
Abstract
Background
The potential causes of miscarriage are very complex, including genetic, immune, infectious, and endocrine factors. 50%-60% of miscarriages are caused by chromosomal abnormalities. Chromosomal microarray analysis (CMA) is a key tool in this context, capable of detecting not only copy number variations (CNV) but also loss of heterozygosity (LOH). CMA has been used as a tool to investigate the genetic reasons for miscarriage.
Methods
In our study, chromosomal microarray analysis (CMA) conducted 1220 miscarriage villous tissues. The results from this technology were used to identify the genetic reasons for miscarriage and evaluated strategies for subsequent pre-pregnancy planning.
Results
Here, the abnormality rate of miscarriage was 56.07%(684/1220). The aneuploidy rate accounted for 81.14%(555/684), and was significantly higher in group >35-year-old age. The second most common genetic reason for miscarriage was polyploidy, accounting for 10.09%(69/684). Additionally, we discovered loss of heterozygosity (LOH) in a small percentage of cases, accounting for 2.20%(15/684) reason for miscarriage genetic reasons, due to the advantage of CMA can detect isodisomy (a kind of uniparental disomy). 45 cases (6.58%) with copy number variants, which due to the CMA can detect copy number variations.
Conclusion
Our study indicated that miscarriage villous tissues should be performed genetic analysis, seek help from professional genetic counseling.
Introduction
Miscarriage, affecting about 10–15% of child-bearing individualsCitation1,Citation2, often has uncertain causes, including genetic, immune, infectious, and endocrine factors.Citation3,Citation4 Chromosomal abnormalities are implicated in 50%-60% of miscarriages,Citation5–7 underscoring the importance of genetic analysis in miscarriage tissues to understand underlying reasons and provide risk assessments for future pregnancies. Chromosomal microarray analysis (CMA) is a key tool in this context, as it can detect not just copy number variations (CNV) but also loss of heterozygosity (LOH). Its applications range from prenatal diagnosis to studying developmental delays, autism, and miscarriage.Citation8–12 The advantage of CMA is its ability to perform without the need for cell culture, providing a comprehensive analysis.Citation13,Citation14 In addition to aneuploidies and CNVs, SNP array data from CMA can reveal uniparental disomy (UPD), consanguinity, mosaicism, zygosity, maternal cell contamination, and parent of origin.Citation15,Citation16 In this study, we involved 1220 miscarriage cases utilizing CMA to investigate the clinical significance of chromosomal abnormalities in these miscarriages.
Methods and Materials
Study Participants and Samples
This study involved 1220 women who experienced spontaneous miscarriages, with samples collected between January 2017 and August 2023 at the Maternal and Child Health Hospital of Shandong Province, China. The age range of participants was 18 to 46 years. These women were categorized into two groups based on age: those under 35 and those 35 or older. When necessary, the partners’ peripheral blood was collected to rule out maternal contamination and facilitate result interpretation. Informed consent was obtained from all participants, our research complies with the Declaration of Helsinki, and the study received approval from the Ethics Committee of the Maternal and Child Health Hospital of Shandong Province.
DNA Extraction
The miscarriage tissues were preserved in 0.9% sodium chloride solution, then separated from maternal blood and decidua to collect clean villous tissues for DNA extraction. The genomic DNA was extracted following the protocol provided by the Qiagen kit (Germany). The process involved adding 200 µL of ATL lysis buffer to the villous tissues in an Eppendorf tube and shaking it at 56°C for 10 minutes, followed by the addition of 200 µL of AL lysis buffer and further shaking at 70°C for another 10 minutes. Subsequently, 200 µL of absolute ethyl alcohol was added to the mixture. The solution was then transferred to a separation column, and the DNA was rinsed with a buffer and eluted using EDTA buffer. The concentration and purity of the extracted DNA were evaluated using a Nanodrop device (USA).
CMA Experiment
The CMA experiment protocol, utilizing the CytoScan 750K microarray from Thermo Scientific (USA), encompasses a comprehensive process for genomic DNA analysis. Based on the DNA Extraction, the procedure includes DNA extraction, digestion, ligation, amplification, purification, fragmentation, labeling, hybridization, washing, staining, and scanning using the Affymetrix CytoScan 750K microarray. The analysis of results is performed with Chromosome Analysis Suite Version 4.2, adhering to the 2019 ACMG guidelines for pathogenicity interpretation. Reference databases used include OMIM (https://www.omim.org/), Decipher (https://decipher.sanger.ac.uk/index), PubMed (https://www.ncbi.nlm.nih.), Clingen (https://dosage.clinicalgenome.org/gov), and DGV (http://dgv.tcag.ca/dgv/app/home). The CNV results are categorized into five classifications: Pathogenic, Likely Pathogenic, Uncertain Clinical Significance, Likely Benign, and Benign.
Immunohistochemical Staining
The process began with routine fixation, dehydration, and paraffin embedding of the tissue. After sectioning, the tissue underwent Hematoxylin and Eosin (H&E) staining to examine its morphology. The immunohistochemistry involved the SP three-step method from Zhongshanjinqiao (Beijing, China), utilizing DAB (Diaminobenzidine) for color development and hematoxylin for contrast staining. The p57kip2 antibody was applied as per the provided instructions. Positive staining was indicated by nuclear brown granules in the decidua tissue and/or intermediate trophoblast, serving as an internal control.
STR Genotyping
For STR genotyping, DNA was first assessed for quality using optical density measurements and then diluted to a concentration of 5~10 ng/μL. The AmpFlSTR®Identifiler™ PCR Amplification Kit (Applied Biosystems) was utilized to detect 15 gene loci alongside a single sex identification locus. The reaction mixture consisted of 10.5 μL of AmpFlSTR® PCR mix, 0.5 μL of AmpliTaq Gold® DNA polymerase, 5.5 μL of AmpFlSTR® Sinofiler™ primer set, 1.0 μL of DNA (5–10 ng/μL), and nuclease-free water to a final volume of 25 μL. PCR cycling conditions were as follows: initial denaturation at 95°C for 15 minutes; 28 cycles of 94°C for 1 minute, 59°C for 1 minute, and 72°C for 1 minute; followed by a final extension at 60°C for 10 minutes. Post-PCR, the products were analyzed using capillary electrophoresis sequencing. The sequence data was then processed and interpreted using Gene-Mapper® ID-X version 1.2 software.
Statistical Analysis
Statistical comparisons between groups were conducted using the chi-square test. A P-value of less than 0.05 was considered indicative of statistical significance. For these analyses, the SPSS Statistics software, version 20.0, was utilized.
Results
Genetic Results of Chromosome Abnormalities
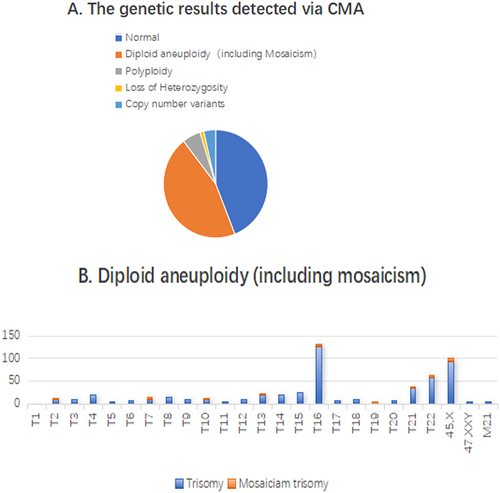
In the analysis of 1220 abortion villous samples, 684 (56.07%) exhibited chromosomal abnormalities. These included 555 cases (81.14%) of diploid aneuploidy, 69 cases (10.09%) of polyploidy, 15 cases (2.20%) showing loss of heterozygosity, and 45 cases (6.58%) with copy number variants. The specific breakdown and details of these findings are illustrated in .
Figure 1 (A) Distribution of normal, diploid aneuploidy, polyploidy, loss of heterozygosity and copy number variants of miscarriage villous genetic results. (B) Distribution of single chromosome aneuploidy among the different human chromosomes.
The Results of Different Types in CMA
In this study of 555 aneuploidy samples, trisomy was identified in 402 cases. Among these, trisomy of chromosome 16 was most prevalent, accounting for 32.84% (132 out of 402 samples), including 5 mosaicisms. Turner syndrome was detected in 102 samples, which represents 18.38% of the total aneuploidy cases (102/555), with 9 of these exhibiting mosaicism. Additionally, 20 samples were diagnosed with double trisomy (3.60%, 20/555), of which 5 included Turner syndrome concomitant with trisomy of another chromosome. There were also 2 instances of triple trisomy, one of which involved Turner syndrome in conjunction with trisomies of chromosomes 20, 21, and 22. The distribution of these aneuploidy cases is depicted in . Mosaicism was present in 31 samples, with Turner syndrome mosaicism being the most common (29.03%, 9 out of 31). There were 4 cases of dual-chromosome mosaicism, 1 chimera, and 1 instance of chromosome 19 trisomy mosaicism, all of which are documented as special cases. Further details are presented in .
Table 1 The Results of Mosaic
In this study, we observed 69 polyploidies among the samples. Specifically, 54 cases were identified as triploidy, constituting 78.26% of the polyploidy cases. Additionally, there were 13 cases of triploid aneuploidy, one hypo-triploid, and one tetraploid case. These findings are detailed in .
Table 2 The Results of Polyploidy
Remarkably, within the 684 samples of abnormal miscarriage villi, we identified 15 instances of loss of heterozygosity (LOH), accounting for 2.20% of the samples. chromosomal irregularities. These results are elaborated in . Out of the abnormal samples, 45 were identified with copy number variants (CNVs). Of these, 21 displayed chromosomal structural abnormalities. We recalled 10 couples for further testing through peripheral blood karyotyping, leading to the discovery that 2 mothers were carriers of balanced translocation, representing 20% of the tested group. Regrettably, we were unable to recall the remaining participants for further analysis. Of the CNV cases, 4 samples (8.89%) involved deletions in the short arm of chromosome 5, corresponding to the cri-du-chat syndrome region, and another 4 (8.89%) involved deletions at chromosome 22q11.2, which includes the TBX1 gene and is associated with haploinsufficiency. Three samples exhibited alterations in regions of known pathogenicity as well as regions of unknown clinical significance, while one sample was identified with a likely pathogenic region and another with an unknown region of significance. The comprehensive details of all CNV cases are presented in Supplemental Table 1 and Supplemental Table 2.
Table 3 The Results of Uniparental Disomy
Prevalence of Chromosomal Aneuploidy in Relation to Maternal Age
This study of 752 cases revealed chromosomal abnormalities, encompassing both aneuploidy and polyploidy. Stratification by maternal age demarcated at 35 years (those younger than 35 vs those 35 or older) highlighted a statistically significant divergence in the incidence rates of these conditions (P < 0.001). In-depth statistical analysis underscored a pronounced disparity predominantly in the prevalence of aneuploidy between the two age groups, with the frequency markedly elevated in women aged 35 and above as opposed to their younger counterparts (P < 0.000) although no significant variance was noted in the occurrence of polyploidy (P > 0.05). elaborates on the specific figures.
Table 4 The Frequency of Abnormality Types in Maternal Age Groups
Notable Special Cases
Within our results, three special cases captured our attention:
Case 1: We identified a sample exhibiting whole genome mosaic uniparental disomy (WGUPD), which is an exceedingly rare occurrence in clinical literature. Subsequent pathological staining and STR analysis suggested a potential paternal hydatid mole, as detailed in .
Figure 2 (A) IHC and H&E stained results illustrated molar mole of the villous (B) STR results showed it was paternal molar mole (C) CMA results showed it was whole genome mosaic uniparental disomy. (D) and (E) showed here that this villous was chimera, its CMA results was arr(X)×1~2,(Y)×0~1,(3)×2~3,(1,2,4–22)×2 chi. STR results revealed that this sample was heterologous chimera from parental and maternal origin. (F and G) showed here that this villous was T19 mosaic.
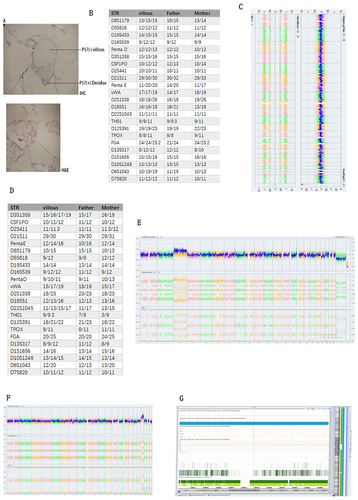
Case 2: The second notable sample was a chimeric individual. CMA analysis indicated arr(X)×1~2,(Y)×0~1,(3)×2~3,(1,2,4–22)×2 chi. Further analysis through both CMA and STR confirmed the chimerism to be of both paternal and maternal origins, as illustrated in .
Case 3: The third case involved a T19 mosaic, a chromosomal anomaly of extreme rarity. The affected individual exhibited a mosaicism range of approximately 60~80%, according to our findings, with comprehensive details presented in .
Discussion
The causes of miscarriage are multifaceted, with chromosomal anomalies being the primary culprit as current evidence suggests.Citation17 Our study aligns with past research,Citation5,Citation18 revealing that 56.07% of the 684 examined samples presented abnormalities, predominantly aneuploidy.Citation19 Notably, trisomy 16, 21, 22, and Turner syndrome were observed more frequently than other chromosomal aneuploidy. Excluding chromosome 1, aneuploidies spanned all chromosomes.Citation20–23 The genesis of aneuploidy might be linked to meiosis in germ cell formation, where non-disjunction of homologous chromosomes leads to numerical chromosomal discrepancies in the embryo.Citation4,Citation23 Furthermore, maternal age is a crucial determinant in the incidence of trisomy. Our data indicates that the occurrence in women aged 35 or above is significantly greater compared to their younger counterparts, underscoring a correlation between advancing maternal age and an increased likelihood of diploid aneuploidy.Citation21,Citation24 This increase may be attributed to impaired chromosome separation during ovarian germ cell formation in older women, coupled with the prolonged arrest of primary oocytes in the first meiotic phase. Triploidy stands as the secondary chromosomal number aberration contributing to miscarriages.Citation23 Our findings suggest that triploidy manifests uniformly across all ages within the childbearing spectrum. In addition to numerical chromosomal abnormalities, CMA is instrumental in detecting LOH and copy number variants through SNP and CNV hybridization probes. Structural chromosome anomalies such as duplications and deletions observed in the miscarried villi suggest the possibility of balanced translocations in one or both parents. These insights have profound implications for genetic counseling, providing valuable direction for their future reproductive decisions.
Our pivotal finding is that LOH accounts for 2.20% of villous abnormalities in miscarriages, aligning with the previously reported range of 0.5–2.7%.Citation25–27 LOH occurs when one allele at a heterozygous locus is lost, resulting in homozygosity. UPD arises when both chromosome homologues are inherited from a single parent. UPD can emerge through several mechanisms, including trisomy rescue, monosomy rescue, gamete complementation, or mitotic errors post-fertilization. The result may be heterodisomy (both homologues present), isodisomy (two copies of a single homologue), or a combination thereof (segmental UPD), depending on meiotic recombination events. Consequently, UPD involving an entire chromosome typically exhibits mixed isodisomic and heterodisomic segments. In our CMA, we detected segmental UPD in 8 samples and chromosome 22 isodisomy in 3 samples. UPD can be classified as maternal or paternal, which has implications for disease manifestation due to imprinted genes. However, SNP-based arrays only detect isodisomy and not heterodisomy due to the absence of LOH regions. Hence, the segmental UPDs we observed may represent undetected isodisomies owing to the method’s inherent constraints. Remarkably, we have identified 2 cases of whole-genome uniparental disomy. Individuals with whole-genome UPD are rare, and they exhibit a chimera of two cell lines: one with normal biparental inheritance and another with uniparental isodisomy across the genome. Our findings include an extremely unusual case of whole-genome mosaic UPD, identified through a combination of pathological diagnosis of a hydatidiform mole and paternal origin confirmed by STR analysis. Several hypotheses exist to explain the occurrence of chimeric whole genome UPD, with the most likely being endoreduplication of a single parental genome prior to the fusion of pronuclei in the zygote. Clinically, UPD is significant due to its association with imprinting disorders stemming from errors in gene methylation, as well as autosomal recessive diseases resulting from isodisomy UPD. Prior research has identified definitive imprinted genes on chromosomes 6, 7, 11, 14, 15, and 20, while disorders linked to other chromosomes are primarily due to homozygous variations in autosomal recessive genes. In our study, no cases of single chromosome isodisomy or segmental UPD were found within definitively pathogenic imprinted regions, suggesting that there may be additional imprinted regions yet to be discovered. Alternatively, recessive mutations within these UPD regions could lead to severe phenotypes or even spontaneous abortions, particularly if the genes involved are crucial for embryogenesis and fetal development. Despite the substantial number of UPD cases detected in our study, both in terms of quantity and sample size, the underlying molecular mechanisms of UPD remain elusive. Therefore, further molecular genetic studies on UPD are necessary to advance our understanding in this area. In the retrospective literature review, it was observed that compared to low-depth sequencing for copy number variations, CMA is capable of detecting triploidy and higher levels of polyploidy, as well as LOH. Our findings suggest that LOH could be a significant factor contributing to miscarriages, which may have been previously overlooked. For the purpose of determining the causes of miscarriage and providing a basis for risk assessment in subsequent pregnancies, genetic testing and counseling are strongly advised for women who have experienced a miscarriage, irrespective of their age. Accurate molecular genetic diagnostics are crucial for guiding reproductive decisions. One limitation of Chromosomal Microarray Analysis is its inability to detect balanced chromosomal rearrangements, gene mutations, low-level mosaicism, and heterodisomy. Nevertheless, CMA can offer a more comprehensive genetic profile and provide a precise diagnosis for the causes of miscarriage, which in turn can inform genetic risk assessments for couples planning future pregnancies.
Conclusion
As a conclusion, our study indicated that miscarriage villous tissue should be performed genetic analysis, provided diagnosis for the reason of miscarriage, which can help couples assess recurrent risk of next pregnancy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
Data Sharing Statement
All data generated during and analyzed during the current study are available upon request by contact the corresponding author.
Additional information
Funding
References
- Rull K, Nagirnaja L, Laan M. Genetics of recurrent miscarriage: challenges, current knowledge, future directions. Front Genet. 2012;3:34. doi:10.3389/fgene.2012.00034
- van den Berg MM, van Maarle MC, van Wely M M, Goddijn M. Goddijn, Genetics of early miscarriage. Biochim Biophys Acta. 2012;1822(12):1951–1959. doi:10.1016/j.bbadis.2012.07.001
- Zheng D, Li C, Wu T, Tang K. Factors associated with spontaneous abortion: a cross-sectional study of Chinese populations. Reprod Health. 2017;14(1):33. doi:10.1186/s12978-017-0297-2
- Woods L, Perez-Garcia V, Kieckbusch J, et al. Decidualisation and placentation defects are a major cause of age-related reproductive decline. Nat Commun. 2017;8(1):352. doi:10.1038/s41467-017-00308-x
- Rai R, Regan L. Recurrent miscarriage. Lancet. 2006;368(9535):601–611. doi:10.1016/S0140-6736(06)69204-0
- Kacprzak M, Chrzanowska M, Skoczylas B, Moczulska H, Borowiec M, Sieroszewski P. Genetic causes of recurrent miscarriages. Ginekol Pol. 2016;87(10):722–726. doi:10.5603/GP.2016.0075
- Warburton D, Susser M, Stein Z, Kline J. Genetic and epidemiologic investigation of spontaneous abortion: relevance to clinical practice. Birth Defects Orig Artic Ser. 1979;15(5a):127–136.
- A. Swedish Council on Health Technology, SBU Systematic Review Summaries. Prenatal Diagnosis Through Chromosomal Microarray Analysis (CMA). Stockholm: Swedish Council on Health Technology Assessment (SBU)Copyright © 2016 by the Swedish Council on Health Technology Assessment; 2016.
- McNamee K, Dawood F, Farquharson RG. Evaluation of array comparative genomic hybridization in recurrent miscarriage. Br J Hosp Med. 2013;74(1):36–40. doi:10.12968/hmed.2013.74.1.36
- Hu T, Tian T, Zhang Z, et al. Prenatal chromosomal microarray analysis in 2466 fetuses with ultrasonographic soft markers: a prospective cohort study. Am J Obstet Gynecol. 2021;224(5):516.e1–516.e16. doi:10.1016/j.ajog.2020.10.039
- Wu XL, Li R, Fu F, et al. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017;17(1):117. doi:10.1186/s12887-017-0863-3
- Brady PD, Vermeesch JR. Genomic microarrays: a technology overview. Prenat Diagn. 2012;32(4):336–343. doi:10.1002/pd.2933
- McQueen DB, Lathi RB. Miscarriage chromosome testing: indications, benefits and methodologies. Semin Perinatol. 2019;43(2):101–104. doi:10.1053/j.semperi.2018.12.007
- Deshpande M, Harper J, Holloway M, Palmer R, Wang R. Evaluation of array comparative genomic hybridization for genetic analysis of chorionic villus sampling from pregnancy loss in comparison to karyotyping and multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers. 2010;14(3):421–424. doi:10.1089/gtmb.2010.0014
- Wenstrom KD. Microarray analysis: elegant, accurate, and expensive. Obstet Gynecol. 2014;124(2 Pt 1):199–201. doi:10.1097/AOG.0000000000000407
- Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):742–745. doi:10.1097/GIM.0b013e3181f8baad
- Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Genet Med. 2005;7(4):251–263. doi:10.1097/01.GIM.0000160075.96707.04
- Tur-Torres MH, Garrido-Gimenez C, Alijotas-Reig J. Genetics of recurrent miscarriage and fetal loss. Best Pract Res Clin Obstet Gynaecol. 2017;42:11–25. doi:10.1016/j.bpobgyn.2017.03.007
- Subramaniyam S, Pulijaal VR, Mathew S. Double and multiple chromosomal aneuploidies in spontaneous abortions: a single institutional experience. J Hum Reprod Sci. 2014;7(4):262–268. doi:10.4103/0974-1208.147494
- Choi TY, Lee HM, Park WK, Jeong SY, Moon HS. Spontaneous abortion and recurrent miscarriage: a comparison of cytogenetic diagnosis in 250 cases. Obstet Gynecol Sci. 2014;57(6):518–525. doi:10.5468/ogs.2014.57.6.518
- Cai M, Lin N, Xu L, Huang H. Comparative clinical genetic testing in spontaneous miscarriage: insights from a study in Southern Chinese women. J Cell Mol Med. 2021;25(12):5721–5728. doi:10.1111/jcmm.16588
- Sugiura-Ogasawara M, Ozaki Y, Katano K, Suzumori N, Kitaori T, Mizutani E. Abnormal embryonic karyotype is the most frequent cause of recurrent miscarriage. Hum Reprod. 2012;27(8):2297–2303. doi:10.1093/humrep/des179
- Pylyp LY, Spynenko LO, Verhoglyad NV, Mishenko AO, Mykytenko DO, Zukin VD. Chromosomal abnormalities in products of conception of first-trimester miscarriages detected by conventional cytogenetic analysis: a review of 1000 cases. J Assist Reprod Genet. 2018;35(2):265–271. doi:10.1007/s10815-017-1069-1
- Heffner LJ. Advanced maternal age--how old is too old? N Engl J Med. 2004;351(19):1927–1929. doi:10.1056/NEJMp048087
- Gogiel M, Begemann M, Spengler S, et al. Genome-wide paternal uniparental disomy mosaicism in a woman with Beckwith-Wiedemann syndrome and ovarian steroid cell tumour. Eur J Hum Genet. 2013;21(7):788–791. doi:10.1038/ejhg.2012.259
- Lalou I, Gkrozou F, Meridis E, Tsonis O, Paschopoulos M, Syrrou M. Molecular investigation of uniparental disomy (UPD) in spontaneous abortions. Eur J Obstet Gynecol Reprod Biol. 2019;236:116–120. doi:10.1016/j.ejogrb.2019.03.004
- Fritz B, Aslan M, Kalscheuer V, et al. Low incidence of UPD in spontaneous abortions beyond the 5th gestational week. Eur J Hum Genet. 2001;9(12):910–916. doi:10.1038/sj.ejhg.5200741