
Abstract
Therapeutic options for acute myeloid leukemia (AML) have remained unchanged for nearly the past 5 decades, with cytarabine and anthracyclines and use of hypomethylating agents for less intensive therapy. Implementation of large-scale genomic studies in the past decade has unraveled the genetic landscape and molecular etiology of AML. The approval of several novel drugs for targeted therapy, including midostaurin, enasidenib, ivosidenib, gemtuzumab–ozogamicin, and CPX351 by the US Food and Drug Administration has widened the treatment options for clinicians treating AML. This review focuses on some of these novel therapies and other promising agents under development, along with key clinical trial findings in AML.
Background
Acute myeloid leukemia (AML) is characterized by blocked myeloid-lineage differentiation and accumulation of myeloid blast cells in the bone-marrow that results in catastrophic bone-marrow failure.Citation1,Citation2 AML is a highly heterogeneous disease driven by different mutations that affect signaling pathways, transcription factors, and epigenetic regulators.Citation3 The current treatment for AML starts with induction chemotherapy (7+3), followed by a few cycles of consolidation chemotherapy or an allogeneic hematopoietic stem–cell transplantation (allo-HSCT).Citation4
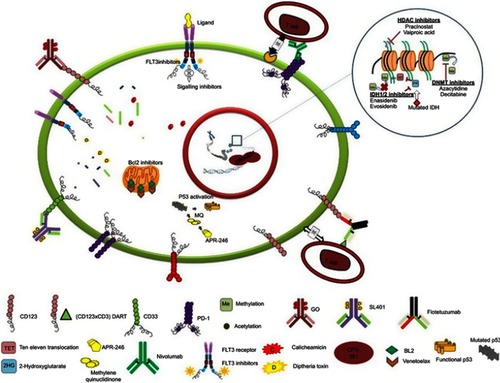
The disease's prognosis and outcomes in adults and the elderly, who account for the majority of AML patients, remains poor.Citation5,Citation6 The ever-growing advancement in genomic technologies allows us to have an unprecedented view of the spectrum and frequency of mutations, their clonal nature, and their evolution during progression of the disease and the epigenetic modulation of the disease. Incorporating “omics” data with ex vivo high-throughput drug screening has the potential to tailor therapy for patients.Citation7 This review provides an overview on novel targeted therapies, approved drugs, and ongoing clinical trials with the aim of personalizing AML therapy ().
Figure 1 Illustration of recently approved and novel drugs in clinical trials targeting the different entities of leukemic cells.
A nucleoside analogue and an antibiotic
Cytarabine, a nucleoside analogue, and anthracyclines (daunorubicin, idarubicin, doxorubicin) derived from antibiotics have been an integral part of the chemotherapy regimen in treating AML for the past 5 decades. Cytarabine as a continuous infusion for 7 days with addition of anthracyclines as a short infusion in the first 3 days forms the basis of chemotherapy (7+3 regimen), drastically reducing the leukemia burden in patients. This, followed by high-dose cytarabine therapy for consolidation or allo-HSCT, has remained the standard treatment for AML patients. We and others have evaluated the mechanisms of resistance to cytarabine and anthracyclines extensively, and have shown how leukemic cells evolve and adapt to become resistant to chemotherapy.Citation8–Citation14 In vitro and preclinical studies with novel agents in combination with standard chemotherapeutic drugs have shown to overcome drug resistance in AML.Citation15–Citation19 This could pave the way to the introduction of promising agents that would help to personalize therapy for AML patients.
CPX351
Cytarabine and daunorubicin have been the backbone of conventional chemotherapy in AML over the past four decades. CPX351, a novel nanoliposomal formulation of cytarabine and daunorubicin in a 5:1 molar ratio, leads to enhanced intracellular uptake of the drug and reduced clearance compared to the conventional 7+3 regimen.Citation20–Citation22 A multicenter phase III trial comparing CPX351 with standard induction and consolidation among elderly patients showed significantly improved overall survival (OS; 9.6 months vs 5.9 months, P=0.005) with CPX351 compared to the standard 7+3 arm (A group of patients receiving a specific treatment). Patients in the CPX351 arm showed a substantial increase in mean terminal half-life compared to the conventional 7+3 infusion (24.5 hours for CPX351 vs 3 hours for cytarabine), although the time to neutrophil and platelet recovery was longer. Also, more patients in the CPX351 arm were eligible for an allotransplant than the 7+3 arm (34% versus 25%).Citation23 CPX351 was approved by the FDA in 2017 for fit elderly patients with treatment-related AML (t-AML) and for de novo AML with myelodysplastic syndrome (MDS).
Hypomethylating agents
The hypomethylating agents azacitidine (Aza) and decitabine are analogues of cytosine with replacement of the fifth carbon atom by a nitrogen atom.Citation24 For the past decade, they have been commonly used in AML patients who are not eligible for cytarabine-based intensive therapy. Aza has a short half-life.Citation25 The new oral formulation of Aza, CC486 is in early-stage trials testing its efficacy. CC486 could possibly enhance antileukemic activity by increasing drug-exposure time. Extended-dosing studies with CC486 have shown it to be well tolerated, with a toxicity profile comparable to Aza.Citation26 Patients who fail therapy with Aza respond well with extended dosing of CC486, indicating involvement of contrasting demethylation patterns due to prolonged exposure.Citation27 Efficacy of CC486 as maintenance therapy post-HSCT (NCT01835587) and after induction/consolidation for AML/MDS patients is under investigation.
Guadecitabine is a next-generation decitabine that is resistant to deamination by cytidine deaminase, thereby increasing its half-life and leading to prolonged exposure during the S-phase and enhanced cell kill.Citation28 Patients ineligible for intensive therapy who are treatment-naïve responded well to guadecitabine in early-phase trials, with >50% of patients achieving composite complete remission (CR).Citation29 A large multicenter phase III trial with guadecitabine or treatment of choice by the physician is under way for previously treated AML (NCT02920008).
Isocitrate dehydrogenase inhibitors
IDH catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate while converting NAD(P)+ to NAD(P)H and maintaining cellular redox homeostasis.Citation30 Mutations in IDH1 and IDH2 are seen in approximately 6%–10% and 9%–13% of patients with AML.Citation31 Mutant IDH starts catalyzing the α-ketoglutarate (αKG) to the oncometabolite D2-hydroxyglutarate (D2HG). This in turn acts as a competitive antagonist for αKG and downregulates the family of αKG-dependent dioxygenases, in particular histone lysine demethylases and the Ten eleven translocation (TET) family of DNA hydroxylases, both in vitro and in vivo.Citation32,Citation33 There is a resultant elevation in histone methylation and 5-methlycytosine, leading to global hypermethylation with lack of global gene expression, causing maturation arrest.Citation34 IDH2 mutations produce high amounts of D2HG with increased cellular expression compared to IDH1-mutated cells. Studies have shown that IDH1-mutated cells depend on the wild-type (WT) IDH1 allele for enhanced D2HG production.Citation35
Enasidenib (previously known as AG221) has been shown to selectively target the IDH2 mutation. In a recent study conducted on relapsed refractory AML (RR-AML) patients with IDH2 mutations treated with enasidenib monotherapy, the overall response rate was 40%, median response duration 5.8 months and median OS 9.3 months. Among the 34 patients who achieved CR with enasidenib, the median OS was 19.7 months.Citation36 Inhibitors of IDH have been shown to produce synergy with Aza, as both drugs reduce DNA methylation and evade blockade in cell differentiation.Citation37 Ivosidenib (AG120) was approved in 2018 for RR-AML with IDH1 mutation. Ivosidenib efficacy was tested in a multicenter trial with 179 RR-AML patients. The overall response rates was 41.6%, with 21.6% achieving CR.Citation38 Treatment using enasidenib and ivosidenib induces differentiation syndrome, owing to the drugs' mechanisms of action.Citation39
FLT3 inhibitors
Approximately a third of adult patients with AML carry mutations in the FMS like tyrosine kinase 3 (FLT3) receptor.Citation40 FLT3 mutation leads to aberrant downstream proliferation signals independently of ligand activation. Internal tandem duplication (ITD) mutations in the FLT3 gene are seen in about 22% of AML patients, and is one of the driver mutations that presents with high leukemic burden and confers a poor prognosis (high incidence of relapse). The remaining 8% of mutations occur in the tyrosine-kinase domain (TKD), with unknown prognosis.Citation41,Citation42 The FLT3-WT vs FLT3-ITD allelic ratio is also an important prognostic factor. According to European LeukemiaNet guidelines, patients with an allelic ratio <0.5 or low ITD with an Nucleophosmin (NPM1) mutations are classified under “good prognosis”, while patients with high allelic ratio (>0.5 or high ITD) and NPM1-WT are grouped under the adverse-risk category.Citation43
Given the adverse clinical outcomes due to the FLT3-ITD mutation in AML patients, induction chemotherapy followed by HSCT is warranted, as it lowers the incidence of relapse and improves survival rates. The incidence of relapse is higher in patients with ITD mutations undergoing allo-HSCT with lower OS compared to FLT3-WT patients.Citation44,Citation45 This has vastly driven attempts to target the FLT3 mutation with kinase inhibitors (KIs) and clinical trials for FLT3 targeted therapy in combination with induction chemotherapy or as maintenance therapy following HSCT.
Midostaurin (Mido), a multi-KI, has been shown to inhibit FLT3 ITD–receptor activity and showed synergistic effects with the standard chemotherapeutic drugs cytarabine and daunorubicin in preclinical studies.Citation46 A phase II trial using Mido as monotherapy for RR-AML/MDS patients with FLT3 mutations showed reduction in peripheral blasts in 70% of patients.Citation47 Subsequently, a phase III randomized trial (RATIFY) that spanned 10 yearsCitation48 comparing Mido and placebo along with standard chemotherapy in AML patients <60 years of age harbouring FLT3 mutations (ITD, TKD) was conducted. Mido treatment resulted in significant improvements in OS (HR 0.78, P=0.009) and event-free survival (HR 0.78, P=0.002) at 4 years, despite an insignificant difference in the rate of CR. OS was also higher for patients in the Mido arm who underwent HSCT in first remissionCitation49 As a multi KI, Mido may also have inhibitory action on the FLT3-WT receptor, as shown by significant blast reduction in FLT3-WT AML patients, based on which a phase III study on FLT3-WT AML patients with Mido has been initiated (NCT003512197).Citation50 In April 2017, Mido in combination with conventional chemotherapy was approved for treatment in newly diagnosed AML patients with FLT3 mutations <60 years of age by the FDA.
Second-generation FLT3 tyrosine-kinase inhibitors
The first generation tyrosine KIs (TKIs) sorafenib, lestaurtinib, and Mido are broad spectrum multi-KIs. The next-generation TKIs quizartinib, crenolanib, and gilteritinib are more selective and potent oral drugs that are currently in clinical trials. These drugs have reduced toxicity compared to the first-generation TKIs.Citation51 There are encouraging clinical data for these second-generation TKIs, as shown in and .Citation52–Citation57 Resistance to TKIs has been a major roadblock in AML therapy, making these TKIs less efficient as single agents. Resistance mechanisms are diverse, and one known mechanisms is acquisition of FLT3 point mutations in TKD during therapy that may mediate resistance.Citation51 Elevated levels of FLT3 ligands in the bone-marrow microenvironment after induction therapy leads to activation of FLT3-WT receptors that are sensitive to ligands but resistant to FLT3 inhibitors.Citation58 In addition, rewired signaling pathways and additional driver mutations in other genes also lead to TKI resistance. One strategy to overcome resistance is to employ combinational therapy that targets different entities of the leukemic cell. FLT3 inhibitors in combination with the hypomethylating agent 5-Aza has been shown to alter methylation patterns in FLT3-mutated cells and overcome stromal protection.Citation59
Table 1 Select clinical studies on second-generation TKIs with induction therapy
Table 2 Select clinical studies on second-generation TKIs as monotherapy
Gemtuzumab ozogamicin
The CD33 antigen is a transmembrane receptor and myeloid-differentiation marker that is expressed in most AMLs. It's extracellular immunoglobulin-like V domain can be exploited using antibody–drug conjugate (anti-CD33-directed) therapy to target CD33-positive leukemic blasts.Citation60,Citation61 Gemtuzumab ozogamicin (GO) is an anti-CD33-directed antibody–drug conjugate comprised of a human mAb linked to calicheamicin. GO binds to the CD33 antigen present on the surface of leukemic blasts and myeloid-precursor cells and localizes into the cell.Citation62,Citation63 The complex breaks after localization and the released calicheamicin binds to DNA, resulting in DNA double-strand breaks and initiates apoptosis. GO was initially approved in 2000 as monotherapy (two doses of 9 mg/m2, 14 days apart) for older patients (60 years) in first relapse with CD33-positive AML who were not eligible for chemotherapy.Citation64 In 2010, during the phase III SWOG S0106 study the drug was withdrawn from the market, due to increased mortality when used in combination with induction and consolidation therapy (single dose of 6 mg/m2 on day 4).Citation65
Subsequent trials (ALFA-0701 phase III study, AML-19 phase III study, MyloFrance 1 and 2 studies) were conducted with lower doses of GO (3 mg/m2 on days 1, 4, and 7, and 6 mg/m2 on day 1 and 3 mg/m2 on day 4) in patients with de novo AML. After a long break, GO was reapproved by the FDA in 2017 for treating CD33-positive newly diagnosed or RR-AML, with a black-book warning for hepatotoxicity and veno-occlusive disease.Citation66,Citation67 A subtype of AML known as core binding factor (CBF) AML responds well to GO.Citation68 The biological significance of CBF-AML blast cells arises from matured committed leukemia-initiating cells with CD33 expression. Multi drug transporter 1 (MDR1) is known to extrude GO from leukemic cells and reduce its efficacy.Citation69 CBF-AML inherently has reduced expression of MDR1,allowing cells to retain GO and enhance cytotoxicity.Citation70–Citation72
Targeting leukemic stem cells
Leukemic stem cells (LSCs) are believed to be responsible for disease relapse in AML, as they escape conventional chemotherapy.Citation70 Targeting LSCs in AML has been a paramount task because of the absence of a consistent surface marker that distinguishes HSCs from LSCs. Many studies have described multiple immunophenotypic differences that may distinguish LSCs from HSCs (CD25, CD44, CD96, CD93, and CD123), but a universal surface marker for LSCs is yet to be identified.Citation73 CD123 has been the only marker under constant investigation to target LSCs compared to the other surface markers. SL401 is the first CD123 monoclonal antibody fused with a catalytic unit of a diphtheria toxin introduced to target LSCs. In a phase I trial conducted on 74 patients with RR-AML or de novo AML, CR was observed in two patients, four patients showed 50% blast reduction in bone marrow, while 14 showed reduction in blast percentage from baseline.Citation74 Capillary-leak syndrome was one of the noted adverse events in this trial, but was manageable with early intervention.Citation75,Citation76 SL401 is now currently being investigated as remission-maintenance therapy for high-risk AML in CR1 or CR2 to improve relapse-free survival (phase II trial NCT02270463)
Flotetuzumab is a bispecific (CD123 × CD3) dual-affinity retargeting novel T cell–redirecting molecule.Citation77 A phase I dose-escalation study in RR-AML/MDS related AML patients demonstrated an overall response of 43% (six of 14 patients), while two patients showed consistent disease with only 20%–25% blast reduction. Consistent activation of CD4 and CD8 T cells was observed in the peripheral blood. Toxicity profiles mainly consisted of cytokine-release syndrome.Citation78 The study continues with cohort expansion (24 RR-AML and 24 MDS-AML [NCT02152956]).
Novel therapies
Mutant TP53 activation
Tumour suppressor p53 (TP53) is one of the commonly mutated genes in malignancies. About 5%–10% of de novo AML and 30% of t-AML harbor TP53 mutations, conferring poor prognosis.Citation79 The mutation leads to misfolded formation of the transcription factor and voiding its ability to bind DNA. The prodrug APR246 spontaneously gets converted to its active compound methylene quinuclidinone at physiological pH. This compound then stabilizes the mutant protein by covalently binding to cysteine residues, leading to refolding and restoration of p53 function.Citation80 Promising initial results were observed from phase IB/II trials using APR246 in combination with Aza conducted on MDS/AML patients with TP53 mutations (n=11). There was a 100% response rate by International Working Group criteria, and nine of eleven patients achieved CR.Citation81
BCL2 inhibitor
B-cell lymphoma 2 (BCL2), an antiapoptotic protein highly expressed in AML cells, confers resistance to conventional therapy by increasing mitochondrial fitness and prevents apoptosis.Citation82 BCL2 is also highly upregulated in the LSC compartment, aiding quiescence and disease relapse.Citation83 Venetoclax (Ven) is a BCL2 inhibitor that blocks its antiapoptotic activity, potentiates the effect of other partnered drugs, and enhances apoptosis. In a phase IB/II trial of Ven in combination with low-dose cytarabine in elderly AML patients unfit for intensive therapy, 62% of patients achieved CR, with median OS of 11.4 months.Citation84 Further promising results were observed combining Ven with Aza in elderly AML patients unfit for therapy, with CR 61% and median OS 17.5 months in early-phase trials.Citation85,Citation86 Phase III randomized trials are ongoing comparing the combination of Aza and Ven versus Aza monotherapy (NCT02993523).
Immune checkpoint inhibitors
Allogenic transplant remains the superior therapy for AML, mainly due to the graft vs. leukemia effect produced by the donor cells.Citation87,Citation88 AML cells express high levels of Programmed death ligand 1 (PDL-1), an inhibitory molecule that suppresses the cytotoxic activity of T cells, leading to immune exhaustion, and increases the chances of relapse.Citation89,Citation90 Immune modulation could effectively work in the AML context, rewiring the immune system. Early-phase trials with nivolumab (PD1 inhibitor) and Aza in RR-AML patients have demonstrated CR of 22% and median OS of 15 months in responders.Citation91 Phase II trials combining nivolumab with conventional 7+3 in newly diagnosed AML showed CR and CR with an incomplete hematologic recovery rate of 72%, and nine patients were eligible for transplant.Citation92 Studies evaluating the efficacy of pembrolizumab (PD1 inhibitor) with other agents are ongoing NCT02996474, NCT02845297. A list of other novel agents in early- and late-phase trials is presented in .
Table 3 Novel agents in clinical trials
Standard chemotherapy to personalizing therapy: a reality?
Over the past few decades of treatment and trials, it has become clear that “one size does not fit all” in terms of treating AML. The best and first example would be acute promyelocytic leukemia. Until 1987, this was treated solely with intensive chemotherapy.Citation93 It was only after 1992 that the combination of all-trans retinoic acid and arsenic trioxide to induce apoptosis and differentiation created a benchmark in the treatment of acute promyelocytic leukemia.Citation94 Years later, the genomic landscape of AML helped us to understand that somatic mutations carry prognostic importance no less than recurrent cytogenetic abnormalities. Only after the discovery of somatic mutations did personalizing therapy for treating AML came into the limelight.Citation95
Though there are studies to substantiate the need and importance of novel therapies targeting different drivers of mutation, there is a setback when this is translated with genomic information alone. Even though genomic testing has been employed in a number of clinical trials on AML patients, it has been hardly used to stratify, due to extended turnaround times, which often delay the treatment.Citation2,Citation3,Citation96 On the other hand, drug-sensitivity testing/ex-vivo high-throughput drug screening can be performed, as it is not time-consuming and a large variety of drugs can be screened within a short time.Citation97–Citation99 Clinical trials like the BEAT AML study aimed to integrate genomic profiling and drug-sensitivity testing in a large cohort of elderly AML patients, which seems promising for making personalization of the treatment regimen for each individual with the disease a reality. We believe that the combination of molecular profiling and ex-vivo drug screening would significantly improve the survival of patients with newly diagnosed and RR-AML (). Further clinical trials are recommended to substantiate the data obtained from both genomic profiling and ex-vivo high-throughput drug screening in patients. However, newer paradigms incorporating novel agents as monotherapy or in combination with standard care still pose a daunting task.
Table 4 Exvivo drug screening–based clinical trials
Acknowledgments
PB (IA/S/15/1/501842) and VM (IA/S/11/2500267) are supported by Wellcome DBT India Alliance Senior fellowships. This study was partly funded by a Centre of Excellence grant from the Department of Biotechnology India (BT/COE/34/SP13432/2015).
Disclosure
SD is supported by a junior research fellowship from the University Grants Commission (UGC), New Delhi, India. The authors report no other conflicts of interest in this work.
References
- Assi SA, Imperato MR, Coleman DJL, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51(1):151–162. doi:10.1038/s41588-018-0270-130420649
- Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi:10.1056/NEJMoa1301689.23634996
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa151619227276561
- Brinda B, Khan I, Parkin B, Konig H. The rocky road to personalized medicine in acute myeloid leukaemia. J Cell Mol Med. 2018;22(3):1411–1427. doi:10.1111/jcmm.1347829327808
- De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. doi:10.1038/bcj.2016.5027367478
- Rubnitz JE. Current management of childhood acute myeloid leukemia. Paediatr Drugs. 2017;19(1):1–10. doi:10.1007/s40272-016-0200-627785777
- Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15(12):747–756. doi:10.1038/nrc401526536825
- Abraham A, Varatharajan S, Karathedath S, et al. RNA expression of genes involved in cytarabine metabolism and transport predicts cytarabine response in acute myeloid leukemia. Pharmacogenomics. 2015;16(8):877–890. doi:10.2217/pgs.15.4426083014
- Abraham A, Devasia AJ, Varatharajan S, Karathedath S, Balasubramanian P, Mathews V. Effect of cytosine arabinoside metabolizing enzyme expression on drug toxicity in acute myeloid leukemia. Ann Hematol. 2015;94(5):883–885. doi:10.1007/s00277-014-2254-225391240
- Varatharajan S, Panetta JC, Abraham A, et al. Population pharmacokinetics of Daunorubicin in adult patients with acute myeloid leukemia. Cancer Chemother Pharmacol. 2016;78(5):1051–1058. doi:10.1007/s00280-016-3166-827738808
- Varatharajan S, Abraham A, Zhang W, et al. Carbonyl reductase 1 expression influences daunorubicin metabolism in acute myeloid leukemia. Eur J Clin Pharmacol. 2012;68(12):1577–1586. doi:10.1007/s00228-012-1291-922562609
- Elsayed AH, Cao X, Crews KR, et al. Comprehensive Ara-C SNP score predicts leukemic cell intracellular ara-CTP levels in pediatric acute myeloid leukemia patients. Pharmacogenomics. 2018;19(14):1101–1110. doi:10.2217/pgs-2018-008630088438
- Mitra AK, Crews KR, Pounds S, et al. Genetic variants in cytosolic 5ʹ-nucleotidase II are associated with its expression and cytarabine sensitivity in HapMap cell lines and in patients with acute myeloid leukemia. J Pharmacol Exp Ther. 2011;339(1):9–23. doi:10.1124/jpet.111.18287321712425
- Schneider C, Oellerich T, Baldauf H-M, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med. 2017;23(2):250–255. doi:10.1038/nm.425527991919
- Garzon R, Savona M, Baz R, et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood. 2017;129(24):3165–3174. doi:10.1182/blood-2016-11-75015828336527
- Martelli MP, Gionfriddo I, Mezzasoma F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125(22):3455–3465. doi:10.1182/blood-2014-11-61145925795919
- Karathedath S, Rajamani BM, Aalam SMM, et al. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS One. 2017;12(5):e0177227. doi:10.1371/journal.pone.017722728505160
- Ganesan S, Alex AA, Chendamarai E, et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2016;30(11):2169–2178. doi:10.1038/leu.2016.22727560113
- Skrtić M, Sriskanthadevan S, Jhas B, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20(5):674–688. doi:10.1016/j.ccr.2011.10.01522094260
- Tardi P, Johnstone S, Harasym N, et al. In vivo maintenance of synergistic cytarabine: daunorubicinratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33(1):129–139. doi:10.1016/j.leukres.2008.06.02818676016
- Lim W-S, Tardi PG, Dos Santos N, et al. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine: daunorubicinformulation, in bone marrow xenografts. Leuk Res. 2010;34(9):1214–1223. doi:10.1016/j.leukres.2010.01.01520138667
- Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5(7):1854–1863. doi:10.1158/1535-7163.MCT-06-011816891472
- Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36(26):2684–2692. doi:10.1200/JCO.2017.77.611230024784
- Glover AB, Leyland-Jones B. Biochemistry of azacitidine: a review. Cancer Treat Rep. 1987;71(10):959–964.2443243
- Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the cancer and leukemia group B. J Clin Oncol. 2006;24(24):3895–3903. doi:10.1200/JCO.2005.05.434616921040
- Savona MR, Gore SD, Kolibaba KS, et al. CC-486 (oral azacitidine) monotherapy in patients with acute myeloid leukemia (AML). Blood. 2015;126(23): 452–452.
- Savona MR, Kolibaba K, Conkling P, et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am J Hematol. 2018;93(10):1199–1206. doi:10.1002/ajh.2521630016552
- Roosendaal J, Wang K, Rosing H, et al. Development and validation of LC-MS/MS methods for the quantification of the novel anticancer agent guadecitabine and its active metabolite β‑decitabine in human plasma, whole blood and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1109:132–141. doi:10.1016/j.jchromb.2019.01.011
- Kantarjian HM, Roboz GJ, Kropf PL, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017;18(10):1317–1326. doi:10.1016/S1470-2045(17)30576-428844816
- Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi:10.1038/nature1086022343901
- Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–281. doi:10.1038/leu.2016.27527721426
- Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi:10.1016/j.ccr.2010.11.01521130701
- Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi:10.1016/j.ccr.2010.12.01421251613
- Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi:10.1016/j.ccr.2010.01.02020171147
- Ward PS, Lu C, Cross JR, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. 2013;288(6):3804–3815. doi:10.1074/jbc.M112.43549523264629
- Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi:10.1182/blood-2017-04-77940528588020
- DiNardo CD, Stein AS, Fathi AT, et al. Mutant isocitrate dehydrogenase (mIDH) inhibitors, enasidenib or ivosidenib, in combination with azacitidine (AZA): preliminary results of a Phase 1b/2 study in patients with newly diagnosed acute myeloid leukemia (AML). Blood. 2017;130(Suppl 1): 639–639.
- DiNardo CD, Stein EM, de Botton S, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med. 2018. doi:10.1056/NEJMoa1716984
- Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a Phase 1/2 study. JAMA Oncol. 2018;4(8):1106–1110. doi:10.1001/jamaoncol.2017.469529346478
- Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99(1):310–318. doi:10.1182/blood.V99.1.31011756186
- Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia. 2005;19(8):1345–1349. doi:10.1038/sj.leu.240383815959528
- Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi:10.1182/blood.v99.12.432612036858
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi:10.1182/blood-2016-08-73319627895058
- Brunet S, Labopin M, Esteve J, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012;30(7):735–741. doi:10.1200/JCO.2011.36.986822291086
- Deol A, Sengsayadeth S, Ahn KW, et al. Does FLT3 mutation impact survival after hematopoietic stem cell transplantation for acute myeloid leukemia? A Center for International Blood and Marrow Transplant Research (CIBMTR) analysis. Cancer. 2016;122(19):3005–3014. doi:10.1002/cncr.3014027315441
- Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–443.12124173
- Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi:10.1182/blood-2004-03-089115345597
- Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4):444–453. doi:10.1182/bloodadvances.201701108029487059
- Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa161435928644114
- Fischer T, Stone RM, DeAngelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. doi:10.1200/JCO.2010.28.967820733134
- Staudt D, Murray HC, McLachlan T, et al. Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: the path to least resistance. Int J Mol Sci. 2018;19:10. doi:10.3390/ijms19103198
- Pratz KW, Cherry M, Altman JK, et al. Updated results from a Phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in subjects with newly diagnosed acute myeloid leukemia (AML). Blood. 2018;132(Suppl1): 564–564. doi:10.1182/blood-2018-99-110975
- Wang ES, Tallman MS, Stone RM, et al. Low relapse rate in younger patients ≤ 60 years old with newly diagnosed FLT3-mutated acute myeloid leukemia (AML) treated with crenolanib and cytarabine/anthracycline chemotherapy. Blood. 2017;130(Suppl 1): 566–566.
- Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–221. doi:10.1002/ajh.2497429139135
- Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061–1075. doi:10.1016/S1470-2045(17)30416-328645776
- Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J Clin Oncol. 2016;34(15_suppl):7008. doi:10.1200/JCO.2016.34.15_suppl.7008
- Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903. doi:10.1016/S1470-2045(18)30240-729859851
- Sato T, Yang X, Knapper S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117(12):3286–3293. doi:10.1182/blood-2010-01-26674221263155
- Al-Jamal HAN, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer. 2015;15:869. doi:10.1186/s12885-015-1695-x26547689
- Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31(9):1855–1868. doi:10.1038/leu.2017.18728607471
- DeStefano CB, Hourigan CS. Personalizing initial therapy in acute myeloid leukemia: incorporating novel agents into clinical practice. Ther Adv Hematol. 2018;9(5):109–121. doi:10.1177/204062071876177829713444
- van der Velden VHJ, Te Marvelde JG, Hoogeveen PG, et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood. 2001;97(10):3197–3204. doi:10.1182/blood.V97.10.319711342449
- Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–1496.11410481
- Larson RA, Boogaerts M, Estey E, et al. Antibody-targeted chemotherapy of older patients with acute myeloid leukemia in first relapse using Mylotarg (gemtuzumab ozogamicin). Leukemia. 2002;16(9):1627–1636. doi:10.1038/sj.leu.240267712200674
- Sperr WR, Florian S, Hauswirth AW, Valent P. CD 33 as a target of therapy in acute myeloid leukemia: current status and future perspectives. Leuk Lymphoma. 2005;46(8):1115–1120. doi:10.1080/1042819050012607516085551
- Jen EY, Ko C-W, Lee JE, et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res. 2018;24(14):3242–3246. doi:10.1158/1078-0432.CCR-17-317929476018
- Raj RV, Abedin SM, Atallah E. Incorporating newer agents in the treatment of acute myeloid leukemia. Leuk Res. 2018;74:113–120. doi:10.1016/j.leukres.2018.10.00830401522
- Gottardi M, Mosna F, de Angeli S, et al. Clinical and experimental efficacy of gemtuzumab ozogamicin in core binding factor acute myeloid leukemia. Hematol Rep. 2017;9(3). doi:10.4081/hr.2017.7028
- Linenberger ML, Hong T, Flowers D, et al. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood. 2001;98(4):988–994. doi:10.1182/blood.v98.4.98811493443
- Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129(12):1577–1585. doi:10.1182/blood-2016-10-69605428159741
- Del Poeta G, Venditti A, Aronica G, et al. P-glycoprotein expression in de novo acute myeloid leukemia. Leuk Lymphoma. 1997;27(3–4):257–274. doi:10.3109/104281997090596829402325
- Lutterbach B, Sun D, Schuetz J, Hiebert SW. The MYND motif is required for repression of basal transcription from the multidrug resistance 1 promoter by the t(8;21) fusion protein. Mol Cell Biol. 1998;18(6):3604–3611. doi:10.1128/mcb.18.6.36049584201
- Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627–1635. doi:10.1182/blood-2016-10-69603928159738
- Assi R, Kantarjian H, Ravandi F, Daver N. Immune therapies in acute myeloid leukemia: a focus on monoclonal antibodies and immune checkpoint inhibitors. Curr Opin Hematol. 2018;25(2):136–145. doi:10.1097/MOH.000000000000040129206680
- Yalcintepe L, Frankel AE, Hogge DE. Expression of interleukin-3 receptor subunits on defined subpopulations of acute myeloid leukemia blasts predicts the cytotoxicity of diphtheria toxin interleukin-3 fusion protein against malignant progenitors that engraft in immunodeficient mice. Blood. 2006;108(10):3530–3537. doi:10.1182/blood-2006-04-01381316882709
- Sweet KL, Pemmaraju N, Lane AA, et al. Lead-in stage results of a pivotal trial of SL-401, an interleukin-3 receptor (IL-3R) targeting biologic, in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN) or acute myeloid leukemia (AML). Blood. 2015;126(23): 3795–3795.
- Campagne O, Delmas A, Fouliard S, et al. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. 2018;24(11):2631–2641. doi:10.1158/1078-0432.CCR-17-226529463552
- Uy GL, Godwin J, Rettig MP, et al. Preliminary results of a Phase 1 study of flotetuzumab, a CD123 x CD3 Bispecific Dart® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood. 2017;130(Suppl 1): 637–637.
- Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45. doi:10.1186/s13045-015-0139-z25952993
- Lambert JMR, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15(5):376–388. doi:10.1016/j.ccr.2009.03.00319411067
- Sallman DA, DeZern A, Sweet K, et al. Abstract CT068: Phase Ib/II combination study of APR-246 and azacitidine (AZA) in patients with TP53 mutant myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Cancer Res. 2018;78(13 Supplement):CT068–CT068. doi:10.1158/1538-7445.AM2018-CT068
- Anstee NS, Bilardi RA, Ng AP, et al. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ. 2018;1. doi:10.1038/s41418-018-0209-1
- Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–341. doi:10.1016/j.stem.2012.12.01323333149
- Thomas XG, Dmoszynska A, Wierzbowska A, et al. Results from a randomized phase III trial of decitabine versus supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed AML. J Clin Oncol. 2011;29(15_suppl): 6504–6504. doi:10.1200/jco.2011.29.15_suppl.6504
- DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-86875230361262
- Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–1866. doi:10.1038/s41591-018-0233-130420752
- Ganapule A, Nemani S, Korula A, et al. Allogeneic stem cell transplant for acute myeloid leukemia: evolution of an effective strategy in India. J Glob Oncol. 2017;3(6):773–781. doi:10.1200/JGO.2016.00665029244983
- Lipof JJ, Loh KP, O’Dwyer K, Liesveld JL. Allogeneic hematopoietic cell transplantation for older adults with acute myeloid leukemia. Cancers. 2018;10(6):179. doi:10.3390/cancers10060179
- Brodská B, Fuchs O, Otevřelová P, Salek C, Kuželová K. PD-L1 Is frequently expressed in acute myeloid leukemia patients with leukocytosis. Blood. 2016;128(22): 5229–5229.
- Haroun F, Solola SA, Nassereddine S, Tabbara I. PD-1 signaling and inhibition in AML and MDS. Ann Hematol. 2017;96(9):1441–1448. doi:10.1007/s00277-017-3051-528643044
- Daver N, Garcia-Manero G, Basu S, et al. Nivolumab (Nivo) with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML) or frontline elderly AML. Blood. 2017;130(Suppl 1): 1345–1345.
- Ravandi F, Daver N, Garcia-Manero G, et al. Phase 2 study of combination of cytarabine, idarubicin, and nivolumab for initial therapy of patients with newly diagnosed acute myeloid leukemia. Blood. 2017;130(Suppl 1): 815–815.
- Parmar S, Tallman MS. Acute promyelocytic leukaemia: a review. Expert Opin Pharmacother. 2003;4(8):1379–1392. doi:10.1517/14656566.4.8.137912877645
- Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–121. doi:10.1056/NEJMoa130087423841729
- Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi:10.1016/j.cell.2012.06.02322817890
- Metzeler KH, Herold T, Rothenberg-Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–698. doi:10.1182/blood-2016-01-69387927288520
- Eriksson A, Österroos A, Hassan S, et al. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015;5(4):e307. doi:10.1038/bcj.2015.3125885427
- Lee S-I, Celik S, Logsdon BA, et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun. 2018;9(1):42. doi:10.1038/s41467-017-02465-529298978
- Azzam D, Volmar C-H, Hassan -A-A, et al. A patient-specific ex vivo screening platform for personalized acute myeloid leukemia (AML) therapy. Blood. 2015;126(23): 1352–1352.