Abstract
Oral administration of sapropterin hydrochloride, recently approved for use by the US Food and Drug Administration and the European Commission, is a novel approach for the treatment of phenylketonuria (PKU), one of the most common inborn errors of metabolism. PKU is caused by an inherited deficiency of the enzyme phenylalanine hydroxylase (PAH), and the pathophysiology of the disorder is related to chronic accumulation of the free amino acid phenylalanine in tissues. Contemporary therapy is based upon restriction of dietary protein intake, which leads to reduction of blood phenylalanine levels. This therapy is difficult to maintain throughout life, and dietary noncompliance is commonplace. Sapropterin dihydrochloride is a synthetic version of tetrahydrobiopterin, the naturally occurring pterin cofactor that is required for PAH-mediated phenylalanine hydroxylation. In a subset of individuals with PAH deficiency, sapropterin administration leads to reduction in blood phenylalanine levels independent of dietary protein. For these individuals, sapropterin is an effective novel therapy for PKU.
Introduction
Dietary restriction of phenylalanine intake has been the mainstay of therapy for phenylketonuria (PKU), one of the most common inborn errors of metabolism, for the past 50 years. With the recent US Food and Drug Administration (FDA) and European Commission approvals of sapropterin dihydrochloride (Kuvan®, BioMarin Corporation, Tiburon, CA), a new era in PKU treatment has begun. Sapropterin dihydrochloride is a synthetic version of the naturally occurring pterin cofactor, tetrahydrobiopterin (BH4). In a subset of individuals with PKU, sapropterin treatment substantially reduces blood phenylalanine levels independent of dietary phenylalanine intake. For these so-called “BH4-responsive” patients, sapropterin therapy provides the benefit of improved disease control and may even eliminate the need for dietary therapy completely in individuals with milder forms of hyperphenylalaninemia.
Biology of phenylketonuria and tetrahydrobiopterin
PKU has an annual incidence of about 1:16,000 births in the US.Citation1 PKU is caused by recessively inherited mutations in the PAH gene that encodes the enzyme phenylalanine hydroxylase (PAH). PAH is predominantly expressed in liver (but also kidney and pancreas) and catalyzes the irreversible hydroxylation of phenylalanine, an essential amino acid, to tyrosine (). PAH is a homotetramer that requires iron and molecular oxygen as well as the unconjugated pterin cofactor, BH4, for catalytic activity. BH4 is synthesized de novo from guanosine triphosphate in several tissues, including liver, but is also recycled after phenylalanine hydroxylation through enzymatically-catalyzed reduction.Citation2 Inherited deficiency of BH4 synthesis or recycling enzymes is the cause of hyperphenylalaninemia in approximately 2% of infants detected through newborn screening. Treatment of these children frequently requires chronic oral or parenteral BH4 administration in addition to dietary phenylalanine restriction.
Figure 1 Phenylalanine hydroxylation. Phenylalanine is hydroxylated to tyrosine through the catalytic activity of phenylalanine hydroxylase, which requires the presence of the unconjugated pterin cofactor, tetrahydrobiopterin (BH4). Sapropterin is a synthetic form of BH4 that augments the endogenous BH4 supply. During phenylalanine hydroxylation, BH4 is oxidized to quinonoid dihydrobiopterin (qBH2). Fully active BH4 is regenerated through the sequential action of pterin-4a-carbinolamine dehydratase and dihydropteridine reductase (DHPR) or may be synthesized de novo from guanosine triphosphate (GTP).Citation2
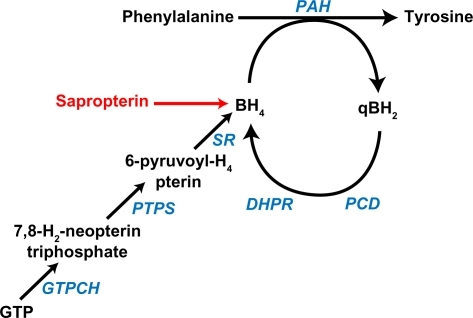
PAH deficiency leads to accumulation of phenylalanine in all tissues of the body and relative deficiency of tyrosine. Untreated PKU is associated with chronic hyperphenylalaninemia, microcephaly, mental retardation, epilepsy, relative hypopigmentation, growth delay, and eczema. Fortunately, all infants in the US and most Western countries are screened for hyperphenylalaninemia at 24–72 hours of life. Initiation of therapy early in life prevents the major manifestations of the disease. Contemporary therapy is based upon the restriction of dietary phenylalanine intake through the reduction of dietary protein. Under this regimen, synthetic medical foods must be consumed in order to provide adequate intake of total energy and amino acids other than phenylalanine. Intellectual outcome is directly related to success in reducing blood phenylalanine levels,Citation3 and restriction of dietary phenylalanine is recommended throughout the life of the individual with PKU.Citation4 However, this therapeutic approach is complicated, unpalatable, and difficult to maintain through adolescence and into adulthood. Loss of dietary control may be associated with deterioration of cognitive performance and attention span, resulting from development of a neurodegenerative disease of white matter,Citation5 and in pregnant women with PKU and elevated blood phenylalanine, severe teratogenesis with fetal growth failure, microcephaly, mental retardation, and congenital heart disease, ie, the so-called maternal PKU syndrome.Citation6 For all of these reasons, novel alternatives to dietary therapy are being sought.
History of tetrahydrobiopterin responsiveness in phenylketonuria
Individuals with inherited PAH deficiency have normal biopterin content in blood and urine, but Kure et al reported that oral administration of additional BH4 to some individuals with mild hyperphenylalaninemia led to a significant reduction in blood phenylalanine levels without altering dietary phenylalanine content.Citation7 Several other investigators have subsequently confirmed this observation.Citation8–Citation17 The basis of BH4 responsiveness may be related to different molecular mechanisms. Increased liver BH4 content may simply stimulate the activity of mutant partially active PAH enzymeCitation18 or it may act as a chemical chaperone to stabilize mutant PAH monomers.Citation19 Proof of this latter mechanism is provided by investigation of other small molecules that act as chaperones to stabilize mutant PAH.Citation20 Some specific PAH mutations are known to affect the affinity of the PAH enzyme for its biopterin cofactor.Citation21 In patients with these specific mutations, oral biopterin administration may overcome this block and restore enzymatic activity. In reality, only a few such mutations have been shown to be often (but not invariably) associated with BH4 responsiveness in PKU patientsCitation22,Citation23 while other mutations, such as the R408W mutation, are consistently associated with nonresponsiveness. BH4 responsiveness in an individual PKU patient cannot always be accurately predicted from their PAH genotype.Citation22,Citation24
Clinical trials of sapropterin in phenylketonuria
After the discovery that some PKU patients could benefit from BH4 supplementation, few patients were able to avail themselves of this treatment option because the worldwide supply of BH4 was limited and detailed pharmacokinetic studies had not been performed. In the USA, BH4 was approved by the FDA only for use as an investigational drug. To overcome this problem, BioMarin Corporation (Novato, CA) initiated a specific effort to develop and intensively study sapropterin dihydrochloride ((6R)-2-amino-6-[(1R, 2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydro-4(3H)-pteridinone dihydrochloride, ), a biologically active synthetic form of BH4, in subjects with PKU. Sapropterin dihydrochloride is supplied in airtight blister packs as 100 mg tablets that may be administered whole or dissolved in water or apple juice. The tablets contain ascorbic acid to maintain the reduction status of sapropterin and allowing storage at room temperature. In patients with PKU who are responsive to sapropterin, daily oral administration of sapropterin leads to a sustained decrease in blood phenylalanine levels without alteration of dietary phenylalanine intake. The mean half-life of elimination of sapropterin from the blood is 6.7 (range 3–17) hours in PKU patients.Citation25 The specific mechanism of elimination for sapropterin dihydrochloride has not been published, but abundant data concerning the fate of orally administered natural BH4 in humans and animals demonstrate that BH4 is quickly sequestered in the liver or excreted in urine.Citation26
Figure 2 Sapropterin dihydrochloride.
To determine the actual prevalence of BH4 responsiveness, an open-label, screening study was performed in 490 individuals with PKU enrolled at 30 treatment centers in the US and Europe.Citation27 Blood phenylalanine levels were greater than 450 μM (normal range 35–150 μM) in all subjects at the beginning of the study, signifying less than ideal disease control on standard dietary therapy. Sapropterin dihydrochloride, 10 mg/kg body weight, was administered orally once per day for eight days. Subjects were instructed not to alter their typical dietary protein intake during the study. Blood phenylalanine levels were measured prior to sapropterin treatment and again on day 8, and “BH4 responsiveness” was defined as a 30% reduction in blood phenylalanine. Overall, 20% of study subjects (96/485 subjects who completed the entire eight-day trial) responded to sapropterin treatment. Among all responders, blood phenylalanine levels decreased to 391.8 ± 185.3 μM (mean ± 1 standard deviation) after eight days of sapropterin administration. When stratified by blood phenylalanine level prior to initiating therapy, a higher proportion of individuals with mild PKU responded to sapropterin. In these subjects, 54% and 24% with initial blood levels of 450–600 μM or 600–900 μM, respectively, responded to sapropterin. Somewhat surprisingly, 10% of subjects with initial phenylalanine levels greater than 900 μM also responded, with at least a 30% reduction in blood phenylalanine. For standard dietary therapy in young children with PKU, the target range for blood phenylalanine during treatment is commonly 120–360 μM. Approximately 10% of subjects with PKU in this screening trial achieved a reduction in blood phenylalanine to the accepted treatment target range on sapropterin alone without any change in dietary phenylalanine intake. An additional 10% of the study population achieved a clinically significant reduction in blood phenylalanine, but phenylalanine levels remained above 360 μM. No severe adverse effects of sapropterin therapy were reported.
Once the prevalence of BH4-responsive hyperphenylalaninemia was established, a Phase III, randomized, double-blind, placebo-controlled trialCitation28 was initiated to evaluate fully the efficacy of sapropterin treatment. Sapropterin-responsive subjects identified in the Phase II trial were randomly assigned to receive either sapropterin 10 mg/kg once per day or placebo for six weeks. At the end of the trial, no serious adverse events were reported in either study group, and the incidence of minor events was equal in the two study groups. By the end of the six-week trial, blood phenylalanine in the sapropterin treatment group (n = 40 subjects) decreased from 842.7 ± 299.6 μM to 606.9 ± 377.0 μM on average. The mean change in blood phenylalanine was −235.9 ± 257.0 μM over six weeks on sapropterin compared with a change of only +2.9 ± 239.5 μM in the control group (n = 47 subjects). The estimated difference between the two groups was −245 ± 52.5 μM (P = 0.0002). Over the course of the study, 44% of subjects on sapropterin had a sustained reduction in blood phenylalanine greater than 30% from baseline compared with only 9% of controls. Prior to initiation of the trial, only 17% of subjects exhibited blood phenylalanine levels below 600 μM. In the sapropterin treatment group, 54% of subjects achieved blood phenylalanine below 600 μM. Blood phenylalanine decreased to less than 360 μM in 32% of subjects.
BH4 responsiveness has been predicted to occur only in patients who carry at least one mutant PAH allele that yields some residual PAH enzyme activity.Citation18 Among sapropterin-responsive subjects in the Phase III trial, PAH genotyping was available for 17 subjects, and non-null PAH mutations were detected on at least one allele in 16 of 17 subjects. However, in the seventeenth subject, both PAH alleles carried severe mutations that would not appear to permit expression of residual PAH activity, yet this subject responded to sapropterin treatment. Sapropterin responsiveness in the study subjects was not linked to any specific PAH mutation. Two reviews suggest that genotype can predict to some degree whether a patient with PKU will be responsive to BH4 but that ultimately only BH4 challenge will fully ascertain BH4 responsiveness.Citation23,Citation24
In a third clinical trial, 80 sapropterin-responsive subjects participated in a multicenter, open-label extension study.Citation29 The initial phase of the trial was a forced dose titration study, with subjects receiving doses of 5, 20, and 10 mg/kg/day of sapropterin consecutively for two weeks each. This was followed by a four-week period of 10 mg/kg/day and then a 12-week fixed dose extension. In the last phase, subjects received one of three different sapropterin doses (5, 10, or 20 mg/kg/day) based upon their individual responses in the dose titration phase of the study. In the first phase, although plasma phenylalanine levels decreased from week 0 at all sapropterin doses, a dose-response relationship was demonstrated, with significantly lower plasma phenylalanine levels on average during treatment with 10 or 20 mg/kg/day than when the subjects received only 5 mg/kg/day. During the fixed-dose extension phase, six subjects (8%) responded to 5 mg/kg/day, while 37 subjects (46%) received 10 mg/kg/day, and 37 subjects (46%) received 20 mg/kg/day. The choice of dose and the reduction in phenylalanine levels achieved by 22 weeks clearly reflect a dichotomy in the severity of PKU in the study subjects. Individuals with higher phenylalanine levels at baseline required more drug to effect a decrease in blood phenylalanine, and the drop in phenylalanine level achieved was not as satisfactory as that demonstrated by subjects responding to either 5 or 10 mg/kg/day. Overall, plasma phenylalanine decreased by 190.5 μM (±355.7) in all dose groups after 22 weeks of drug. The mean plasma phenylalanine level at the end of 22 weeks in the 20 mg/kg dose group was 895.7 (±407.2) μM while mean phenylalanine levels in the 5 and 10 mg/kg dose groups were 437.8 (±260.5) μM and 449.9 (±193.1) μM, respectively. Individuals with a lower baseline phenylalanine level (a measure of dietary phenylalanine tolerance) showed greater responsiveness to the drug. Only 50%, 49%, and 42% of subjects in the 5, 10, and 20 mg/kg dose groups, respectively, achieved a 30% decrease in plasma phenylalanine from baseline at the end of the 22 weeks. Recall that all study subjects had demonstrated a 30% reduction in plasma phenylalanine over eight days in the initial screening study. Unintended changes in dietary protein intake over the course of the trials (further protein restriction during the screening trial or liberalization of dietary protein intake during the extension trial) would have affected these results. There were no minor or severe adverse events attributable to sapropterin treatment in the trial. This extension study demonstrates that sapropterin treatment may safely reduce plasma phenylalanine concentrations in a dose-dependent manner in BH4-responsive individuals. Furthermore, empiric use of the drug for one month or more may be needed.
The effectiveness of sapropterin treatment has also been examined in 90 children aged 4–12 years with PKU while they continued on a protein-restricted diet.Citation30 Of these subjects, 56% experienced a 30% reduction in blood phenylalanine levels after eight days of sapropterin treatment. After the challenge period, all responders were offered participation in a placebo-controlled trial during which dietary phenylalanine intake was gradually increased. In the group receiving sapropterin, dietary phenylalanine tolerance doubled on average, with no significant increase in blood phenylalanine levels in comparison with phenylalanine tolerance prior to sapropterin treatment, while dietary phenylalanine tolerance increased only slightly in the placebo group. Again, no significant safety issues were revealed.
Based on the accumulated study data, the FDA approved sapropterin dihydrochloride for clinical use in December 2007. Marketing authorization for sapropterin has subsequently been granted in Japan (July 2008) and Europe (December 2008), and is under expedited review in Canada. In Europe, sapropterin has been licensed for use in children with PAH deficiency over the age of four years, and also in individuals of all ages with hyperphenylalaninemia due to congenital BH4 deficiency. In the US, sapropterin treatment is approved in all age groups and in pregnant women, even though sapropterin has not been formally studied in children under four years age or during pregnancy. Limited experience with administration of other commercially available forms of BH4 during pregnancy suggests that no adverse effects are to be expected.Citation31 In the case of the pregnant woman with BH4-responsive PKU, the well documented risks to the fetus associated with maternal hyperphenylalaninemia likely outweigh any putative risks of sapropterin treatment. Still, these issues deserve further study and BioMarin has initiated a voluntary registry system to accumulate data on the use of sapropterin in young children and during pregnancy.
Post-approval clinical use of sapropterin
Now that sapropterin has gained FDA approval, the task ahead is to determine which PKU patients will benefit most from this novel therapeutic approach and how to best introduce this medication into a specific patient’s dietary treatment regimen. Sapropterin has been formally proven in research studies to lower blood phenylalanine levels in individuals who are responsive, but limited data are available concerning the effect of sapropterin treatment on actual protein tolerance. Can the sapropterin-treated patient safely increase their dietary protein intake and maintain blood phenylalanine levels within a designated treatment range? Also, long-term safety and cognitive outcomes should be evaluated in patients who will probably be treated with sapropterin for many years.
The determination of whether a specific patient with PKU is truly BH4-responsive is clearly key to the decision about adding sapropterin to the patient’s treatment regimen. To some degree, the definition of “responsiveness” is an arbitrary one. For the purposes of the screening study, responsiveness was defined as a 30% decrease in plasma phenylalanine after an eight-day trial of oral sapropterin at 10 mg/kg/day. As we have seen, subsequent evaluations have revealed both false-positive and false-negative results from this screening procedure. Some individuals who met criteria for responsiveness during the screening trial subsequently did not maintain a therapeutic effect from sapropterin over the longer term. Some individuals who did not meet criteria for responsiveness with a sapropterin dose of 10 mg/kg/day were later found to be responsive to the higher dose of 20 mg/kg/day. It has been suggested that BH4 responsiveness would require a patient with PKU to carry at least one PAH mutant allele that allows for residual enzyme activity.Citation18 However, in the clinical trial, a few individuals who were found to be sapropterin-responsive carried PAH mutations that were predicted to yield severe PAH deficiency. A recent comprehensive review suggests that genotype can predict to some degree whether a patient with PKU will be responsive to BH4, but that ultimately only BH4 challenge will fully ascertain BH4 responsiveness.Citation24
In addition to the eight-day screening trial employed in the clinical trial,Citation27 several other BH4 challenge protocols have been described in the literature, and these have recommended differing BH4 doses, different durations of challenge, determination of blood phenylalanine levels at different times, varying percentage reductions in blood phenylalanine required to define BH4 responsiveness and, in some cases, challenging the subject with a standardized amount of dietary phenylalanine along with BH4. This topic has been reviewed.Citation32 Recent published data have revealed that the decrease in blood phenylalanine following a single BH4 dose obeys a first-order kinetic model only over the first eight hours after BH4 administration.Citation33 Accordingly, a single BH4 loading test (20 mg/kg) with monitoring of blood phenylalanine at 0, 8, 12, and 24 hours has been proposed as an evaluation of responsiveness.Citation34 If blood phenylalanine dropped more than 20% in the short term, then a several-week trial with BH4 dose adjustment to maintain optimal phenylalanine levels would be warranted.
A rational algorithm for testing BH4 responsiveness in a routine clinical setting has been proposedCitation35 and a similar protocol has been endorsed by clinicians in Europe.Citation36 This algorithm evaluates both the short-term response to BH4 administration (allowing for upward adjustment of dietary phenylalanine intake if blood phenylalanine levels fall dramatically after BH4 treatment) and the longer-term efficacy of sapropterin treatment. In this algorithm, blood phenylalanine is measured prior to sapropterin administration and on days 1, 7, 14, and 28 days following sapropterin 20 mg/kg/day. The sapropterin dose may be decreased or dietary phenylalanine increased if blood phenylalanine levels decrease below the target range. Responsiveness is not defined by any specific percentage decrease in phenylalanine but rather by whether the decrease in phenylalanine is thought by the clinician and patient to be clinically meaningful. Continued sapropterin treatment is warranted if blood phenylalanine levels are lowered into a specific target range defined by clinician and patient and is particularly relevant if dietary phenylalanine restriction can be relaxed without increasing blood phenylalanine. This testing algorithm allows for the individualization of therapy and flexibility in the assessment of BH4 responsiveness.
Cost of sapropterin
The cost of sapropterin therapy versus the PKU diet must also be considered. As a drug with an orphan product designation, the costs of drug development for sapropterin are distributed among a relatively small patient population. The cost of daily sapropterin therapy at the highest dose of 20 mg/kg/day is US $100,000 to $150,000 for the average adult patient versus the cost of the phenylalanine-restricted diet, including the use of medical foods, which is typically US $15,000 to $20,000 per year. The PKU patient and their clinician must consider these factors when deciding whether to employ sapropterin therapy.
Conclusions
The systematic evaluation of sapropterin responsiveness in a PKU population and the subsequent commercial availability of a validated form of BH4 have now provided a new therapeutic option for phenylketonuria. As discussed, continuance of strict dietary therapy throughout life is recommended but is practically difficult. Compliance with this recommendation in adults is often poor. For a subset of individuals with PKU, sapropterin will be essential as sole therapy. For other patients, sapropterin treatment may not completely eliminate the need for dietary therapy, but may increase a patient’s dietary protein tolerance, allowing a significant improvement in daily quality of life. The introduction of sapropterin for widespread clinical use represents an exciting time in the evolution of therapy for PKU.
Disclosure
The author reports no conflict of interest in this work.
References
- DonlonJLevyHScriverCRHyperphenylalaninemia: Phenylalanine hydroxylase deficiencyValleDBeaudetALVogelsteinBThe Metabolic and Molecular Bases of Inherited DiseaseNew York, NYMcGraw-Hill2008
- ThonyBAuerbachGBlauNTetrahydrobiopterin biosynthesis, regeneration and functionsBiochem J2000347Pt 111610727395
- AzenCGKochRFriedmanEGIntellectual development in 12-year-old children treated for phenylketonuriaAm J Dis Child1991145135391985428
- National Institutes of Health Consensus Development Conference Statement: Phenylketonuria: Screening and management, 2000 Oct 16–18Pediatrics2001108497298211581453
- ThompsonAJSmithIBrentonTNeurological deterioration in young adults with phenylketonuriaLancet199033687206026051975386
- PlattLDKochRHanleyWBThe international study of pregnancy outcome in women with maternal phenylketonuria: Report of a 12-year studyAm J Obstet Gynecol2000182232633310694332
- KureSHouDCOhuraTTetrahydrobiopterin-responsive phenylalanine hydroxylase deficiencyJ Pediatr1999135337537810484807
- LindnerMHaasDMayatepekEZschockeJBurgardPTetrahydrobiopterin responsiveness in phenylketonuria differs between patients with the same genotypeMol Genet Metab200173110410611350190
- BerneggerCBlauNHigh frequency of tetrahydrobiopterin responsiveness among hyperphenylalaninemias: A study of 1,919 patients observed from 1988 to 2002Mol Genet Metab200277430431312468276
- MuntauACRoschingerWHabichMTetrahydrobiopterin as an alternative treatment for mild phenylketonuriaN Engl J Med2002347262122213212501224
- CeroneRSchiaffinoMCFantasiaARLong-term follow-up of a patient with mild tetrahydrobiopterin-responsive phenylketonuriaMol Genet Metab200481213713914741196
- ShintakuHKureSOhuraTLong-term treatment and diagnosis of tetrahydrobiopterin-responsive hyperphenylalaninemia with a mutant phenylalanine hydroxylase genePediatr Res200455342543014681498
- FiegeBBonafeLBalhausenDExtended tetrahydrobiopterin loading test in the diagnosis of cofactor-responsive phenylketonuria: A pilot studyMol Genet Metab200586Suppl 1S91S9516290003
- FioriLFiegeBRivaEGiovanniniMIncidence of BH4-responsiveness in phenylalanine-hydroxylase-deficient Italian patientsMol Genet Metab200586Suppl 1S67S7416198137
- HennermannJBBührerCBlauNVetterBMönchELong-term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuriaMol Genet Metab200586Suppl 1S86S9016051511
- MatalonRMichals-MatalonKKochRGradyJTyringSStevensRCResponse of patients with phenylketonuria in the US to tetrahydrobiopterinMol Genet Metab200586Suppl 1S17S2116143554
- MitchellJJWilckenBAlexanderITetrahydrobiopterin-responsive phenylketonuria: The New South Wales experienceMol Genet Metab200586Suppl 1S81S8516091307
- BlauNErlandsenHThe metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiencyMol Genet Metab200482210111115171997
- DobrowolskiSFPeyALKochRBiochemical characterization of mutant phenylalanine hydroxylase enzymes and correlation with clinical presentation in hyperphenylalaninaemic patientsJ Inherit Metab Dis2009321102118937047
- PeyALYingMCremadesNIdentification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuriaJ Clin Invest200811882858286718596920
- ErlandsenHPeyALGamezACorrection of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutationsProc Natl Acad Sci U S A200410148169031690815557004
- KaracicIMeiliDSarnavkaVGenotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiencyMol Genet Metab200997316517119394257
- TrefzFKScheibleDGötzHFrauendienst-EggerGSignificance of genotype in tetrahydrobiopterin-responsive phenylketonuriaJ Inherit Metab Dis2009321222618956252
- ZurfluhMRZschockeJLindnerMMolecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiencyHum Mutat200829116717517935162
- FeilletFClarkeLMeliCPharmacokinetics of sapropterin in patients with phenylketonuriaClin Pharmacokinet2008471281782519026037
- HardingCONeffMWildKThe fate of intravenously administered tetrahydrobiopterin and its implications for heterologous gene therapy of phenylketonuriaMol Genet Metab2004811525714728991
- BurtonBKGrangeDKMilanowskiAThe response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): A phase II, multicentre, open-label, screening studyJ Inherit Metab Dis200730570070717846916
- LevyHLMilanowskiAChakrapaniAEfficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: A phase III randomised placebo-controlled studyLancet2007370958650451017693179
- LeePTreacyEPCrombezESafety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuriaAm J Med Genet A2008146A222851285918932221
- TrefzFKBurtonBKLongoNEfficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: A phase III, randomized, double-blind, placebo-controlled studyJ Pediatr2009154570070719261295
- KochRMoseleyKGuttlerFTetrahydrobiopterin and maternal PKUMol Genet Metab200586Suppl 1S139S14116338627
- FiegeBBlauNAssessment of tetrahydrobiopterin (BH4) responsiveness in phenylketonuriaJ Pediatr2007150662763017517248
- LangenbeckUClassifying tetrahydrobiopterin responsiveness in the hyperphenylalaninaemiasJ Inherit Metab Dis2008311677218210214
- BlauNDefining tetrahydrobiopterin (BH4)-responsiveness in PKUJ Inherit Metab Dis20083112318327672
- LevyHBurtonBCederbaumSScriverCRecommendations for evaluation of responsiveness to tetrahydrobiopterin (BH(4)) in phenylketomuria and its use in treatmentMol Genet Metab200792428729118036498
- BlauNBelanger-QuintanaADemirkolMOptimizing the use of sapropterin (BH4) in the management of phenylketonuriaMol Genet Metab200996415816319208488