Abstract
Introduction
To date, no effective systemic therapies have been made available for paraganglioma. However, multiple mutations in susceptibility genes have been identified that are potential targets for sorafenib, an oral multitargeted tyrosine-kinase inhibitor.
Case presentation
We report the case of a 69-year-old Chinese man with mediastinal paraganglioma that had metastasized to the bone. The paraganglioma responded to sorafenib, a novel multi-tyrosine kinase inhibitor that targets angiogenesis, the Raf-kinase pathway, the platelet-derived growth factor Ret, and c-Kit. The patient was diagnosed as having paraganglioma after biopsy of the mediastinal mass. We first treated the patient with radiotherapy. Then he tolerated an etoposide-and-cisplatin chemotherapy regimen. Subsequently, he received 6 months of maintenance treatment with sorafenib (400 mg twice daily). A dramatic reduction in tumor volume was observed. At present, the patient has achieved a partial response, and his clinical status remains unchanged.
Conclusion
We suggest that sorafenib should be further investigated in the management of patients with paraganglioma.
Introduction
Paragangliomas are rare neuroendocrine tumors, occurring in two to eight persons per million, with a maximum incidence in the third or fourth decade of life.Citation1 They can arise anywhere from the upper neck to the pelvic floor along the paraganglion system. Paragangliomas are frequently hereditary and/or multifocal. Paragangliomas could be benign or malignant; there is no histologic criterion to differentiate between the two. Only metastases, occurring in approximately 10% of patients, present evidence of malignancy. Paragangliomas can metastasize to bone, the lungs, and lymph nodes.Citation2 No more than 30 cases of mediastinal paraganglioma have been reported in the literature.
To date, no effective systemic therapy has existed for patients with paraganglioma. Surgical excision is the main treatment of choice. Tyrosine-kinase inhibitors (TKIs), such as sunitinib, already approved for kidney cancer, proved to be a treatment option for refractory paragangliomas.Citation3 In 2012, Gunaldi et al reported a case of a woman with metastatic paragangliomas who showed complete response to another TKI – sorafenib.Citation4 Sorafenib (Nexavar®; Bayer HealthCare Pharmaceuticals, Montville, NJ, USA) is an oral multitargeted TKI that mainly targets the receptor tyrosine kinases, vascular endothelial growth factor receptors (VEGFRs) 1–3, platelet-derived growth-factor (PDGF) receptor, and Raf kinases, as well as the non-receptor serine threonine kinases B-Raf proto-oncogene serine/threonine-protein kinase and C-Raf proto-oncogene serine/threonine-protein kinase.Citation5,Citation6 Sorafenib has been shown to improve overall survival in patients with advanced hepatocellular carcinoma (HCC) and renal cell carcinoma (RCC).Citation7,Citation8
Case presentation
In this report, we discuss a case of paraganglioma that arose in the anterior mediastinal compartment. In December 2009, a 69-year-old patient was referred to our unit, complaining of chest pain and tightness. He had an Eastern Cooperative Oncology Group performance status of 2. A computed tomography (CT) scan showed an anterior mediastinal mass (87.1 × 75.0 mm, ), accompanied by multiple mediastinal lymph-node metastases, hydropericardium, and lumbar vertebrae metastases. Histologic examination revealed a solid cellular proliferation with an alveolar and focally diffuse architecture. The examination further revealed groups of cells with characteristic Zellballen nested appearance (). Immunohistochemical staining was positive for Ki-67, CD56, chromogranin A, and synaptophysin. On the basis of the histologic and immunohistochemical features, a diagnosis of paraganglioma was proposed (). The cell groups were composed of polyhedral cells with granular amphophilic cytoplasm. Plasma cortisol and adrenocorticotropic hormone levels were normal. Because multiorgan metastases were present, surgery was not indicated.
Figure 1 (A) Baseline computed tomography (CT) scan of the chest with contrast on initial evaluation. (B) Follow-up CT scan at 1 month after radiotherapy. (C) A CT scan taken 2 months after chemotherapy indicating stable disease. (D) A CT scan taken 60 days later showing dramatic improvement in the mediastinal tumor after treatment with oral sorafenib 400 mg twice per day.
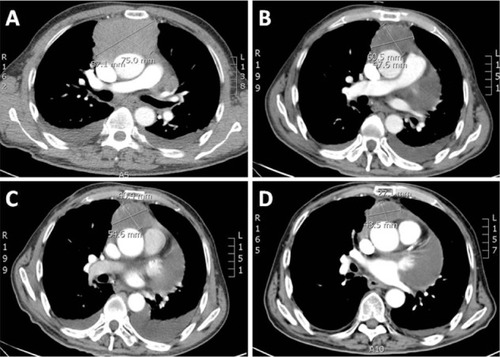
Figure 2 Hematoxylin and eosin staining of the biopsied specimen. Groups of cells have a characteristic Zellballen nested appearance. The groups of cells were composed of polyhedral cells with granular amphophilic cytoplasm.
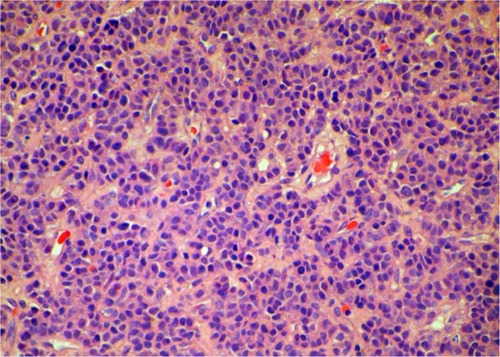
Figure 3 (A–D) Immunohistochemical staining of the biopsied specimen. Immunohistochemical staining was positive for Ki-67 (A), CD56 (B), chromogranin A (C), and synaptophysin (D), demonstrating sustentacular cells around the periphery of the cell nests and supporting the diagnosis of paraganglioma (immunohistochemistry, EnVision).
In general, chemotherapy and radiotherapy have not demonstrated convincing results for patients with unresectable or metastatic disease. We first treated our patient with radiotherapy. The clinical target volume (CTV) included the mass and mediastinal lymph nodes (LNs). The planning target volume (PTV) included the CTV with margins extended by 0.5–1.0 cm. The prescribed dose for the PTV was 50.0 Gy in 28 fractions with 6 MV X-ray. A month later, a chest CT scan showed that the size of the mass had shrunk to 60.5 × 47.6 mm (). Our patient then received palliative chemotherapy with etoposide and cisplatin (EP) for four cycles. The EP regimen comprised of etoposide (100 mg/m2 intravenously) and cisplatin (75 mg/m2 intravenously) on day 1 every 3 weeks. Two months after chemotherapy, a CT scan indicated stable disease, with a mass size of 54.6 × 40.4 mm ().
After extensive discussions with the patient’s family about the risks and benefits of therapy, the patient began off-label use of sorafenib. Sorafenib was initiated at a starting dose of 400 mg twice daily. After 60 days, there was a dramatic improvement in the mediastinal tumor: the mass size decreased to 43.5 × 27.1 mm (). Our patient was followed up monthly. He remained well, with stable disease. Overall, tolerance of sorafenib therapy was good. A mild rash developed on the patient’s back. Sorafenib also resulted in marked fatigue as an initial side effect. Because the dramatic positive clinical response that we observed was accompanied by significant fatigue within 6 months of treatment, we decreased the sorafenib dose to a maintenance dose of 200 mg daily. As per the Response Evaluation Criteria in Solid Tumors evaluation criteria, a partial response was described in accordance with a 30% or greater decrease in the sum of diameters of target lesions.Citation9
Written informed consent was obtained from the patient for publication of this case report and all accompanying images. A copy of the written consent is available for review by the editor-in-chief of this journal.
Discussion
Paragangliomas are tumors derived from the adrenal medulla or extra-adrenal ganglia. They are rare and often benign tumors that are associated with high morbidity and mortality due to mass effect and high circulating catecholamines. Although most paragangliomas are thought to be sporadic, over one-third are associated with susceptibility genes. A germ-line mutation in a paraganglioma susceptibility gene was identified in the SDHx genes,Citation10,Citation11 VHL,Citation12,Citation13 RET,Citation14 and TMEM127.Citation15–Citation17 Recently, the Myc-associated factor X geneCitation14 and VEGFR2 have also been identified as related to paragangliomas.Citation16–Citation18 To diagnose paraganglioma, histologic and immunohistochemical evaluations are necessary.
The ideal treatment for metastatic paragangliomas remains elusive. Targeted therapy has not been evaluated sufficiently in patients with paragangliomas. Sorafenib, an oral multitargeted TKI, is currently approved by the US Food and Drug Administration for the treatment of advanced and unresectable HCC and RCC. Sorafenib has activity against Raf kinase, VEGFR2, PDGF, Flt-3, Ret, and c-Kit.Citation19 A review of the literature shows treatment of such TKIs as sunitinib showed partial response for refractory paragangliomas,Citation3 and a case of a woman with metastatic paragangliomas showed complete response to sorafenib.Citation4 In the sorafenib case, the mass was first excised; after local recurrence and metastases, she received chemotherapy and subsequent targeted therapy with sorafenib. Our patient had metastasis to the bone in primary diagnosis and could not be excised, but after radiotherapy and chemotherapy, and subsequent targeted therapy with sorafenib, we also attained a truly stable disease – a major clinical benefit. This confirmed previous reports of successful use of targeted therapy in paraganglioma. Skin rash is a common adverse reaction to sorafenib, occurring within weeks of the start of treatment. However, our patient only developed a mild rash. It has been suggested that sorafenib may be efficacious for paragangliomas because it targets several different cellular pathways. Because these tumors are extremely variable in terms of the molecules they express, a broader approach to these targets seems reasonable.
The surprising success of sorafenib treatment also raises some questions and could be a matter of debate. For instance, one could assume that mutations or alterations to the genomic profile of our patient’s tumor made it especially sensitive to sorafenib. The surprising outcome of our patient also demonstrates that more research is needed to understand the molecular mechanisms involved in paragangliomas and to find potential targets for future therapy development.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of China (81071664 and 81272714 to Qi Li) and the Shanghai Pujiang Programme (13PJD025 to Qi Li, PKJ2010-Y09 to Yun Lin, and PWRq2011–16 to Qun Li).
Disclosure
The authors report no conflicts of interest in this work.
References
- DeLellisRALloydRVHeitzPUEngCWorld Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Endocrine OrgansLyonIARC2004
- BryantJFarmerJKesslerLJTownsendRRNathansonKLPheochromocytoma: the expanding genetic differential diagnosisJ Natl Cancer Inst2003951196120412928344
- DettinoAAGuimaraesGCFormigaMPheochromocytoma (PC)/paraganglioma (PG) and sunitinib: Review of reported cases and literature and case report contributionJ Clin Oncol201129Suppl 740621172892
- GunaldiMKaraIOAfsarCUDumanBBSorafenib in the treatment of metastatic paraganglioma: a case reportJ Clin Oncol201230Supple13523
- StrumbergDClarkJWAwadaASafety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumorsOncologist20071242643717470685
- Abou-AlfaGKSchwartzLRicciSPhase II study of sorafenib in patients with advanced hepatocellular carcinomaJ Clin Oncol2006244293430016908937
- LlovetJMRicciSMazzaferroVSorafenib in advanced hepato-cellular carcinomaN Engl J Med200835937839018650514
- ChengALKangYKChenZEfficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trialLancet Oncol200910253419095497
- EisenhauerEATherassePBogaertsJNew response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1)Eur J Cancer20094522824719097774
- DahiaPLHaoKRogusJNovel pheochromocytoma susceptibility loci identified by integrative genomicsCancer Res2005659651965816266984
- López-JiménezEGómez-LópezGLeandro-GarcíaLJResearch resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomasMol Endocrinol2010242382239120980436
- DelmanKAShapiroSEJonaschEWAbdominal visceral lesions in von Hippel-Lindau disease: incidence and clinical behavior of pancreatic and adrenal lesions at a single centerWorld J Surg20063066566916617420
- BoedekerCCErlicZRichardSHead and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2J Clin Endocrinol Metab2009941938194419336503
- Comino-MéndezIGracia-AznárezFJSchiaviFExome sequencing identifies MAX mutations as a cause of hereditary pheochromocytomaNat Genet20114366366721685915
- BuffetAVenisseANauVA decade (2001–2010) of genetic testing for pheochromocytoma and paragangliomaHorm Metab Res20124435936622517557
- FishbeinLNathansonKLPheochromocytoma and paraganglioma: understanding the complexities of the genetic backgroundCancer Genet201220511122429592
- NoltingSGrossmanABSignaling pathways in pheochromocytomas and paragangliomas: prospects for future therapiesEndocr Pathol201223213322391976
- EisenhoferGTischlerASde KrijgerRRDiagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratificationEndocr Pathol20122341422180288
- WilhelmSCarterCLynchMDiscovery and development of sorafenib: a multikinase inhibitor for treating cancerNat Rev Drug Discov2006583584417016424