, the BRAFi treatment led to a fast and strong decrease of tumor outgrowth (blue line). More strikingly, single MEKi treatment (green line) completely arrested tumor growth. Similar to observations in patients, MEKi-treated mice developed serious skin toxicity (between day 18 and 66) and eventually were omitted from treatment. When combining the BRAFi and MEKi (gray line), melanoma growth was controlled to the same extent as with the single MEKi treatment. Moreover, drug administration could now be continued safely for at least 5 months due to reduced incidence of MEKi-associated skin toxicity in these mice. However, cure was not achieved, as mice that were removed from combination treatment quickly relapsed (purple line).
Figure 1 Synchronous BRAFi and MEKi allows long-term tumor control in a murine melanoma model.
Abbreviations: MEK, mitogen-activated protein kinase kinase; MEKi, MEK inhibitor; BRAFi, BRAF inhibitor; GSK212, GSK1120212.
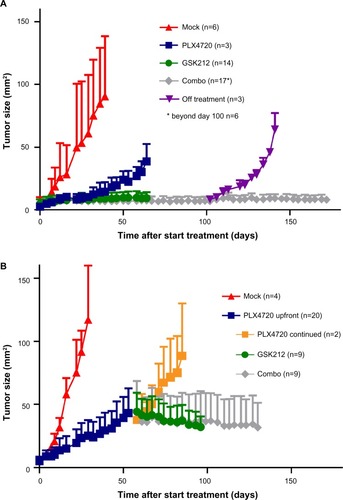
Subsequently, we assessed the effect of combined BRAFi and MEKi treatment after a period of single BRAFi treatment, mimicking the clinical setting in which treatment is altered upon BRAFi escape. We analyzed tumor control in mice that received single BRAFi treatment for 53 days (dark blue line) followed by MEKi (green line), BRAFi (orange line), or combination (gray line) treatment (). In this setting, we observed that the high dose of MEKi could induce tumor control, but again, only the combination of BRAFi and MEKi treatment allowed long-term drug administration due to the reduced MEKi-associated skin toxicity.
Synchronous BRAFV600E and MEK inhibition reduces MEKi induced skin toxicity
Irrespective of preceding BRAFi treatment, the MEKi-induced skin toxicity generally occurred as early as in the third week of treatment, whereas none of the mock- or BRAFi-treated mice showed skin toxicity ( and ). The MEKi-associated skin toxicity was reduced in its incidence and delayed in its kinetics when BRAFi treatment was added synchronously. In detail, within the studied time frame, toxicity occurred in 100% versus 30% of mice for the single MEKi- and combination-treated mice, respectively. The 30% incidence rate of skin toxicity was reached at 28 versus 145 days of treatment ().
Figure 2 MEKi-associated skin toxicity can be reduced by addition of BRSFi.
Abbreviations: MEK, mitogen-activated protein kinase kinase; MEKi, MEK inhibitor; BRAFi, BRAF inhibitor; GSK212, GSK1120212.
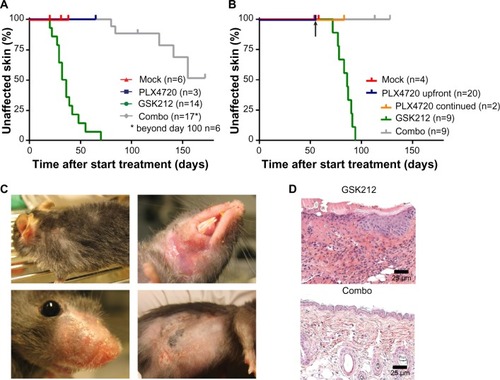
Skin toxicities in MEKi-treated patients are predominantly observed in sebum-rich areas, such as the face and upper torso.Citation22 The murine skin toxicity mainly occurred in areas surrounding the mouth, nose, and neck. Similar to the clinical setting in humans, infections in the study mice occurred at affected skin sites. Affected skin was also observed on the flank, but only in the region irritated by shaving for tumor measurements (). Histology of the affected skin at later time points demonstrated the occurrence of necrotic dermatitis, as characterized by inflammatory cell infiltrates and epidermal ulceration ().
Apart from skin toxicities, MEKi treatment can lead to severe diarrhea in patients.Citation5–Citation7 None of the MEKi-treated mice developed diarrhea. However, most of the mice treated with MEKi demonstrated red-colored feces after 3 weeks of treatment. Pathological examination performed on the GI tract at later time points revealed that most mice suffered from necrotic cecitis (data not shown). Both observations were irrespective of synchronous BRAFi treatment.
Dose reduction of MEKi prevents the occurrence of skin toxicity, but also reduces tumor control
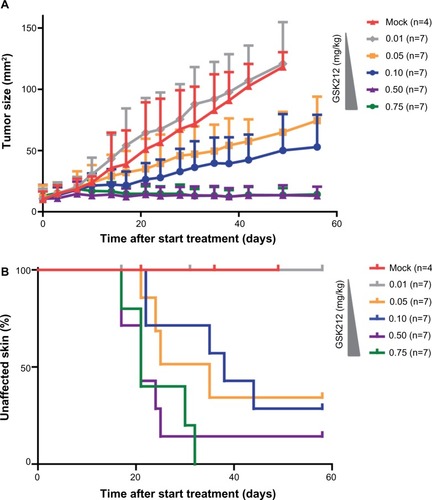
In the clinical setting, the occurrence of skin toxicities upon use of the MEKi often leads to a reduction of the drug dose.Citation5,Citation8 Although such a dose reduction usually alleviates the side effects, tumor control often also becomes reduced (personal observation of CU Blank during Phase II trial testing MEK162 2011-2013 at the Netherlands Cancer Institute). To test whether the MEKi could be dosed at levels that prevent the occurrence of skin toxicity while still retaining tumor control, we treated tumor-bearing mice with 0.01, 0.05, 0.1, 0.5, or 0.75 mg/kg MEKi ( and ). We observed that the two highest doses of MEKi equally inhibited tumor outgrowth. These MEKi dose levels induced skin toxicity, whereas treating with lower doses of MEKi led to reduction in the occurrence of skin toxicity, but also concomitant loss of tumor control.
Figure 3 MEKi dose reduction does not achieve reduced skin toxicity without impairing tumor control.
Abbreviations: MEK, mitogen-activated protein kinase kinase; MEKi, MEK inhibitor; BRAFi, BRAF inhibitor; GSK212, GSK1120212.
Reduced MEKi-associated skin toxicity upon addition of BRAFi treatment is not associated with restored levels of pERK1/2 in skin
The MEKi-induced skin toxicity has been suggested to be mediated by inhibition of ERK activity, which is known to be crucial for the maturation of keratinocytes.Citation7,Citation23 BRAFi treatment has been associated with paradoxical ERK activation in cells that have preexisting RAS activation. This paradoxical ERK activation may restore the reduced levels of pERK upon MEKi treatment.Citation13,Citation14,Citation24 Therefore, we analyzed the levels of pERK1/2 in skin from mice treated with BRAFi, MEKi, or their combination after 14 days of treatment using immunohistochemistry (prior to the occurrence of toxicity to avoid bias resulting from infections) (). Loss of pERK1/2 expression in keratinocytes was observed in mice treated with MEKi, irrespective of the addition of BRAFi. More quantitative analysis for pERK1/2 levels were performed by Western blot ( and ).
Figure 4 Skin pERK levels do not correlate with reduced MEKi-induced skin toxicity upon synchronous BRAFi.
Abbreviations: MEK, mitogen-activated protein kinase kinase; MEKi, MEK inhibitor; BRAFi, BRAF inhibitor; GSK212, GSK1120212.
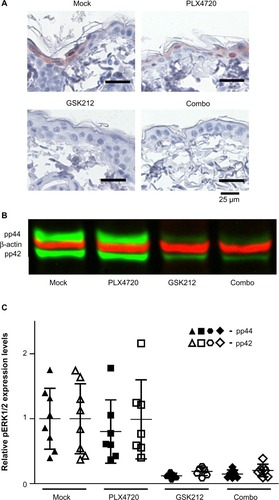
The relative quantification by Western blot of the pERK1/2 levels in skin of 7–8 individual mice per group confirmed that MEKi treatment indeed strongly decreased the levels of pERK1/2 in the skin, but these levels were not restored to baseline when synchronous BRAFi treatment was used (). An identical pattern of pERK1/2 level alteration was observed when examining the skin at week 5 (data not shown).
Discussion
BRAFi and MEKi as single treatments
Combining BRAFi and MEKi treatment has been suggested as an approach to overcome BRAFi resistance.Citation25,Citation26 Additionally, such a combination was expected to reduce the skin toxicity observed with either treatment, as these drugs are thought to have antagonistic effects on ERK activation in keratinocytes.Citation13 Indeed, this hypothesis is supported by data from clinical Phase I and II studies combining BRAFi and MEKi treatment.Citation15,Citation16 The reduced incidence of MEKi-induced skin toxicities that was observed in these trials was not statistically significant but may support the investigation of dosing MEKi beyond the MTD. This increased dosing of MEKi could further improve the efficacy of the combination therapy. Therefore, we tested the effect of BRAFi at a fixed dose, MEKi treatment up to levels beyond MTD and their combination on melanoma control and occurrence of skin toxicity.
We found that the treatment of murine BrafV600E/+/Pten−/− melanomas with single MEKi led to strong tumor control, but also induced severe skin toxicity, as observed in the clinical studies testing MEKi.Citation6,Citation9 Reducing the dose of the MEKi led to less skin toxicity, but also concomitantly reduced tumor control. Our findings therefore suggest that increasing the dose of MEKi beyond the MTD can increase the efficacy of the treatment.
Treating the mice with the BRAFi decreased the outgrowth of the melanomas, but the tumor control was less strong than that observed for mice treated with MEKi dosed beyond the MTD. In contrast to patients treated with single BRAFi, the BRAFi-treated mice did not develop skin lesions such as keratoacanthomas, squamous cell carcinomas, or rash.Citation3,Citation4 This is most likely because the mice were not exposed to ultraviolet (UV) radiation. Hence, they did not develop the phototoxicity-induced rash and lacked preexisting RAS-activating mutations in the keratinocytes, shown to be present in the majority of BRAFi-associated skin lesions.Citation14
BRAFi and MEKi combination treatment
Combining BRAFi and MEKi treatment decreased MEKi-induced skin toxicity in the Tyr::CreERT2;PtenLoxP/LoxP;BrafCA/+ mice, and therefore high and long-term dosing of the MEKi was possible, resulting in long-term tumor control. Because of the absence of the BRAFi-induced skin toxicity in the mice, we could not recapitulate the notion that the MEKi treatment can also protect against the BRAFi-induced skin lesions, as shown in the clinical Phase II study, although this effect was not shown to be statistically significant.Citation15 In addition, we did not observe paradoxical ERK1/2 activation upon single BRAFi in the murine skin, possible due to absence of preexisting UV-induced RAS activation in the mice.
Interestingly, we never observed tumor outgrowth in mice receiving high doses of MEKi treatment during the maximal observation periods of 70 and 172 days for single and combination treatment, respectively. Since evident escape from BRAFi or MEKi did not occur during our observation period, we could not determine whether either treatment prevents resistance to the other. However, the clinical Phase II study demonstrated improved PFS upon combination treatment, indicating that the combination therapy could help to prevent escape.Citation15 As the PFS and overall survival was further increased upon doubling the MEKi dosing, this trial argues for the MEKi being mainly responsible for patients’ improved outcome.
Mechanism of MEKi-associated skin toxicity reduction
To date, the exact mechanism driving the observed reduction in skin-associated adverse events is unknown. It has been postulated that MEKi treatment leads to skin toxicities by inducing downregulation of pERK levels in keratinocytes, whereas the BRAFi-mediated skin toxicity is believed to result from paradoxical upregulation of pERK levels in these cells.Citation13 The combination of BRAFi and MEKi is postulated to balance the pERK signaling in keratinocytes, resulting in reduction of the skin-associated adverse events observed with either drug alone.Citation13
The relative quantification of pERK1/2 levels in the skin of treated mice showed that although MEKi-associated skin toxicity was reduced, the decreased pERK1/2 levels upon MEKi treatment were not restored when synchronous BRAFi treatment was used. Thus, whereas reduced levels of pERK1/2 in skin may still contribute to the MEKi-induced skin toxicity, our data indicate that restoring these levels of pERK1/2 to baseline levels is not required to alleviate MEKi-associated skin toxicity. This finding implies that the mechanism by which BRAFi treatment reduces MEKi-induced skin toxicity is distinct from mechanisms suggested previously.Citation13 The exact mechanism that mediates the MEKi-associated skin toxicity remains unclear. Signaling proteins downstream of pERK might be involved in the skin toxicity mechanism as well. Although speculative, concomitant BRAFi may be able to reduce the skin toxicity not by restoring pERK levels, but rather by acting on other downstream signaling proteins to restore their disturbed activation pattern. Alternatively, the MEKi-induced skin toxicity could be caused by the MAPK intrapathway feedback loops that can lead to paradoxical MEK accumulation.Citation27 The BRAFi may be able to reduce such a potential MEKi-induced MEK accumulation, decreasing the MEKi-induced skin toxicity. The exact mechanism leading to MEKi-associated skin toxicity needs to be unraveled to ascertain whether any of such speculative notions are correct.
Study limitations
A potential limitation of our study is that the Tyr::CreERT2; PtenLoxP/LoxP;BrafCA/+ melanoma model is biased toward resistance to MAPK pathway inhibition as PTEN loss, and thus PI3K pathway activation has been associated with resistance to BRAF and MEK inhibition.Citation28,Citation29 Therefore, our suggestion to increase MEKi dosing when combined with BRAFi may only be beneficial for patients developing BRAFi escape via PI3K pathway activation. Testing such increased MEKi dosing in the Pten wild type but BrafV600E mutant mouse melanoma models may help to determine the effect of increased MEKi dosing for other melanoma patient groups.
Clinical implications
Nevertheless, and in line with suggestions by Flaherty et al,Citation15 we believe that the combination of BRAFi and MEKi treatment warrants further clinical evaluation. Using our murine melanoma model, we could reproduce the clinical finding that the combined BRAFi and MEKi treatment resulted in a trend toward less skin toxicity as compared with single MEKi treatment.Citation5,Citation15 In addition, our data indicate that MEKi can be dosed beyond its MTD when combined with BRAFi treatment. This elevated MEKi dosing could improve the response rates and PFS of melanoma patients. However, it cannot be excluded that in such a setting, adverse events other than skin toxicities will become dose-limiting.
The MEKi was not dosed beyond the MTD in the clinical Phase II study, but treatment efficacy was clearly dependent on MEKi dosing in that study.Citation15 Notably, doubling the MEKi dose increased the response rate of the combination therapy from 50% to 76% while the toxicity remained comparable. This observation and our findings suggest that the MEKi dose could potentially be increased beyond the MTD, which most likely will increase the response rates to combination therapy. Because in such a protocol new dose-limiting toxicities could develop, additional clinical Phase I dose-escalation studies should be performed to test the effect of combined BRAFi and MEKi when dosing beyond the single drug MTD.
Acknowledgments
We thank Dr Andrew Kaiser, Dr Daniel Peeper, and Dr Ton Schumacher for their scientific input. Furthermore, we are grateful to Plexxikon Inc and GlaxoSmithKline for supplying us with PLX4720 and trametinib, respectively.
Disclosure
The authors have no potential conflicts of interest to disclose.
References
- HodisEWatsonIRKryukovGVA landscape of driver mutations in melanomaCell2012150225126322817889
- SmalleyKSUnderstanding melanoma signaling networks as the basis for molecular targeted therapyJ Invest Dermatol20101301283719571822
- ChapmanPBHauschildARobertCImproved survival with vemurafenib in melanoma with BRAF V600E mutationN Engl J Med2011364262507251621639808
- HauschildAGrobJDemidovLPhase III, randomized, open-label, multicenter trial (BREAK-3) comparing the BRAF kinase inhibitor dabrafenib (GSK2118436) with dacarbazine (DTIC) in patients with BRAFV600E-mutated melanomaAnnual Meeting Program, American Society of Clinical OncologyJune 1–5, 2012Chicago, IL, USA Abstract LBA 8500
- FlahertyKTRobertCHerseyPMETRIC Study GroupImproved survival with MEK inhibition in BRAF-mutated melanomaN Engl J Med2012367210711422663011
- KirkwoodJMBastholtLRobertCPhase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanomaClin Cancer Res201218255556722048237
- LoRussoPMKrishnamurthiSSRinehartJJPhase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancersClin Cancer Res20101661924193720215549
- FalchookGSLewisKDInfanteJRActivity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trialLancet Oncol201213878278922805292
- SullivanRJFlahertyKTResistance to BRAF-targeted therapy in melanomaEur J Cancer20134961297130423290787
- GregerJGEastmanSDZhangVCombinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutationsMol Cancer Ther201211490992022389471
- CorcoranRBDias-SantagataDBergethonKIafrateAJSettlemanJEngelmanJABRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutationSci Signal20103149ra8421098728
- LittleASBalmannoKSaleMJAmplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cellsSci Signal20114166ra1721447798
- PoulikakosPISolitDBResistance to MEK inhibitors: should we co-target upstream?Sci Signal20114166e16
- SuFVirosAMilagreCRAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitorsN Engl J Med2012366320721522256804
- FlahertyKTInfanteJRDaudACombined BRAF and MEK inhibition in melanoma with BRAF V600 mutationsN Engl J Med2012367181694170323020132
- GonzalezRRibasADaudAPhase IB Study of vemurafenib in combination with the MEK inhibitor, GDC-0973, in patients (pts) with unresectable or metastatic BRAFV600 mutated melanoma (BRIM7)Presented at: European Society for Medical Oncology CongressSeptember 28–October 2, 2012 Abstract LBA28_PR. Available from: http://abstracts.webges.com/esmo2012/myitinerary
- HooijkaasAIGadiotJvan der ValkMMooiWJBlankCUTargeting BRAF(V600E) in an inducible murine model of melanomaAm J Pathol2012181378579422796458
- HooijkaasAGadiotJMorrowMStewartRSchumacherTBlankCSelective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanomaOncoImmunology201216715
- DankortDCurleyDPCartlidgeRABraf(V600E) cooperates with Pten loss to induce metastatic melanomaNat Genet200941554455219282848
- GilmartinAGBleamMRGroyAGSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibitionClin Cancer Res2011175989100021245089
- TsaiJLeeJTWangWDiscovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activityProc Natl Acad Sci U S A200810583041304618287029
- BalagulaYBarth HustonKBusamKJLacoutureMEChapmanPBMyskowskiPLDermatologic side effects associated with the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886)Invest New Drugs20112951114112120978926
- DumesicPASchollFABarraganDIKhavariPAErk1/2 MAP kinases are required for epidermal G2/M progressionJ Cell Biol2009185340942219414607
- CichowskiKJännePADrug discovery: inhibitors that activateNature2010464728735835920237552
- BlankCUHooijkaasAIHaanenJBSchumacherTNCombination of targeted therapy and immunotherapy in melanomaCancer Immunol Immunother201160101359137121847631
- ParaisoKHFedorenkoIVCantiniLPRecovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapyBr J Cancer2010102121724173020531415
- FridayBBYuCDyGKBRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteinsCancer Res200868156145615318676837
- ByronSALochDCWellensCLSensitivity to the MEK inhibitor E6201 in melanoma cells is associated with mutant BRAF and wildtype PTEN statusMol Cancer2012117523039341
- ParaisoKHXiangYRebeccaVWPTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expressionCancer Res20117172750276021317224