Abstract
Chronic obstructive pulmonary disease (COPD) is a complex chronic inflammatory disease involving a wide variety of cells and inflammatory mediators. The most important etiological factor in the development of this disease is cigarette smoking. Much of the research into the mechanisms of COPD has been concerned with the induction of inflammation and the role of neutrophils and macrophages in the pathophysiology of the disease. The possible contribution of the epithelium to the development of COPD has only recently become apparent and remains unclear. In this article we review research into the effect of cigarette smoke on the pulmonary epithelium with particular emphasis on oxidative stress, proteolytic load, pro-inflammatory cytokine and chemokine profile and epithelial secretions. In addition, we have also reviewed how cigarette smoke may affect epithelial damage and repair processes.
Introduction
COPD is a major cause of morbidity and mortality and is the fourth most common cause of death in the USA. It is estimated that over one million individuals in the UK, 6% of men and 4% of women, suffer from COPD (CitationBarnes 1998) with some 30,000 deaths per year. The global initiative for chronic obstructive lung disease (GOLD) defines COPD as a “disease state characterized by airflow limitation that is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases” (CitationPauwels et al 2001).
Smoking is the most important causative factor in the development of COPD. COPD is the culmination of three pathological disorders, chronic bronchitis, small airways disease and emphysema, which can exist separately or in combination. Historically, infiltrating leukocytes were thought to be central to the pathology whilst the epithelium was believed to be merely an observer and target of the damage. However, recent research shows that the epithelium is in fact a rich source of molecules involved in modulating inflammation and lung defense mechanisms.
Airway epithelial structure
The airway epithelium is one of the first targets of cigarette smoke. The cellular profile and function of the airway epithelium adapts to protect the lung from the effects of smoking and the inflammatory response but, in smokers who develop COPD, these changes contribute to irreversible pathological changes in lung structure and function. The complex pathology of COPD involves distinct cellular responses of different regions of the respiratory tract to cigarette smoke which in turn reflects the need for the airways to respond to both acute and chronic exposure.
Large airways
The epithelial layer of the cartilaginous airways comprises a diverse array of pseudostratified columnar cells. The ciliated, serous, Clara and goblet cells are essential to mucociliary defense. In the trachea the ciliated cells predominate, making up as much as 60% of the total cell number (CitationWanner et al 1996) and amongst these, approximately 20% are mucus secreting goblet cells (CitationLozewicz et al 1990). By the 5th generation, ciliated cell numbers fall to approximately 15% and in further generations, ciliated and goblet cell number diminish as they are replaced by serous and Clara cells. The sub-mucosal region of the cartilaginous airways contains glands that are forty times more frequent than goblet cells and release sero-mucus secretions.
Chronic bronchitis is defined as “the presence of chronic cough and recurrent increases in bronchial secretions sufficient to cause expectoration. The secretions are present on most days for a minimum of three months a year, for at least two successive years, and cannot be attributed to other pulmonary or cardiac causes” (CitationSiafakas et al 1995). Cigarette smoke induces epithelial changes associated with development of bronchitis which may or may not be associated with COPD, which depends on the degree of epithelial inflammation (CitationMaestrelli et al 2001; CitationVestbo and Hogg 2006). There is goblet cell hyperplasia and submucosal gland hypertrophy associated with loss of ciliated epithelial cell numbers and function leading to reduced mucociliary clearance and mucus plug formation. Increased numbers of goblet cells are also found in the small bronchi and bronchioles, where there are normally very few (CitationCosio et al 1980). Under scanning electron microscopy a continuous layer of mucus can be seen covering the airways, unlike in healthy subjects where it is only seen in discrete patches (CitationJeffery 1998).
Small airways
The small airways are a significant site of cigarette smoke-induced pathology. Hogg and colleagues observed that non-cartilaginous airways of 2 millimeters or less in diameter provide only a small part of the total airway resistance, approximately 25%, in healthy patients, whereas in those with COPD it becomes the principal site of increased airway resistance (CitationHogg et al 1968). Thus, in smokers with mild emphysema, despite no change in total airway resistance, there was a four fold increase in peripheral airway resistance. More severe cases of emphysema displayed an increase in total airway resistance due almost entirely to the increase in the peripheral airway component. Even though there may be minimal signs of emphysema, the peripheral airways of smokers exhibit a significantly greater number of bronchioles with a diameter less than 0.4mm (CitationCosio et al 1980). Studies of young smokers (CitationNiewoehner et al 1974) showed denuded epithelium in peripheral bronchioles associated with an increased number of inflammatory cells. In addition to inflammatory cell infiltration (CitationSaetta 1998), metaplasia of goblet cells (CitationSaetta et al 2000) and fibrosis (CitationVlahovic et al 1999) have been described, resulting in increased thickness of walls, which all contribute to increased airways resistance due to airways obstruction. The appearance of (usually absent) goblet cells in the bronchioles is a primary characteristic of this disease, and leads to production of mucus at a site in the lung where there is no mucociliary transport to clear the accumulating secretions. This increase in mucus provides a prime site for chronic bacterial colonization (CitationSethi 2000), further aggravating the condition.
Respiratory units
The alveolar airspaces form a complex network of alveolar ducts tightly associated with a rich capillary network. The alveolar epithelial layer consists of two cell types, the type 1 (AT1) and type 2 (AT2) epithelial cells. These cells form tight junctions and lie on a thin fused basement membrane that is shared with, and separates them from, the capillary endothelium. The AT2 cell makes up the majority of the alveolar epithelial cells by number, but only a fraction of the surface area, as only a small projection on the apical side of the cell is apparent between the attenuated AT1 cells. Thus, although there are twice as many AT2 cells, the AT1 cells cover over 90% of the alveolar surface.
AT2 cells are the secretory cells of the alveolus, where they synthesize and secrete surfactant (CitationFrerking et al 2001) to maintain surface tension and prevent alveolar collapse, as well as producing defense molecules such as collectins and antioxidants. The AT2 cell is also a stem cell. Following damage to the alveolar epithelium, AT2 cells proliferate and migrate into the damaged area where they can differentiate into AT1 cells and restore the tight barrier to protect the interstitium and capillary bed from environmental materials (CitationKim et al 1997). AT1 cells are highly attenuated, flattened epithelial cells which provide a large surface area with a thin gas-permeable barrier of approximately 0.2μm in depth to allow rapid, free gas exchange between the alveolar airspace and the underlying capillary network. Relatively little is known about AT1 cells (especially human) as it is only recently that methods have been established to isolate these cells and identify specific AT1 cell markers so that these cells can be recognised and investigated in vivo and in vitro (CitationMcElroy and Kasper 2004). AT1 cells have been described as terminally differentiated, but as their turnover rate is 40–120 days it is difficult to observe or monitor AT1 cell division (CitationWilliams 2003), and in vitro studies suggest that AT1 cells may sometimes revert to the AT2 cell phenotype, although there is as yet no evidence that this occurs in vivo (CitationBorok et al 1998).
It is at this site where the third component of COPD, emphysema, occurs. It is a destructive disease described as ”a loss of lung elasticity and abnormal enlargement of the air spaces distal to the terminal bronchioles, with destruction of the alveolar walls and capillary beds” (CitationPorth 2005). An early event is the destruction of the elastic fibers that provide the natural recoil of the lung, leading to deformation of the alveoli and respiratory bronchioles (CitationSaetta et al 1985). This in turn results in a decreased number of functional alveoli and leads to one symptom that is often a strong indicator of emphysema, shortness of breath. Two subtypes of emphysema exist. The first, centriacinar emphysema, occurs in smokers and is confined to the apical regions of the lung. It is characterized by destruction of the central zone of the acinus leading to enlarged respiratory bronchioles. The second, panacinar emphysema, involves destruction of the whole of the acinus almost uniformly and tends to affect the lower lobes of the lung more than the upper lobes (CitationSnider 1992). Panacinar emphysema occurs in smokers with alpha 1-antitrypsin deficiency but is also present in smokers with normal alpha 1-antitrypsin levels and advanced emphysema.
Epithelial antioxidants
Non-enzymatic antioxidants
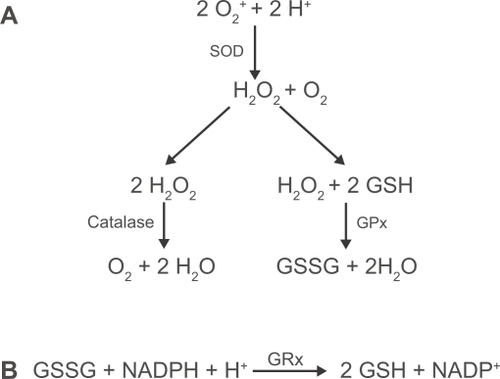
The lung employs a number of enzymatic and non-enzymatic antioxidant mechanisms to neutralize the deleterious effects of ROS. Numerous non-enzymatic antioxidants are present in the lung lining fluid, including uric acid, ascorbic acid, glutathione and α-tocopherol. Of these, glutathione (GSH) is synthesized locally in the lungs, is critical to the protection of the airspaces and the intracellular compartment of the airway epithelium and is found in increased amounts in secretions from smokers (CitationCantin et al 1987). It is a tripeptide consisting of glutamate, cysteine and glycine and contains a thiol group which is able to neutralize oxidative activity by acting as a target for ROS causing oxidation of the thiol group and dimerisation to form GSSG. This dimer can then be reduced back to active GSH units by the activity of glutathione reductase (), which is also found both extracellularly, in lung lining liquid, and intracellularly. GSH is synthesized and released by pulmonary epithelial cells and macrophages and the increased levels of GSH in smokers’ lung secretions may be due to smoking-associated increases in glutamate cysteine ligase, a rate-limiting enzyme crucial to cellular GSH synthesis (CitationNeurohr et al 2003), and hence subsequent release. In vitro studies of the A549 adenocarcinoma cell line and in vivo studies in rats have shown however, that acute exposure to cigarette smoke is associated with a decrease in GSH (CitationRahman et al 1995). This work has been confirmed in other species where it was hypothesized that the decrease in glutathione levels may be due to decreased synthesis or conjugation with electrophilic cigarette smoke components (CitationJoshi et al 1988). Why there is a disparity between the effect of acute and chronic exposure to cigarette smoke is not fully understood. Increased extracellular GSH in chronic smokers and COPD subjects may reflect active release of intracellular GSH or release due to increased cell permeability and/or cell death. Since epithelial cells synthesize and release significant quantities of GSH, they are an important source of GSH, playing a role in antioxidant defense against cigarette smoke.
Figure 1 (A) Enzymatic clearance of reactive oxygen species. Superoxide anions undergo dismutation by superoxide dismutase (SOD) leading to the generation of hydrogen peroxide. This in turn is processed by catalase and glutathione peroxidase (GPx). (B) Reduced glutathione (GSH) is regenerated from its oxidized form (GSSG), using NADPH, by glutathione reductase (GRx).
Enzymatic antioxidants
In addition to GSH, the respiratory epithelium produces a number of enzymes with powerful antioxidant activity, including the superoxide dismutases (SOD), catalase and enzymes associated with glutathione regulation (CitationBowler, Barnes, and Crapo 2004). Of the superoxide dismutases, copper zinc SOD (CuZnSOD) and extracellular SOD (ECSOD) are expressed throughout the respiratory tract epithelium, whereas manganese SOD (MnSOD) is mainly expressed by the AT2 cells (CitationDobashi et al 1993). This class of enzyme is the only one to have activity against superoxide radicals () produced by activated leukocytes and studies in humans and animals have shown that SOD expression is sensitive to cigarette smoke exposure. Studies in rats have revealed that MnSOD expression is rapidly and transiently up-regulated following exposure to cigarette smoke (CitationStringer et al 2004). Its expression is also found to be up-regulated by 30%–50% in the lungs of smokers, in particular in the AT2 cell (CitationHarju et al 2004), suggesting an important role combating the oxidative effects of cigarette smoke.
Catalase, glutathione peroxidase (GPx), glutathione reductase and glutamate cysteine ligase are all important enzymes in the clearance of hydrogen peroxide () and lipid peroxides from the lung and are found to be expressed throughout the respiratory tract epithelium (CitationHackett et al 2003). Catalase is most highly expressed in AT2 cells and is widely considered to be the most important enzyme in the clearance of hydrogen peroxide (CitationFarioli-Vecchioli et al 2001). Studies of human lung tissue have shown that, in the COPD lung, mRNA expression of catalase is significantly reduced compared to non-smokers’ lung (Tomaki et al). Of the enzymes involved in glutathione regulation, GPx has been the most actively researched. However, how cigarette smoke affects GPx remains unclear. Studies of human and animal tissue have shown that the primary sites of GPx production are the epithelium and alveolar macrophage (CitationAvissar et al 1996; CitationComhair et al 2001). Studies using macrophages and epithelial cells obtained by BAL and bronchial brushings from humans have shown that smokers have increased levels of mRNA for GPx (CitationComhair et al 2001; CitationHackett et al 2003). Conversely, in gene micro-array studies of lung tissue from subjects with advanced emphysema, GPx expression was found to be down-regulated (CitationGolpon et al 2004). This difference in results may reflect alterations occurring in the lung tissue structure and cell profile during disease progression where GPx is important for maintaining antioxidant status but where loss of epithelial structure and/or function, eg during emphysema, crucially limits GPx.
Thus, the pulmonary epithelium is a significant source of pulmonary antioxidants, which may be stimulated or inhibited, acutely or chronically, depending on the smoking status, history and lung pathology of each individual. It seems likely that, in susceptible individuals, antioxidant defenses may initially be effective but may ultimately be overwhelmed leading to oxidative stress-induced inflammation and tissue damage.
Oxidative stress
Exogenous sources
The lung is constantly exposed to oxidants which may either be produced endogenously, by metabolic reactions, or derived exogenously, for example, in air pollutants and cigarette smoke. This, coupled with the high oxygen environment and large surface area of the lung, means the lung could be highly susceptible to injury by reactive oxygen species (ROS). It is estimated that there are more than 1014 oxidative free radicals per puff of cigarette smoke (CitationChurch and Pryor 1985). As well as the more common short-lived oxidants such as the superoxide radical (O2•−) and nitric oxide (NO), there a number of oxidative molecules which can persist in the lung for longer periods of time such as tar-semiquinone (CitationPryor and Stone 2007). This oxidant can generate the highly potent hydroxyl radical (•OH) and hydrogen peroxide in the presence of iron via the Fenton reaction. This is particularly important in the smoker’s lung, where there are also elevated levels of free iron (CitationMateos et al 1998).
Cellular sources
In addition to exogenous sources of oxidative stress, activated macrophages and neutrophils are known to release high levels of reactive oxygen species, in particular, O2• −, hydrogen peroxide and •OH (CitationBowler et al 2004). This, coupled with the greater numbers of these cells in the lungs of smokers, further compounds the situation. In vivo studies have shown that cigarette smoking induces a transient increase in neutrophil sequestration in the lungs (Citationvan der Vaart et al 2005). Further in vitro studies suggest that this increase in neutrophil numbers may be, at least in part, oxidant-dependent as cigarette smoke-induced oxidative stress in neutrophils leads to polymerization of F actin filaments in the cytosol which lead to a decrease in the deformability of the cells (CitationDrost et al 1992), which consequently become trapped in the capillaries prior to migrating into the airway lumen. In addition, epithelial cells, as well as leukocytes, may generate extracellular ROS via NADPH oxidase. Such ROS can play an important role in gene transcription.
Role of oxidants in gene transcription
The airway epithelium is one of the first cellular targets of cigarette smoke. ROS cause lipid peroxidation, which can impair cellular function in a number of ways including disruption of the cellular membrane and inactivation of membrane-bound receptors and enzymes (CitationRahman 2005). A number of products of oxidative stress, including those of lipid peroxidation, have been shown to inactivate histone deacetylase-2, an enzyme which is integral to the regulation of pro-inflammatory gene transcription (CitationRahman et al 2004). HDACs are a family of enzymes which, along with histone acetylases, control gene transcription. Acetylation of lysine residues on histones neutralizes their charge, causing the chromatin to unwind and thus increase accessibility for transcription factors and subsequent up-regulation of gene transcription. Conversely, deacetylation of lysine residues on the histone causes conformational changes which allow the chromatin structure to close and become inaccessible to transcription factors (CitationBarnes et al 2005) thus preventing gene transcription. One of the anti-inflammatory mechanisms of corticosteroids is to up-regulate HDAC activity which in turn down regulates transcription of pro-inflammatory mediators. HDACs may be particularly relevant to COPD as oxidative inactivation of HDAC-2 results in steroid resistance, a phenomenon well recognised in COPD which has made treating the disease extremely difficult. The activity of HDACs −2, −3, −5 and −8 have all been demonstrated to be reduced in COPD subjects (CitationRahman, Marwick, and Kirkham 2004; CitationIto et al 2005).
Oxidative stress can also activate the redox sensitive transcription factors, nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1). NF-κB was the first transcription factor shown to be regulated by oxidative stress. It has been demonstrated that a variety of stimuli known to activate NF-κB, including interleukin- 1 (IL-1), tumor necrosis factor-α (TNF-α), UV irradiation and LPS, also induce a transient increase in ROS. This increase in ROS leads to NF-κB being released from its inhibitory complex with IκB, allowing it to translocate to the nucleus (CitationSchulze-Osthoff et al 1995) where it triggers expression of inflammatory mediators. In vitro studies using the A549 adenocarcinoma and H1299 cell lines and primary human bronchial epithelial cells have shown that cigarette smoke condensate can induce expression of mRNA for a number of cytokines including, most notably, IL-6 and CXCL8 (IL-8). This, they showed, correlated with a concomitant increase in NF-κB activity (CitationKode et al 2006) as a result of increased oxidative stress. A consequence of cigarette smoke-induced activation of epithelial AP-1 is increased mucin secretion, addressed in more detail later in this review.
Epithelial antiproteases
Serine protease inhibitors – SERPINs
The association of an increased protease burden with development of emphysema was established in the 1960’s when it was shown that a deficiency in alpha 1-antitrypsin, a serpin (SERine Proteinase INhibitor) and the major serum neutrophil elastase inhibitor, was associated with early onset emphysema (CitationLaurell and Eriksson 1963). However, the epithelium is not the major source of pulmonary alpha 1-antitrypsin (a 54KDa protein synthesized in the liver and secreted into the circulation) which is largely serum-derived, diffusing into the lung tissue and airways. Until recently, alpha 1-antitrypsin was the only antiprotease known to be genetically linked to COPD. Interestingly, another serpin, SERPINE2, which codes for a thrombin and urokinase inhibitor, has recently been associated with development of COPD (CitationZhu et al 2007). This 44 kDa protein is expressed by pulmonary epithelium and is up-regulated in emphysema (CitationDemeo et al 2006). The antiprotease has not been shown to inhibit neutrophil elastase and its exact functional role in COPD has not been established; it is possible that its effects are unrelated to antiprotease activity.
Low molecular weight inhibitors of serine proteases
The pulmonary epithelium is a major site of synthesis and release of the low molecular weight serine protease inhibitors, secretory leukocyte protease inhibitor (SLPI; also known as antileukoprotease or mucus proteinase inhibitor) and elafin (also known as elastase-specific inhibitor and skin-derived antileukoprotease, SKALP). All the secretory cells, including goblet cells, serous cells, Clara cells and AT2 cells can synthesize and release SLPI and elafin. SLPI is 11.7kDa; elafin (6kDa) is a cleavage product of pre-elafin (also called trappin-2), which is 9.9kDa. These are both cationic, non-glycosylated, acid stable, serine proteinase inhibitors that are also important in host defense and tissue repair (CitationTetley 2006). Both antiproteases are potent, reversible inhibitors of neutrophil elastase; SLPI also inhibits cathepsin G, trypsin, chymotrypsin and chymase, while elafin also inhibits proteinase-3 (CitationTetley 2006). SLPI is constitutively expressed, released at high levels into the lung lumen and is also found interstitially, bound to extracellular matrix, due to its cationic properties, where it is a very effective inhibitor of matrix-bound neutrophil elastase (unlike alpha 1-antitrypsin). In contrast, epithelial-derived elafin is not normally synthesized at high levels but it can be up-regulated, for example during infection or following corticosteroids, to supplement baseline antiprotease levels. In vitro studies show that elafin is synthesized in response to inflammatory mediators such as cytokines, eg TNF-α, IL-1β (CitationBingle et al 2001; CitationSallenave 2002), as well as lipopolysaccharide (LPS) and, interestingly, elafin is also up-regulated by neutrophil elastase itself (CitationReid et al 1999), which may be a feedback mechanism. SLPI is usually present in lung secretions at higher concentrations than elafin, but is also upregulated by the same mediators.
SLPI is the most abundant serine protease inhibitor in the large and central airway secretions and contributes a significant proportion of the serine protease inhibitory capacity in the respiratory units. Although goblet cell metaplasia and hypersecretion leading to increased levels of SLPI might be expected during bronchitis, there is an inverse relationship between sputum SLPI and increased neutrophil elastase, possibly due to proteolysis of SLPI or re-location to other lung compartments (eg interstitium). Loss of alveolar epithelium in emphysema is also likely to reduce absolute levels of SLPI and elafin. Latent adenoviral infection of bronchial and alveolar epithelial cells and elevated epithelial expression of the adenoviral trans-activating protein (E1A) in the airways of COPD subjects could also lead to reduced SLPI and elafin expression, as shown by in vitro studies of E1A transfected lung epithelial cells where production of SLPI and elafin is significantly reduced (CitationHigashimoto et al 2006).
Tissue inhibitors of metalloproteinases
Another crucial family of antiproteases are the tissue inhibitors of matrix metalloproteases (TIMPs). As their name suggests, this class of inhibitor targets the matrix metalloproteinases (MMPs) that are thought to be a central component of the pathogenesis of COPD, as discussed later. Studies of secretions from human subjects have shown that TIMP-1 is elevated in smokers and COPD subjects (CitationBeeh et al 2003). Further in vitro studies utilizing primary epithelial cells cultured from healthy normal and COPD subjects, and ex vivo studies of tissue from a rat model of COPD, have immunolocalized TIMP-1 to the respiratory epithelium (CitationLi et al 2002, Citation2005) and shown that it is elevated in COPD indicating that, under chronic inflammatory conditions where proteases are elevated, the epithelium may act to regulate localized proteolytic activity in order to protect itself. This may also reflect elevated epithelial MMP production and co-release of TIMP, as described previously (CitationTetley 2005)
Inactivation of antiprotease activity
Oxidation of the methionine residue at the reactive site of alpha 1-antitrypsin, SLPI and elafin interferes with their inhibitory activity (CitationTaggart et al 2000; CitationTetley 2006). Although this is an important “switch” to modulate antiprotease activity as necessary, for example to allow pericellular proteolysis, excessive oxidative inactivation of pulmonary serine protease inhibitors due to cigarette smoke exposure or via release of high levels of cellular oxidative free radicals could result in a reduced antiprotease screen and increased proteolytic load, rendering the lung susceptible to the consequences of excessive proteolytic activity, which triggers tissue injury and exacerbates inflammation (CitationTetley 2006). Oxidative stress may also play an important role in modulating the activity of TIMPs in the lung. In vitro studies utilizing recombinant TIMP-1 have shown that peroxynitrite (ONOO−), an oxidant generated by the interaction of superoxide (O2 .-) with nitric oxide (NO), is able to inactivate TIMP-1 in a dose dependent manner (CitationFrears et al 1996), possibly through disruption of the disulphide bonds that hold TIMP-1 in its complex tertiary structure. Similar studies have also shown that products of the nitric oxide pathway can degrade TIMP-1 activity in cultures of stromal fibroblasts (CitationBrown et al 2004).
In addition to oxidative regulation of antiproteases, numerous studies have shown that proteases themselves may be able to cross-regulate inhibitors of other classes of protease (CitationTetley 2005). Okada and colleagues have shown that serine proteases are able to degrade TIMP-1, resulting in inactivation of their antiprotease activity (CitationOkada et al 1988), whilst MMPs and cysteine proteases, such as the cathepsins, have been shown to degrade and inactivate serine protease inhibitors (CitationJohnson et al 1986; CitationDesrochers et al 1991; CitationLiu et al 2000; CitationTaggart et al 2001; CitationNie and Pei 2004).
Proteases
There is a considerable amount of evidence that infiltrating phagocytic leukocytes, namely neutrophils and macrophages, contribute to increased proteolytic load in the lungs of smokers and both these cell types secrete a broad spectrum of enzymes, including serine proteases (neutrophil elastase, proteinase 3, cathepsin G) (CitationTetley 2005; CitationPham 2006), cysteine proteases (Cathepsins B, H, L, K and S) (CitationChapman et al 1997; CitationTetley 2005), and a range of MMPs, into the lungs of smokers (CitationRussell et al 2002; CitationTetley 2002; CitationVernooy et al 2004). Evidence that epithelial-derived proteases might contribute to the increased proteolytic load is more recent.
Matrix metalloproteinases in COPD and inflammation
We now know that the pulmonary epithelium is a rich source of MMPs. These are perhaps the largest family of proteases implicated in the pathogenesis of COPD with over 25 members identified. Collectively, this family of enzymes is able to degrade all components of the extracellular matrix and was originally broadly classified on the basis of substrate specificity. Thus, MMP-1, −8 and −13 are collagenolytic, MMP-2 and −9 are gelatinolytic, MMP-3, −10 and −11 are stromelysins, while MMP-7 and −12 are elastinolytic. In addition there are membrane type MMP’s (MT-MMPs, MMP-14, −15, −16 and −17) which are anchored to the surface membrane of cells which can orchestrate the activity of other MMPs, for example MMP-14 cleaves, and activates, latent pro-MMP-2 in the vicinity of the cell.
Immunohistochemical studies have shown that MMP-1 and MMP-2 are up-regulated in the AT2 epithelial cells of COPD subjects and in vitro studies suggest that this increase in expression may be due to cigarette smoke activating mitogen-activated protein kinases (MAPK) and subsequent up-regulation of the MMP gene promoter region (CitationSegura-Valdez et al 2000; CitationMercer et al 2006). Furthermore, studies using both primary and immortal human bronchial epithelial cells have shown that cigarette smoke exposure stimulates MMP-9 and MMP-12 expression (CitationHan et al 2003; CitationLavigne and Eppihimer 2005). Increased epithelial cell MMP expression following cigarette smoke exposure may reflect the known role of MMPs in repair and remodeling; clearly very important in an acute situation but, in a chronic situation, increased epithelial MMP synthesis and release is likely to contribute to tissue pathology. Another sub-group of MMPs synthesized by pulmonary epithelial cells, known as the ADAMs (A Disintegrin and Metalloproteinase) family, have recently emerged and are being implicated in a number of diseases. Recent studies showing an association of ADAM 33 with bronchial hyper-reactivity in asthmatics (CitationVan et al 2002) have been extended to subjects with COPD where there is a close linkage between this molecule and airway inflammation in the lungs of COPD subject (CitationGosman et al 2007). The exact role of this family of metalloproteinases, however, is yet to be fully understood.
MMPs can also regulate cytokine and chemokine activity. MMP-2 and MMP-9 have both pro- and anti-inflammatory effects. For example, considering chemokines (classified below), MMP-2 can generate a truncated anti-inflammatory form of CCL7 (MCP-3) from the pro-inflammatory form, while MMP-9 can inactivate CXCL1 (GRO-α). Conversely, MMP-9 can generate a truncated form of CXCL8 that is ten times more active than the parent molecule (CitationVan den Steen et al 2000). Cytokines that are proteolytically processed and activated by MMP-9 include membrane-bound TNF-α (CitationGearing et al 1994) and transforming growth factor-β (TGF-β) (CitationYu and Stamenkovic 2000). Furthermore both MMP-2 and MMP-9 proteolytically activate latent, pro-IL-1β (CitationSchonbeck et al 1998). MMP-1 inactivation of the chemokines, CCL2, −8 and −13 (MCP-1, −2 and −4) generates cleaved forms that bind to the MCP receptors CCR-2 and −3 without eliciting leukocyte migration, thereby acting as endogenous inhibitors (CitationMcQuibban et al 2002).
Epithelial cathepsins
Epithelial cells from the bronchial and alveolar regions also secrete cathepsins K, L and H (CitationBuhling et al 2004). This class of proteases has been shown to be potent in its ability to degrade collagen and elastin (CitationCardozo et al 1992; CitationNovinec et al 2007), as well as inactivating SLPI and α1-antitrypsin as described above. Very little is known of the role of epithelial cell-derived cathepsins in smoking. However, macrophages from smokers demonstrate elevated levels of a number of cathepsins including cathepsins K, L and H (CitationReilly et al 1991; CitationTakahashi et al 1993). It is therefore possible that cigarette smoke also modulates the synthesis and release of epithelial cathepsins, but to our knowledge this has not yet been described.
Epithelial integrity and repair
An important event in the development of emphysema is loss of the alveolar structure leading to enlarged alveoli and reduced surface area. In small airways disease there is also a significant amount of remodeling. The initial damage is likely to be a combination of factors, including the direct effects of cigarette smoke as well as the effects of locally produced mediators. Whilst detrimental in itself, disruption of the epithelial layer without adequate repair will enhance the likelihood of exposure of the extracellular matrix and interstitial cells to external agents eg cigarette smoke, particles, micro-organisms as well as locally produced mediators such as oxidants, cytokines, chemokines and proteases described above which may have detrimental effects on normal repair processes.
Cell survival and death
Research into the exact mechanism by which cigarette smoke causes loss of the epithelial layer has generated a number of hypotheses. Exposure of human bronchial epithelial cells to cigarette smoke in vitro caused DNA damage but also stimulated synthesis of the DNA repair enzyme poly (ADP-ribose) polymerase (PARP). Thus, cells were able to recover from cigarette smoke-induced DNA damage and continued to proliferate with no signs of apoptosis or necrosis (CitationLiu et al 2005); thus a defect in this repair mechanism may contribute to epithelial damage in COPD. In contrast, other studies have shown that exposure of the A549 adenocarcinoma cell line to cigarette smoke can cause both apoptosis and necrosis. At low doses, cigarette smoke appeared to induce apoptosis via a caspase-independent pathway whereas at higher doses necrosis was initiated. Instigation of the apoptotic pathway was dependent on components of the volatile phase of the cigarette smoke and could be attenuated by antioxidants and aldehyde scavengers (CitationHoshino et al 2001) supporting a mechanistic role for oxidants. Similarly, another study of A549 cells showed that higher doses of cigarette smoke induced necrosis and in addition prevented apoptosis by inhibiting caspase-3 activation. However, in contrast to the previous study, there was no apoptosis detected at lower concentrations of cigarette smoke (CitationWickenden et al 2003). Other in vivo studies and studies using primary alveolar epithelial cells in vitro show that cigarette smoke induces senescence, a state of irreversible growth arrest (CitationTsuji et al 2004), a feature akin to the ageing process. Further studies by the same group confirmed that epithelial senescence is an element of emphysema following investigation of lung tissue obtained from patients with emphysema, where they showed alveolar epithelial cell accumulation of senescence-associated cyclin-dependent kinase inhibitors, p16INK4a and p21CIP1/WAF1/Sdi1 (CitationTsuji et al 2006). These studies suggest that there is a fine balance between the amount/concentration of cigarette smoke exposure and induction of cell death, which may also depend on the epithelial phenotype. Furthermore, alveolar epithelial cell senescence may contribute to the loss of tissue structure and function in emphysema.
Epithelial layer permeability
Another mechanism by which cigarette smoke can disrupt the integrity of the epithelial layer is by disrupting the tight junctions which tether cells together to form an impermeable barrier. These tight junctions, consisting of strands of claudin and occludin, are positioned near the apical surface and form a belt around the cell. Studies using gut epithelium have shown that the phosphorylation state of the tight junction proteins drastically affects their ability to maintain a tight barrier. Phosphorylation of occludin serine/threonine residues has been shown to increase tight junction integrity (CitationSakakibara 1997) whereas as phosphorylation of tyrosine residues is associated with increased permeability (CitationWard 2002). In relation to the lung, Olivera and colleagues have investigated the effect of cigarette smoke on an airway cell line in vitro (CitationOlivera et al 2007). In these studies it has been shown that, following exposure to cigarette smoke, there is a transient decrease in transepithelial resistance associated with increased macromolecular permeability. These changes in tight junction integrity were dependent on the activity of Rho kinase and protein tyrosine kinases, indicating that cigarette smoke affects airway permeability in a regulated manner and is not a purely cytotoxic response. In addition, Li and colleagues have demonstrated a role for antioxidants in regulation of epithelial cell permeability (CitationLi et al 1994, Citation1996). Following instillation of cigarette smoke condensate in to rat lungs it was shown that increased epithelial cell permeability was associated with a concomitant decrease in glutathione levels. Further in vitro studies showed that the increase in epithelial permeability could be reversed by the addition of glutathione to the growth media, confirming that oxidative mechanisms play a role in modulating permeability.
Role of vascular endothelial growth factor in epithelial repair
In addition to triggering cell death and increasing epithelial layer permeability, cigarette smoke interferes with cell repair mechanisms, further compounding its direct contribution to tissue damage during progression of COPD. In vitro studies of primary human and bovine bronchial epithelial cells showed that cigarette smoke extract inhibits a number of repair processes, including chemotaxis, proliferation and extracellular matrix production, and this was shown to be dependent on the volatile components of the cigarette smoke (CitationLannan et al 1994; CitationWang et al 2001). One mediator that may be central to epithelial repair in smokers is vascular endothelial growth factor (VEGF). As its name suggests this growth factor was once thought to be involved primarily in vascular growth. However, more recent studies have discovered that VEGF is expressed by a number of non-vascular cells and has properties that are important in tissue homeostasis (CitationVoelkel et al 2006). In vitro studies using cell lines representing the bronchial and alveolar epithelium have shown that VEGF expression is increased in response to cigarette smoke exposure (CitationKoyama et al 2002), while in studies comparing VEGF expression levels in COPD subjects and non-smokers, bronchiolar and alveolar epithelial VEGF expression was found to be significantly higher in COPD subjects compared to non-smokers (CitationKranenburg et al 2005) and this was inversely related to FEV1. Increased levels of epithelial VEGF in COPD lungs may simply reflect the up-regulatory effects of cigarette smoke but may also be an attempt to repair damage to the epithelium and sub-epithelium. However, and in contrast, in other studies of severely emphysematous tissue, disease severity was associated with a concomitant decrease in VEGF expression in the endothelial cells in the alveolar region. This decreased expression was also associated with reduced epithelial VEGF and increased alveolar epithelial cell apoptosis (CitationKasahara et al 2001), possibly as a direct result of a reduction in the anti-apoptotic effects of VEGF. The latter study is supported by experimental animal investigations of genetic deletion of VEGF receptors-1 and −2 (CitationTang et al 2004), where the animals developed emphysema, suggesting that VEGF is important in maintaining alveolar structure (CitationKasahara et al 2001). Decreased levels of VEGF in emphysematous regions of human lung associated with apoptosis may be as a result of the cigarette smoke overcoming the body’s defense mechanisms leading to loss of the very cells that produce VEGF. As well as being a pro-survival factor for the alveolar unit, VEGF may also play an important role in the clearance of apoptotic cells. Alveolar macrophages are known to express VEGF receptors and it has been shown that blockade of these receptors drastically reduces the ability of the macrophages to phagocytose apoptotic cells (CitationVoelkel et al 2006). Reduced VEGF may explain why there are increased numbers of apoptotic cells in the sputum of COPD subjects. Impaired clearance of these cells may further aggravate the situation, as apoptosis itself has been shown to stimulate VEGF release (CitationGolpon et al 2004). Thus the exact role of epithelial cell-derived VEGF in COPD is unclear, although a significant body of evidence suggests that reduced VEGF contributes to development of emphysema (CitationVoelkel et al 2006). It seems likely that epithelial cell-derived VEGF exhibits differential activity at different levels of the respiratory tract and at different stages of the disease process.
Bacterial triggers of epithelial responses in COPD
Sources of bacterial products
A number of studies have shown that, in addition to the potential inflammatory stimulus of the oxidative components of cigarette smoke, it is a source of bacterial products, notably, endotoxin. Approximately 1% of the total endotoxin content of a cigarette can survive combustion and be detected in smoke generated from a lit cigarette. Despite being such a low proportion of the total endotoxin present in the unlit cigarette it has been concluded that the smoke inhaled from smoking one pack of cigarettes contains as much active endotoxin as that found to cause adverse health effects in cotton textile workers (CitationHasday et al 1999). It is not only the filtered, inhaled smoke that may have a deleterious effect on the health of smokers. The quality of indoor air from a room in which cigarettes have been smoked contains 120-fold more endotoxin compared to a room in which no cigarettes have been smoked (CitationLarsson 2004). In addition, it is estimated that approximately 50% of all subjects with COPD have a persistent colonization of the lower respiratory tract by bacteria such as Streptococcus pneumoniae, Haemophilus infuenzae and Moraxella catarhallis (CitationZalacain et al 1999), which are often associated with periods of exacerbation leading to an accelerated decline in lung function.
Role of Toll-like receptor-4 activation
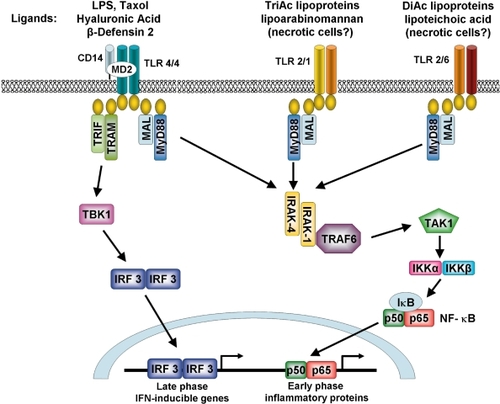
LPS exerts its effect through Toll-like receptor-4 (TLR-4) a member of a superfamily of pattern recognition receptors (PRR). PRRs are activated by specific pathogen-associated molecular patterns (PAMP); thus, activation of TLR-4 and subsequent downstream signaling ultimately leads to transcription of a number of genes involved in host defense (). Amongst these are genes for chemokines, cytokines and major histocompatibility complex (MHC). TLRs, whilst being selective for their ligand of activation, share a number of similar intracellular signaling pathways, as well as having specific pathways of their own. Signaling of these pathways leads to activation of the transcription factors NF-κB and AP-1, via phosphatidylinositol 3-kinase (PI3-K) and MAPKs (CitationAkira and Takeda 2004). TLR4 has been located to bronchial epithelium from a number of species (CitationWassef et al 2004) including healthy human lung, being reduced in cystic fibrosis (CitationHauber et al 2005). LPS induces expression of TLR4 in bronchial epithelium of rat lung (CitationJanardhan et al 2006) and TLR4 mRNA expression is increased in the bronchial epithelium from horses with airflow obstruction (CitationBerndt et al 2007). To our knowledge there are as yet no similar studies of human lung tissue.
Figure 2 Signaling pathways initiated by TLR-2 and TLR-4 activation. Upon ligation of the TLR-4 receptor complex by exogenous (eg, LPS) or endogenous (eg, hyaluronic acid and β-defensin 2) ligands, MyD88-dependent and MyD88-independent signaling pathways are initiated leading to activation of the transcription factors NF-κB and IRF3. TLR-2 heterodimerises with either TLR-1 or -6 in order to recognise distinct groups of exogenous ligands and signal through a MyD88-dependent pathway. TLR-1/2 recognizes triacyalted (TriAc) lipoproteins and lipoarabinomannan whereas TLR-2/6 recognizes diacylated (DiAc) lipoproteins, lipoteichoic acid and zymosan. TLR-2 may also initiate responses to endogenous products released by necrotic cells.
Previous studies by us demonstrated human AT2 epithelial cell expression of TLR-4 (CitationArmstrong et al 2004). Following treatment with LPS, AT2 cells released a broad spectrum of cytokines and chemokines including IL-1β, TNF-α, IL-6, CCL2, CXCL8, CXCL1 and CCL20 (MIP-3α) (CitationThorley et al 2005, Citation2007). In addition, release of chemokines from AT2 cells was far greater than that observed by alveolar macrophages from the same subjects under the same conditions of LPS exposure. The possible central role of TLR4 activation of AT2 cells in alveolar chemokine release has been further supported by in vivo studies in mice which found that, following LPS, the AT2 cell is a predominant source of neutrophil chemoattractant chemokines, dependent upon TLR4 signaling mechanisms (CitationJeyaseelan et al 2005).
Newly emerging studies are showing that TLRs may also be able to respond to exogenous stimuli other than those from microbial sources such as cigarette smoke as well as endogenous ligands generated during inflammation, such as necrotic cells, heat shock proteins and β-defensins (CitationTobias and Curtiss 2005). A recent study using human monocyte derived macrophages showed that cigarette smoke exposure stimulated a number of cytokines, including CXCL8. This was mediated via TLR-4 and was independent of any LPS there may have been in the cigarette smoke (CitationKarimi et al 2006). In addition, animal studies have shown that TLR-4 defective mice have an impaired response to cigarette smoke exposure, with significantly less infiltrating leukocytes observed and lower levels of cytokines and chemokines in the BAL (CitationMaes et al 2006). In addition, loss of TLR4 expression (TLR4 deficiency) causes emphysema over time in mice; this was ascribed to increased NADPH oxidase activity leading to oxidative stress and increased elastolytic activity (CitationZhang et al 2006). These studies suggest that in COPD, TLR4 signaling is important in the inflammatory process and abnormalities may contribute to loss of lung structural integrity.
Role of Toll-like receptor-2 activation
In addition to expressing TLR-4, studies by us (CitationArmstrong et al 2004) and preliminary unpublished research carried out in our laboratory have shown that human AT2 epithelial cells express mRNA for TLR-2 and respond to treatment with TLR-2 specific ligands by releasing a broad spectrum of cytokines and chemokines. TLR-2 recognises a number of microbial products such as peptidoglycan from Gram-positive bacteria, bacterial lipoproteins, lipoarabinomannan from mycobacteria and yeast cell walls. Its broad range of ligand specificity may be accountable to its ability to heterodimerise with two other TLRs, TLR-1 (CitationWyllie et al 2000) and TLR-6 (CitationTakeuchi et al 2001) (). This group of TLRs may play a critical role in the inflammatory response of the COPD lung as one of the most common infections in COPD subjects is Moraxella Catarrhalis, a Gram-positive bacteria. It is also possible that TLR-2-active ligands are present in tobacco smoke in a similar fashion to LPS; however, no studies have as yet addressed this. Investigations of a variety of epithelial cells, both primary and immortal, from the bronchial and alveolar regions have shown that TLR-2 is expressed throughout the respiratory tract (CitationDroemann et al 2003; CitationMayer et al 2007; CitationSlevogt et al 2007) and is rapidly up-regulated following exposure to microbial products (CitationSaito et al 2005). Following stimulation with TLR-2 ligands, airway epithelial cells also release a similar profile of mediators to those discussed earlier with respect to LPS (CitationHertz et al 2003; CitationRitter et al 2005; CitationSchmeck et al 2006). This may be explained by further studies that have shown that, whilst the accessory proteins associated with the two classes of receptor may differ, ultimately, they activate the same transcription factors (CitationTrinchieri and Sher 2007). Preliminary work by us shows that human alveolar epithelial cells express a range of TLRs. The role of TLRs in the pathogenesis of COPD is an active area of research and the precise role, if any, of each TLR remains to be established.
Cytokines and chemokines
Interleukin-1β and tumor necrosis factor-α
As mentioned above, activation of PRRs initiates synthesis and release of a variety of mediators including cytokines and chemokines. There is now good evidence that these receptors can also be activated by endogenous and other ligands such as β-defensin-2, heat shock proteins, components of hyaluronic acid and fibrinogen, heparin sulfate and oxidative stress (CitationTobias and Curtiss 2005); such processes could be highly relevant in COPD, which develops in an environment of continual exposure to oxidants and tissue remodeling, and these factors may contribute to the continued cycle of inflammation that takes place even after cessation from smoking. Furthermore, once released, mediators such as cytokines and chemokines can perpetuate the inflammatory response.
A number of cytokines and chemokines have been implicated in the pathogenesis of COPD (.). Amongst the most actively investigated cytokines are the early response cytokines IL-1β and TNF-α. Studies of sputum and cultures of primary bronchial epithelial cells from COPD subjects have shown that both of these cytokines are present in high concentrations compared to non-smokers (CitationKeatings et al 1996), (CitationRusznak et al 2000). The interleukin-1 (IL-1) superfamily consists of two agonists, IL-1α and β, and one antagonist, IL-1 receptor antagonist (IL-1Ra). IL-1α is predominantly found tethered to the cell surface whereas as IL-1β is found in a soluble secreted form. Both isotypes are secreted by a wide variety of cells, including pulmonary epithelial cells, and exert their actions through the same IL-1 type I receptor to elicit similar responses (CitationDinarello 1998). TNF-α exists in a transmembrane form that is activated into its soluble form by tumor necrosis factor (TNF)-converting enzyme (TACE). Evidence suggests that TNF-α is a crucial mediator of the pathogenesis of COPD (CitationChurg et al 2002, Citation2003). Like IL-1, it is produced by a wide variety of lung cells (CitationZiegenhagen et al 1997; CitationNoda et al 2003; CitationProfita et al 2003) in response to a broad spectrum of insults (CitationDriscoll et al 1995; CitationChurg et al 2002; CitationErmert et al 2003). In the case of infection/exacerbation it is believed that the pro-inflammatory response is not only as a result of the direct activity of endotoxin itself but also of the primary cytokines that are subsequently released. Thus, LPS-induction of primary human AT2 cell release of CCL-2 over 24 hours was dependent on the autocrine effects of LPS-induced IL-1 and TNF-α release (CitationThorley et al 2007). Exposure of primary human bronchial epithelial cells from healthy non-smokers, healthy smokers and smokers with COPD to gaseous cigarette smoke caused increased IL-1 release from COPD cells compared to healthy smokers’ cells; the cigarette smoke also induced a marked reduction in cellular GSH, which was suggested to exacerbate IL-1 release (CitationRusznak et al 2000). Furthermore, cigarette smoke extract stimulated both IL-1 and TNF-α secretion by primary human small airway epithelial cells, at least partly via induction of the early growth response gene (Egr-1), a hypoxia-sensitive gene that is highly expressed in the lungs of smokers with COPD (CitationReynolds et al 2006). As these cytokines have marked effects on all inflammatory and structural lung cells, stimulating production of a myriad of mediators, release of these cytokines by pulmonary epithelium, one of the first cellular targets, following induction by cigarette smoke is likely to be an important contributory mechanism to the development of COPD in susceptible subjects.
Table 1 Cytokines and chemokines implicated in the pathogenesis of COPD
Interleukin-6
IL-6 has also been shown to be elevated in the secretions of COPD subjects although its role in COPD remains unclear due to its pleiotropic effects. A variety of cells including alveolar epithelial cells, alveolar macrophages, fibroblasts and endothelial cells are capable of producing IL-6 (CitationSad et al 1995; CitationKoyama et al 1998; CitationFrampton et al 1999; CitationArcangeli et al 2001; CitationYu et al 2002). Whilst the pro-inflammatory effects of IL-6 are widely known, its anti-inflammatory activity is often disregarded. Although it induces expression of many pro-inflammatory mediators in a similar way to TNF-α and IL-1, it does not up-regulate mediators of inflammation such as the prostaglandins and matrix metalloproteinases. Furthermore, LPS-induced IL-6 can inhibit the release of IL-1 and TNF-α, following LPS exposure, by autocrine, feed back mechanisms. It is suggested that IL-6 has a modulatory mechanism that dampens the acute-phase response, preventing development of a chronic inflammatory state (CitationBarton 1997). Bronchiolar and alveolar epithelial cell division/repair was reduced in cigarette smoke exposed IL-6 knock out mice compared to WT controls, suggesting that IL-6 is important in the repair of epithelial damage following acute injury. Interestingly, exposure of human small airway epithelial cells to cigarette smoke induces IL-6 release (CitationKode et al 2006) and is important in preventing DNA damage and cell death on exposure to low levels of cigarette smoke (CitationYu et al 2002). The evidence suggests that this is due to the autocrine activity of IL-6 which activates signal transducer and activator of transcription 3 (STAT3), a transcription factor that has been shown to be important in cell proliferation, differentiation, apoptosis, inflammation and wound repair. Thus cigarette smoke-induced IL-6 release by epithelial cells may be a significant anti-inflammatory and repair mechanism.
Chemokines: monocyte and neutrophil recruitment
Recruitment of different classes of leukocytes is controlled by distinct groups of chemokines. CXC chemokines containing the ELR amino acid motif, such as CXCL8 and CXCL1, are traditionally considered to be the primary chemokines responsible for recruitment of neutrophils, although there is evidence that they also have chemotactic activity for monocytic cells. The CC chemokines, such as CCL2, CCL4 (Macrophage Inflammatory Protein-1β; MIP-1β) and CCL5 (Regulated Upon Activation Normal T cells Expressed and Secreted; RANTES) are also elevated following infection and during pulmonary inflammatory disorders and are key to the recruitment of mononuclear cells. Both CXC and CC chemokines are elevated in COPD (CitationBarnes 2004). In studies of induced sputum from COPD subjects, CCL2 and CXCL1 levels were found to be elevated (CitationTraves et al 2002) as is CXCL8 (CitationPesci et al 1998), which is further elevated during periods of exacerbation (CitationAaron et al 2001). These chemokines are likely to be derived from a number of cellular sources and there has been much emphasis on the macrophage in this process. However, examination of human lung tissue highlights the epithelium as a significant source. For example, de Boer and colleagues discovered that CCL2 and CXCL8 mRNA and protein expression was significantly higher in bronchiolar epithelium from ex-smokers with COPD compared to ex-smokers without COPD (Citationde Boer et al 2000). Subsequent studies (CitationFuke et al 2004) using laser-capture microdissection techniques confirmed this finding with CXCL8 and CCL2 and, in addition, showed that CCL3 (MIP-1α) mRNA expression was higher in bronchiolar epithelium from smokers with airflow limitation and/or emphysema compared to smokers without these conditions and never smokers; furthermore, there were no differences between any of these subject groups in macrophage chemokine expression. This latter observation is interesting in light of the aforementioned investigation of human alveolar epithelial cells and macrophages isolated from the same subjects where the epithelial cells were demonstrated to secrete significantly higher levels of chemokines in response to LPS exposure under identical conditions (CitationThorley et al 2007)), supporting the concept that the epithelium quantitatively and qualitatively modulates leukocyte trafficking, possibly under the influence of cytokine release by macrophages. In vitro studies have also shown that epithelial cells secrete CXCL8 (CitationDeForge et al 1993; CitationKode et al 2006; CitationThorley et al 2007) and CXCL1 (CitationSchulz et al 2004; CitationThorley et al 2007) in response to a number of mediators associated with smoking and COPD eg pro-inflammatory cytokines, bacterial products, oxidative stress and cigarette smoke extract. Cigarette smoke stimulation of primary human bronchial epithelial cell CXCL8 mRNA expression and protein secretion has been attributed to various components of tobacco smoke, including acrolein, acetaldehyde (CitationMio et al 1997) and nicotine (CitationTsai et al 2006), while oxidative stress triggers CXCL8 release by small airway epithelial cells (CitationKode et al 2006). Up-regulation of the MAPK pathway in human bronchial epithelial cells by nicotine, through extracellular signal-related kinase 1 / 2 and c-Jun-NH(2)-terminal kinase, but not p38 MAPK signaling (CitationTsai et al 2006), and increased levels of the transcription factor, NF-κB, have been observed (CitationKode et al 2006). In contrast, other in vitro studies of the effects of cigarette smoke on CXCL8, CXCL1 and CCL2 release by primary human AT2 cells suggest that it is inhibitory, due to oxidative stress; this could be due to inhibition of the transcription factor, AP-1 (CitationLaan et al 2004). Differences between studies will reflect the source of epithelial cells (eg primary or cell lines) (CitationWitherden et al 2004; CitationKode et al 2006) as well as the region of the respiratory tract from which they were isolated. Unlike the stimulatory effects of cigarette smoke that occur with large airway epithelial cells, subjugation of alveolar epithelial responses by cigarette smoke may contribute to emphysema due to inadequate inflammatory response and reduced repair. A counterbalance to this in COPD subjects could be the observed increased sensitivity/capacity of pulmonary epithelium from COPD subjects, compared to subjects with normal lung function, to secrete CXC chemokines in response to TNF-α (CitationPatel et al 2003; CitationSchulz et al 2004); thus, TNF-α release by stimulated inflammatory macrophages (CitationThorley et al 2007) could activate COPD airway epithelium to release pathologically relevant quantities of chemokines.
Chemokines: dendritic cell recruitment
Pulmonary epithelial cells are also a rich source of CCL20 (CitationReibman et al 2003; CitationThorley et al 2005), which is a potent dendritic cell chemokine. Human bronchial epithelial cells release CCL20 in response to cytokines (TNF-α, IL1-β, IL-4, IL-13), environmental particulate matter and diesel exhaust particles (CitationReibman et al 2003), while LPS stimulates human AT2 cells, but not macrophages, to release CCL20 (CitationThorley et al 2005). Furthermore, while exposure of dendritic cells to diesel exhaust failed to stimulate phenotypic or functional maturation, when dendritic cells were co-incubated with diesel exhaust-exposed human bronchial epithelial cells, or conditioned media from these cells, they underwent both phenotypic and functional maturation (CitationBleck et al 2006). Epithelial cell-derived granulocyte-macrophage colony stimulating factor was found to be important in this process. Of particular significance to COPD is the recent discovery that Langerhans positive dendritic cells accumulate in the airways of lung tissue from COPD subjects and this increases with the severity of the disease (CitationDemedts et al 2007). In parallel studies of CCL20 mRNA in total lung and CCL20 protein in induced sputum, there was again an increase in the COPD samples; interestingly, CCL20 levels were inversely correlated with FEV1. Furthermore, when freshly isolated pulmonary dendritic cells were examined for CCR6, the receptor for CCL20, myeloid dendritic cells expressed this receptor, suggesting that myeloid dendritic cell recruitment is orchestrated via epithelial release of CCL20. In this respect, unpublished studies by us show that cigarette smoke induces CCL20 release by primary human AT2 cells, directly linking CCL20-induced dendritic cell recruitment to the stimulatory effect of cigarette smoke on epithelial cells.
Mucus and surfactant
The airway surface fluid overlying the ciliated epithelium is composed of a periciliary fluid layer in which the cilia reside, and a mucus layer which lies on top of this. The respiratory mucus is comprised of a mixture of salts, protein, glycoprotein and water and is derived from the secretions of cells from several areas including the alveolus (CitationDanahay and Jackson 2005). In the peripheral and alveolar regions of the lung, the epithelium is covered by a thin film of surfactant, a complex mixture of phospholipids and proteins secreted by a number of different cells (CitationFrerking et al 2001). As such, the lung lining fluid is one of the first points of contact for inhaled matter and studies have shown that it is more than just a physical barrier.
Mucin
As well as keeping the epithelial layer hydrated and the airways humidified, respiratory mucus acts as a filtration barrier for the airway surface fluid and is essential to local defense. Inhaled particles adhere to the mucus layer due to its complex glycoprotein structure and once embedded within the mucus layer are cleared by the mucociliary escalator (CitationGirod et al 1992; CitationHoutmeyers et al 1999). Their production can be induced by a variety of pro-inflammatory stimuli such as cigarette smoke and cytokines and hyperproduction of mucins plays a key part in the obstruction of the airways observed in COPD (CitationRogers 2005). In vitro and in vivo studies have shown that cigarette smoke-induction of mucin secretion can synergize with bacterial cell wall products to amplify mucin secretion (CitationBaginski et al 2006). This has powerful implications in a disease such as COPD where as many 50% of subjects may have persistent bacterial colonization of the lung (CitationPatel et al 2002).
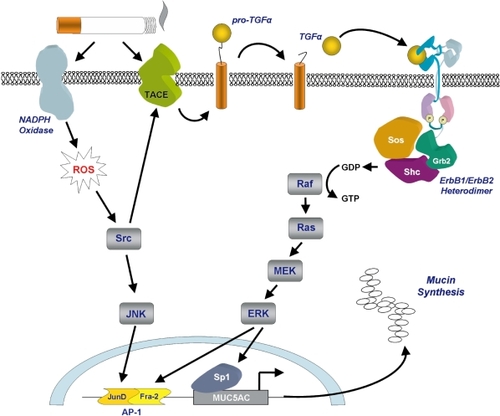
How cigarette smoke induces mucin secretion appears to be dependent on its oxidative potential. Recent studies have shown that increased mucin secretion in the airways is closely related to activation of the epidermal growth factor receptor, a member of the ErbB family of receptors (CitationBurgel and Nadel 2004). Using airway cell lines, Gensch and colleagues showed that expression of MUC5AC was up-regulated by cigarette smoke-induced EGF receptor- dependent and independent activation of ERK and JNK which led to the activation of a smoke response transcription element upstream of the MUC5AC gene (CitationGensch et al 2004). This transcription element was shown to be dependent on the AP-1 JunD/Fra-2 heterodimer for activation (). Increased mucin secretion may be caused by cigarette smoke-induced accumulation of MUC5AC mRNA which has been suggested to be due to enhanced/prolonged gene promoter activity. Furthermore, reactive oxygen species generated by NADPH oxidase in response to cigarette smoke are thought to activate the signaling kinase Src which in turn activates TNF-α converting enzyme (TACE), a member of the ADAMs family. One of the targets of TACE activity is membrane bound TGF-α, a growth factor that is one of the main ligands for the EGF receptor (CitationShao et al 2004).
Figure 3 Cigarette smoke-induced secretion of mucins. Mucin secretion is stimulated following cigarette smoke-induced activation of NADPH oxidase and TACE. NADPH oxidase generates intracellular reactive oxygen species leading to activation of the transcription factor AP-1. TACE cleaves pro-TGFα to generate the active ligand which initiates signaling through the ErbB receptor complex leading to activation of the transcription factors AP-1 and Sp1. Diagram a kind gift of Samir Nuseibeh.
In addition to stimulating increased mucin release via activation of ErbB1 receptors, clinical studies of the ErbB3 receptor show that it is up-regulated on mucin secreting cells in the airway epithelium of long term smokers (CitationO’Donnell et al 2004). This has led to the hypothesis that this receptor, too, may play a role in mucin expression, although its functional role has yet to be demonstrated.
Surfactant
As previously mentioned, surfactant is a mixture of lipids, mainly phospholipids (approximately 80%), and proteins (approximately 10%) secreted primarily by the AT2 cell and, to a lesser extent, Clara cells and bronchial submucosal glands. As well as maintaining the patency of the alveolar unit, by maintaining reduced surface tension, a number of studies have demonstrated that surfactant also plays an important role in the host defense. Surfactant proteins A and D (SP-A and -D) bind to surface motifs on a wide variety of bacteria and act as opsonins, facilitating phagocytosis of bacteria by macrophages (CitationKabha et al 1997), as well as damaging the bacteria directly by disrupting the bacterial envelope (CitationWu et al 2003).
Studies investigating the effect of smoking on lung surfactant have proved inconclusive. Early studies showed that total surfactant levels in bronchoalveolar lavage (BAL) fluid were decreased (CitationFinley and Ladman 1972) whereas later studies showed that there was no difference between non-smokers and healthy smokers (CitationLow and 1978; CitationMancini et al 1993). The lower levels detected in the first study, however, may have been due to the low BAL recovery volume. Measurement of specific surfactant proteins indicates that both SP-A and SP-D levels and activity are decreased in the lungs of smokers and subjects with COPD (CitationCook and Webb 1966; CitationSchmekel et al 1992; CitationHonda et al 1996). Decreased levels of these important antimicrobial collectins could, in theory, render the lung vulnerable to bacterial colonization and may explain, in part, the observation of increased and persistent bacterial load in the lungs of COPD subjects (CitationSethi et al 2006). Very recent population studies have shown that a single nucleotide polymorphism (SNP) in the gene encoding SP-D is associated with development of COPD and a decline in forced expiratory volume in one second (FEV1). In addition, it was also shown that a SNP in SP-A was associated with the rate of decline in FEV1 within subjects with established COPD (Citationvan Diemen et al 2006). This is the first study to show a genetic linkage between surfactant proteins and disease development. However, the relevance of these polymorphisms to the function of the proteins is unknown, but they are likely to be important in the development of COPD.
Summary
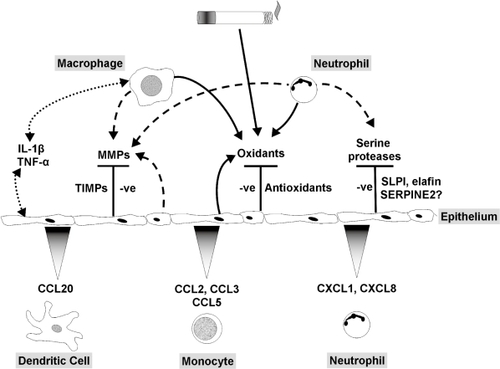
This article aims to highlight the dynamic nature of the pulmonary epithelium in relation to cigarette smoke exposure and the pathophysiology of COPD. We have focused on areas of particular interest to us. A first line of defense against inhaled foreign organic and inorganic material, the epithelium is not only a tight structural membrane barrier but is also a potent metabolic powerhouse, communicating between the luminal, interstitial and vascular compartments (). Here we have emphasized its protective role, for example, against noxious particles and chemicals, combating oxidative stress and excessive proteolytic activity. In addition, we have illustrated some aspects of the crucial role of the epithelium in modulating inflammation, repair and remodeling of the lung. Cigarette smoke has an enormous impact on the epithelium; indeed, it is perhaps surprising that only a small proportion of smokers develop COPD. We suggest that one reason for this is that the pulmonary epithelium plays a central role countering many of the toxic effects of cigarette smoke to limit lung damage and maintain adequate lung function.
Figure 4 Role of the pulmonary epithelium in cigarette smoke-induced inflammation. The pulmonary epithelium combats leukocyte-derived oxidants and free radicals in cigarette smoke via release of antioxidants. Serine proteases are blocked by low molecular weight inhibitors, SERPINS and TIMPs. Induction of cytokine release by macrophages and epithelial cells and autocrine/paracrine activation of the epithelium stimulates chemokine release and recruitment of monocytes, neutrophils and dendritic cells. In COPD the epithelial defense mechanisms are overwhelmed, leading to increased oxidative stress and proteolytic load, together with leukocyte recruitment, resulting in a chronic cycle of inflammation that may be independent of cigarette smoke exposure.
References
- AaronSDAngelJBLunauM2001Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary diseaseAmerican Journal of Respiratory and Critical Care Medicine1633495511179105
- AkiraSTakedaK2004Toll-like receptor signallingNat Rev Immunol449951115229469
- ArcangeliGCupelliVGiulianoG2001Effects of silica on human lung fibroblast in cultureSci Total Environ270135911327386
- ArmstrongLMedfordARLUppingtonKM2004Expression of functional Toll-like receptor-2 and −4 on alveolar epithelial cellsAmerican Journal of Respiratory Cell and Molecular Biology31241515044215
- AvissarNFinkelsteinJNHorowitzS1996Extracellular glutathione peroxidase in human lung epithelial lining fluid and in lung cellsAJP - Lung Cellular and Molecular Physiology270L17382
- BaginskiTKDabbaghKSatjawatcharaphongC2006Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuliAmerican Journal of Respiratory Cell and Molecular Biology351657416543607
- BarnesPJ1998New therapies for chronic obstructive pulmonary diseaseThorax53137479624300
- BarnesPJAdcockIMItoK2005Histone acetylation and deacetylation: importance in inflammatory lung diseasesEuropean Respiratory Journal255526315738302
- BarnesPJ2004Mediators of chronic obstructive pulmonary diseasePharmacological Reviews565154815602009
- BartonBE1997IL-6: Insights into novel biological activitiesClinical Immunology and Immunopathology8516209325064
- BeehKMBeierJKornmannO2003Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjectsRespir Med97634912814147
- BerndtADerksenFJVentaPJ2007Elevated amount of Toll-like receptor 4 mRNA in bronchial epithelial cells is associated with airway inflammation in horses with recurrent airway obstructionAJP - Lung Cellular and Molecular Physiology292L9364317158595
- BingleLTetleyTDBingleCD2001Cytokine-mediated induction of the human elafin gene in pulmonary epithelial cells is regulated by nuclear factor-kappa BAmerican Journal of Respiratory Cell and Molecular Biology25849111472979
- BleckBTseDBJaspersI2006Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturationThe Journal of Immunology1767431716751388
- BorokZLubmanRLDantoSI1998Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin-5Am J Respir Cell Mol Biol18554619533944
- BowlerRPBarnesPJCrapoJD2004The role of oxidative stress in chronic obstructive pulmonary diseaseCOPD12557717136992
- BrackeKRD’HulstAIMaesT2006Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient miceThe Journal of Immunology1774350916982869
- BrownDJLinBChwaM2004Elements of the nitric oxide pathway can degrade TIMP-1 and increase gelatinase activityMol Vis10281815105792
- BucchioniEKharitonovSAAllegraL2003High levels of inter-leukin-6 in the exhaled breath condensate of patients with COPDRespiratory Medicine97129930214682411
- BuhlingFWaldburgNReisenauerA2004Lysosomal cysteine proteases in the lung: role in protein processing and immunoregulationEuropean Respiratory Journal23620815083765
- BurgelPRNadelJA2004Roles of epidermal growth factor receptor activation in epithelial cell repair and mucin production in airway epitheliumThorax59992615516478
- CantinAMNorthSLHubbardRC1987Normal alveolar epithelial lining fluid contains high levels of glutathioneJournal of Applied Physiology6315273040659
- CardozoCKurtzCLesserM1992Degradation of rat lung collagens by cathepsin B. J Lab Clin.Med11916975
- ChapmanHARieseRJShiGP1997Emerging roles for cysteine proteases in human biology. Annu.Rev Physiol596388
- ChurchTPryorWA1985Free-radical chemistry of cigarette smoke and its toxicological implications. Environ.Health Perspect6411126
- ChurgADaiJTaiH2002Tumor necrosis factor-{alpha} is central to acute cigarette smoke-induced inflammation and connective tissue breakdownAmerican Journal of Respiratory and Critical Care Medicine1668495412231496
- ChurgAWangRDTaiH2003Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-{alpha} releaseAmerican Journal of Respiratory and Critical Care Medicine1671083912522030
- ComhairSAABhathenaPRFarverC2001Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cellsThe FASEB Journal1570811149894
- CookWWebbW1966Surfactant in chronic smokersAnn Thorac Surg2327335954467
- CosioMGHaleKANiewoehnerDE1980Morphologic and morphometric effects of prolonged cigarette smoking on the small airwaysAm Rev Respir Dis122265717416603
- DanahayHJacksonAD2005Epithelial mucus-hypersecretion and respiratory diseaseCurr Drug Targets Inflamm Allergy46516417305521
- de BoerWISontJKvan SchadewijkA2000Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPDJ Pathol1906192610727989
- DeForgeLEPrestonAMTakeuchiE1993Regulation of interleukin 8 gene expression by oxidant stressJournal of Biological Chemistry26825568768244994
- DemedtsIKBrackeKRVan PottelbergeG2007Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary diseaseAmerican Journal of Respiratory and Critical Care Medicine175998100517332482
- DemeoDLMarianiTJLangeC2006The SERPINE2 gene is associated with chronic obstructive pulmonary diseaseAm J Hum Genet782536416358219
- DesrochersPEJeffreyJJWeissSJ1991Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activityJ Clin Invest872258651645757
- DignettiP2002Cytokine concentrations in induced sputum of mild COPD patients. Correlation to clinical response to inhaled corticosteroidsEuropean Respiratory Journal20Suppl 38309s
- DinarelloCA1998Interleukin-1{beta}, interleukin-18, and the interleukin-1{beta} converting enzymeAnnals of the New York Academy of Sciences8561119917859
- DobashiKAsayamaKHayashibeH1993Immunohistochemical study of copper-zinc and manganese superoxide dismutases in the lungs of human fetuses and newborn infants: developmental profile and alterations in hyaline membrane disease and bronchopulmonary dysplasiaVirchows Arch A Pathol Anat Histopathol423177848236811
- DriscollKEHassenbeinDGCarterJM1995TNF alpha and increased chemokine expression in rat lung after particle exposureToxicol Lett82834839
- DroemannDGoldmannTBranscheidD2003Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lungHistochem. Cell Biol119103812610729
- DrostEMSelbyCLannanS1992Changes in neutrophil deformability following in vitro smoke exposure: mechanism and protectionAm J Respir Cell Mol Biol6287951311595
- ErmertMPantazisCDunckerH-R2003In situ localization of TNF[alpha]/[beta], TACE AND TNF receptors TNF-R1 and TNF-R2 in control and LPS-treated lung tissueCytokine228910012849708
- Farioli-VecchioliSNardacciRFalciatoriI2001Catalase immunocytochemistry allows automatic detection of lung type II alveolar cellsHistochem. Cell Biol115333911405062
- FinleyTNLadmanAJ1972Low yield of pulmonary surfactant in cigarette smokersN Engl J Med28622375066599
- FramptonMWGhioAJSametJM1999Effects of aqueous extracts of PM10 filters from the Utah Valley on human airway epithelial cellsAJP - Lung Cellular and Molecular Physiology277L9607
- FrearsERZhangZBlakeDR1996Inactivation of tissue inhibitor of metalloproteinase-1 by peroxynitriteFEBS Lett3812148641430
- FrerkingIGuntherASeegerW2001Pulmonary surfactant: functions, abnormalities and therapeutic optionsIntensive Care Medicine27169971711810113
- FujimotoKYasuoMUrushibataK2005Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary diseaseEuropean Respiratory Journal25640615802337
- FukeSBetsuyakuTNasuharaY2004Chemokines in bronchiolar epithelium in the development of chronic obstructive pulmonary diseaseAmerican Journal of Respiratory Cell and Molecular Biology314051215220136
- GearingAJBeckettPChristodoulouM1994Processing of tumour necrosis factor-alpha precursor by metalloproteinasesNature37055578052310
- GenschEGallupMSucherA2004Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNKJournal of Biological Chemistry279390859315262961
- GirodSZahmJMPlotkowskiC1992Role of the physiochemical properties of mucus in the protection of the respiratory epitheliumEuropean Respiratory Journal5477871563506
- GolponHFadokVATaraseviciene-StewartL2004Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growthFASEB J1817161815345697
- GolponHAColdrenCDZamoraMR2004Emphysema lung tissue gene expression profilingAmerican Journal of Respiratory Cell and Molecular Biology3159560015284076
- GosmanMMEMarike BoezenHvan DiemenCC2007A disintegrin and metalloprotease 33 and chronic obstructive pulmonary disease pathophysiologyThorax62242717090574
- HackettNRHeguyAHarveyBG2003Variability of antioxidant-related gene expression in the airway epithelium of cigarette smokersAmerican Journal of Respiratory Cell and Molecular Biology293314312702543
- HanZJunxuZhongN2003Expression of matrix metalloproteinases MMP-9 within the airways in asthmaRespir Med97563712735676
- HarjuTKaarteenaho-WiikRSirvioR2004Manganese superoxide dismutase is increased in the airways of smokers’ lungsEuropean Respiratory Journal247657115516670
- HasdayJDBascomRCostaJJ1999Bacterial endotoxin is an active component of cigarette smokeChest1158293510084499
- HauberHPTulicMKTsicopoulosA2005Toll-like receptors 4 and 2 expression in the bronchial mucosa of patients with cystic fibrosisCan Respir J12131815776129
- HertzCJWuQPorterEM2003Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human {beta} defensin-2The Journal of Immunology1716820614662888
- HigashimotoYYamagataYItohH2006Complex effect of adenovirus early region proteins on innate immune systemInflamm. Allergy Drug Targets52293717168793
- HoggJCMacklemPTThurlbeckWM1968Site and nature of airway obstruction in chronic obstructive lung diseaseN. Engl J Med2781355605650164
- HondaYTakahashiHKurokiY1996Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokersChest109100698635323
- HoshinoYMioTNagaiS2001Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell lineAJP - Lung Cellular and Molecular Physiology281L5091611435227
- HoutmeyersEGosselinkRGayan-RamirezG1999Regulation of mucociliary clearance in health and diseaseEuropean Respiratory Journal1311778810414423
- ItoKItoMElliottWM2005Decreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med35219677615888697
- JanardhanKSMcIsaacMFowlieJ2006Toll like receptor-4 expression in lipopolysaccharide induced lung inflammationHistol Histopathol216879616598667
- JefferyPK1998Structural and inflammatory changes in COPD: a comparison with asthmaThorax531299624299
- JeyaseelanSChuHWYoungSK2005Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injuryInfection and Immunity7317546315731076
- JohnsonDABarrettAJMasonRW1986Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site regionJournal of Biological Chemistry26114748513490478
- JoshiUMKodavantiPRMehendaleHM1988Glutathione metabolism and utilization of external thiols by cigarette smoke-challenged, isolated rat and rabbit lungsToxicol. Appl Pharmacol96324353194918
- KabhaKSchmegnerJKeisariY1997SP-A enhances phagocytosis of Klebsiella by interaction with capsular polysaccharides and alveolar macrophagesAJP - Lung Cellular and Molecular Physiology272L34452
- KarimiKSarirHMortazE2006Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophagesRespir Res76616620395
- KasaharaYTuderRMCoolCD2001Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysemaAmerican Journal of Respiratory and Critical Care Medicine1637374411254533
- KeatingsVMCollinsPDScottDM1996Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthmaAmerican Journal of Respiratory and Critical Care Medicine153530348564092
- KimHJHenkeCASavikSK1997Integrin mediation of alveolar epithelial cell migration on fibronectin and type I collagenAJP - Lung Cellular and Molecular Physiology273L13441
- KodeAYangSRRahmanI2006Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cellsRespir Res713217062156
- KoyamaSSatoETsukadairaA2002Vascular endothelial growth factor mRNA and protein expression in airway epithelial cell lines in vitroEuropean Respiratory Journal2014495612503703
- KoyamaSSatoENomuraH1998Bradykinin stimulates type II alveolar cells to release neutrophil and monocyte chemotactic activity and inflammatory cytokinesAmerican Journal of Pathology1531885939846978
- KranenburgARde BoerWIAlagappanVKT2005Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary diseaseThorax601061315681497
- KuschnerWGD’AlessandroAWongH1996Dose-dependent cigarette smoking-related inflammatory responses in healthy adultsEuropean Respiratory Journal91989948902455
- LaanMBozinovskiSAndersonGP2004Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cellsThe Journal of Immunology17341647015356167
- LannanSDonaldsonKBrownD1994Effect of cigarette smoke and its condensates on alveolar epithelial cell injury in vitroAJP - Lung Cellular and Molecular Physiology266L92100
- LarssonLSzponarBPehrsonC2004Tobacco smoking increases dramatically air concentrations of endotoxinIndoor Air14421415500635
- LaurellCBErikssonS1963The electrophoretic alpha-1-globulin pattern of serum in alpha-1-antitrypsin deficiencyScand J Clin Lab Invest1513240
- LavigneMCEppihimerMJ2005Cigarette smoke condensate induces MMP-12 gene expression in airway-like epitheliaBiochem Biophys Res Commun33019420315781250
- LiHCuiDTongX2002The role of matrix metalloproteinases in extracellular matrix remodelling in chronic obstructive pulmonary disease rat modelsChinese Journal of Internal Medicine413938
- LiWXuYZhangZ2005Activity of matrix metalloproteinase in airway epithelial cells of COPD patientsJ Huazhong Univ Sci Technolog Med Sci25151416116959
- LiXYDonaldsonKRahmanI1994An investigation of the role of glutathione in increased epithelial permeability induced by cigarette smoke in vivo and in vitroAm J Respir Crit Care Med1491518258004308
- LiXYRahmanIDonaldsonK1996Mechanisms of cigarette smoke induced increased airspace permeabilityThorax51465718711672
- LiuXConnerHKobayashiT2005Cigarette smoke extract induces DNA damage but not apoptosis in human bronchial epithelial cellsAmerican Journal of Respiratory Cell and Molecular Biology33121915845867
- LiuZZhouXShapiroSD2000The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivoCell1026475511007483
- LowRBDavisGSGiancolaMS1978Biochemical analyses of bronchoalveolar lavage fluids of healthy human volunteer smokers and nonsmokersAm Rev Respir Dis11886375736357
- LozewiczSWellsCGomezE1990Morphological integrity of the bronchial epithelium in mild asthmaThorax4512152321171
- MaesTBrackeKRVermaelenKY2006Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammationInt Arch Allergy Immunol1413546816940747
- MaestrelliPSaettaMMappCE2001Remodeling in response to infection and injury. airway inflammation and hypersecretion of mucus in smoking subjects with chronic obstructive pulmonary diseaseAmerican Journal of Respiratory and Critical Care Medicine164S768011734472
- ManciniNMBeneMCGerardH1993Early effects of short-time cigarette smoking on the human lung: a study of bronchoalveolar lavage fluidsLung171277918412308
- MateosFBrockJFPerez-ArellanoJL1998Iron metabolism in the lower respiratory tractThorax535946009797761
- MayerAKMuehmerMMagesJ2007Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cellsThe Journal of Immunology17831344217312161
- McElroyMCKasperM2004The use of alveolar epithelial type I cell-selective markers to investigate lung injury and repairEuropean Respiratory Journal246647315459148
- McQuibbanGAGongJHWongJP2002Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivoBlood1001160712149192
- MercerBBrinckerhoffCD’ArmientoJ2006Activation of the MMP-1 promoter by cigarette smoke in human small airway epithelial cells requires ERK MAP kinase signaling: differential response of the 1G and 2G promoter sequencesProceedings of the American Thoracic Society347716921115
- MioTRombergerDJThompsonAB1997Cigarette smoke induces interleukin-8 release from human bronchial epithelial cellsAm J Respir Crit Care Med155177069154890
- MorrisonDStrieterRMDonnellySC1998Neutrophil chemokines in bronchoalveolar lavage fluid and leukocyte-conditioned medium from nonsmokers and smokersEuropean Respiratory Journal121067729863998
- NeurohrCLenzAGDingI2003Glutamate-cysteine ligase modulatory subunit in BAL alveolar macrophages of healthy smokersEuropean Respiratory Journal2282712882455
- NieJPeiD2004Rapid inactivation of alpha-1-proteinase inhibitor by neutrophil specific leukolysin/membrane-type matrix metalloproteinase 6Exp Cell Res2961455015149845
- NiewoehnerDEKleinermanJRiceDB1974Pathologic changes in the peripheral airways of young cigarette smokersN Engl J Med29175584414996
- NodaEHoshinaHWatanabeH2003Production of TNF-alpha by polymorphonuclear leukocytes during mechanical ventilation in the surfactant-depleted rabbit lungPediatr Pulmonol364758114618638
- NovinecMGrassRNStarkWJ2007Interaction between human cathepsins K,L and S and elastins: mechanism of elastinolysis and inhibition by macromolecular inhibitorsJournal of Biological ChemistryM610107200
- O’DonnellRARichterAWardJ2004Expression of ErbB receptors and mucins in the airways of long term current smokersThorax5910324015563701
- OkadaYWatanabeSNakanishiI1988Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinasesFEBS Lett229157603162216
- OliveraDSBoggsSEBeenhouwerC2007Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitroInhal. Toxicol19132217127639
- PatelISRobertsNJLloyd-OwenSJ2003Airway epithelial inflammatory responses and clinical parameters in COPDEuropean Respiratory Journal2294912882457
- PatelISSeemungalTARWilksM2002Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbationsThorax577596412200518
- PauwelsRABuistASCalverleyPMA2001Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) workshop summaryAm J Respir Crit Care Med163125611316667
- PesciABalbiBMajoriM1998Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary diseaseEuropean Respiratory Journal1238069727789
- PhamCT2006Neutrophil serine proteases: specific regulators of inflammationNat Rev Immunol65415016799473
- PorthMC2005Pathophysiology concepts of altered health states7th edNew YorkLippincott Williams and Wilkins
- ProfitaMChiapparaGMirabellaF2003Effect of cilomilast (Ariflo) on TNF-{alpha}, IL-8, and GM-CSF release by airway cells of patients with COPDThorax58573912832668
- PryorWAStoneK2007Oxidants in cigarette smoke: radicals hydrogen peroxides peroxynitrate and peroxynitriteAnn N Y Acad Sci6861228
- RahmanI2005Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanismsCell Biochem Biophys431678816043892
- RahmanILiXYDonaldsonK1995Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stressAJP - Lung Cellular and Molecular Physiology269L28592
- RahmanIMarwickJKirkhamP2004Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expressionBiochem. Pharmacol6812556715313424
- ReibmanJHsuYChenLC2003Airway epithelial cells release MIP-3{alpha}/CCL20 in response to cytokines and ambient particulate matterAmerican Journal of Respiratory Cell and Molecular Biology2864812760962
- ReidPTMarsdenMECunninghamGA1999Human neutrophil elastase regulates the expression and secretion of elafin (elastase-specific inhibitor) in type II alveolar epithelial cellsFEBS Lett45733710486558
- ReillyJJJrChenPSailorLZ1991Cigarette smoking induces an elastolytic cysteine proteinase in macrophages distinct from cathepsin LAJP - Lung Cellular and Molecular Physiology261L418
- ReynoldsPRCosioMGHoidalJR2006Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cellsAmerican Journal of Respiratory Cell and Molecular Biology353141916601242
- RitterMMennerichDWeithA2005Characterization of Toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory responseJ Inflamm (Lond)21616316467
- RogersDF2005The role of airway secretions in COPD: pathophysiology, epidemiology and pharmacotherapeutic optionsCOPD23415317146999
- RussellREKThorleyACulpittSV2002Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteasesAJP - Lung Cellular and Molecular Physiology283L8677312225964
- RusznakCMillsPRDevaliaJL2000Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary diseaseAmerican Journal of Respiratory Cell and Molecular Biology23530611017919
- SadSMarcotteRMosmannTR1995Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokinesImmunity227197697544
- SaettaM1998CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med15782269517597
- SaettaMGhezzoHKimWD1985Loss of alveolar attachments in smokers: a morphomaetric correlate of lung function impairmentAm Rev Respir Dis1328949004051324
- SaettaMTuratoGBaraldoS2000Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with symptoms of chronic bronchitis and chronic airflow limitationAm J Respir Crit Care Med16110162110712357
- SaitoTYamamotoTKazawaT2005Expression of toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouseCell Tissue Res321758815902499
- SakakibaraA1997Possible involvement of phosphorylation of occludin in tight junction formationThe Journal of Cell Biology13713934019182670
- SallenaveJM2002Antimicrobial activity of antiproteinasesBiochem Soc Trans301111512023836
- SchmeckBHuberSMoogK2006Pneumococci induced TLR- and Rac1-dependent NF-{kappa}B-recruitment to the IL-8 promoter in lung epithelial cellsAJP - Lung Cellular and Molecular Physiology290L730716299055
- SchmekelBBosJHKhanAR1992Integrity of the alveolar-capillary barrier and alveolar surfactant system in smokersThorax4760381412116
- SchonbeckUMachFLibbyP1998Generation of biologically active IL-1{beta} by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1{beta} processingThe Journal of Immunology161334069759850
- SchulzCKratzelKWolfK2004Activation of bronchial epithelial cells in smokers without airway obstruction and patients with COPDChest12517061315136380
- Schulze-OsthoffKLosMBaeuerlePA1995Redox signalling by transcription factors NF-kappa B and AP-1 in lymphocytesBiochem. Pharmacol50735417575632
- Segura-ValdezLPardoAGaxiolaM2000Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPDChest1176849410712992
- SethiS2000Bacterial Infection and the Pathogenesis of COPDChest117286S29110843957
- SethiSMaloneyJGroveL2006Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary diseaseAmerican Journal of Respiratory and Critical Care Medicine173991816474030
- ShaoMXGNakanagaTNadelJA2004Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-{alpha}-converting enzyme in human airway epithelial (NCI-H292) cellsAJP - Lung Cellular and Molecular Physiology287L420715121636
- SiafakasNMVermeirePPrideNB1995Optimal assessment and management of chronic obstructive pulmonary disease (COPD). The European Respiratory Society Task ForceEuropean Respiratory Journal813984207489808
- SlevogtHSeyboldJTiwariKN2007Moraxella catarrhalis is internalized in respiratory epithelial cells by a trigger-like mechanism and initiates a TLR2- and partly NOD1-dependent inflammatory immune responseCell Microbiol969470717054439
- SniderGL1992Emphysema: the first two centuries--and beyond. a historical overview, with suggestions for future research: Part 1Am Rev Respir Dis1461334441443893
- SolerNEwigSTorresA1999Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary diseaseEuropean Respiratory Journal1410152210596683
- StringerKAFreedBMDunnJS2004Particulate phase cigarette smoke increases MnSOD, NQO1, and CINC-1 in rat lungsFree Radic Biol Med3715273315477004
- TaggartCCervantes-LaureanDKimG2000Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activityJournal of Biological Chemistry275272586510867014
- TaggartCCLoweGJGreeneCM2001Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitorJournal of Biological Chemistry276333455211435427
- TakahashiHIshidohKMunoD1993Cathepsin L activity is increased in alveolar macrophages and bronchoalveolar lavage fluid of smokersAm Rev Respir Dis147156288503570
- TakanashiSHasegawaYKanehiraY1999Interleukin-10 level in sputum is reduced in bronchial asthma, COPD and in smokersEuropean Respiratory Journal143091410515406
- TakeuchiOKawaiTMuhlradtPF2001Discrimination of bacterial lipoproteins by Toll-like receptor 6International Immunology139334011431423
- TangKRossiterHBWagnerPD2004Lung-targeted VEGF inactivation leads to emphysema phenotype in miceJournal of Applied Physiology9715596615208295
- TetleyTD2002Macrophages and the pathogenesis of COPDChest121156S159S12010845
- TetleyTD2005Antiprotease therapy In Cazzola M et al eds Therapeutic strategies in COPDOxfordClinical Publishing23345
- TetleyTD2006Proteinase inhibitors/secretory leukoprotease inhibitor and elafinLaurentGJShapiroSDEncyclopedia of respiratory medicineLondonAcademic Press51722
- ThorleyAJFordPAGiembyczMA2007Differential regulation of cytokine release and leukocyte migration by lipopolysaccharidestimulated primary human lung alveolar type II epithelial cells and macrophagesThe Journal of Immunology1784637317182585
- ThorleyAJGoldstrawPYoungA2005Primary human alveolar type II epithelial cell CCL20 (macrophage inflammatory protein-3{alpha})-induced dendritic cell migrationAmerican Journal of Respiratory Cell and Molecular Biology32262715618437
- TobiasPCurtissLK2005Thematic review series: the immune system and atherogenesis. Paying the price for pathogen protection: toll receptors in atherogenesisJournal of Lipid Research464041115654120
- TomakiMSugiuraHKoaraiADecreased expression of anti-oxidant enzymes and increased expression of chemokines in COPD lungPulmonary Pharmacology and TherapeuticsIn Press, Corrected Proof
- TravesSLCulpittSVRussellREK2002Increased levels of the chemokines GRO{alpha} and MCP-1 in sputum samples from patients with COPDThorax57590512096201
- TrinchieriGSherA2007Cooperation of Toll-like receptor signals in innate immune defenceNat Rev Immunol71799017318230
- TsaiJRChongIWChenCC2006Mitogen-activated protein kinase pathway was significantly activated in human bronchial epithelial cells by nicotineDNA Cell Biol253122216716121
- TsujiTAoshibaKNagaiA2004Cigarette smoke induces senescence in alveolar epithelial cellsAmerican Journal of Respiratory Cell and Molecular Biology31643915333326
- TsujiTAoshibaKNagaiA2006Alveolar cell senescence in patients with pulmonary emphysemaAmerican Journal of Respiratory and Critical Care Medicine1748869316888288
- Van den SteenPEProostPWuytsA2000Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intactBlood9626738111023497
- van der VaartHPostmaDSTimensW2005Acute effects of cigarette smoking on inflammation in healthy intermittent smokersRespir Res62215740629
- van DiemenCCPostmaDSVonkJM2006Polymorphisms in surfactant proteins are associated with FEV1 decline and development of COPD in the general populationEur Respir Rev152046
- VanEPLittleRDDupuisJ2002Association of the ADAM33 gene with asthma and bronchial hyperresponsivenessNature4184263012110844
- VernooyJHJLindemanJHNJacobsJA2004Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPDChest12618021015596677
- VestboJHoggJC2006Convergence of the epidemiology and pathology of COPDThorax6186816227325
- VlahovicGRussellMLMercerRR1999Cellular and connective tissue changes in alveolar septal walls in emphysemaAm J Respir Crit Care Med16020869210588633
- VoelkelNFVandivierRWTuderRM2006Vascular endothelial growth factor in the lungAJP - Lung Cellular and Molecular Physiology290L2092116403941
- WangHLiuXUminoT2001Cigarette smoke inhibits human bronchial epithelial cell repair processesAmerican Journal of Respiratory Cell and Molecular Biology25772911726404
- WannerASalatheMO’RiordanTG1996Mucociliary clearance in the airwaysAm J Respir Crit Care Med15418689028970383
- WardP2002Phospholipase C-gamma modulates epithelial tight junction permeability through hyperphosphorylation of tight junction proteinsThe Journal of Biological Chemistry27735760512101180
- WassefAJanardhanKPearceJW2004Toll-like receptor 4 in normal and inflamed lungs and other organs of pig, dog and cattleHistol Histopathol191201815375763
- WickendenJAClarkeMCHRossiAG2003Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosisAmerican Journal of Respiratory Cell and Molecular Biology295627012748058
- WilliamsMC2003Alveolar type I cells: molecular phenotype and developmentAnnual Review of Physiology6566995
- WitherdenIRVanden BonEJGoldstrawP2004Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastaseAmerican Journal of Respiratory Cell and Molecular Biology30500915033639
- WuHKuzmenkoAWanS2003Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeabilityJournal of Clinical Investigation111158960212750409
- WyllieDHKiss-TothEVisintinA2000Evidence for an accessory protein function for Toll-like receptor 1 in anti-bacterial responsesThe Journal of Immunology16571253211120843
- YuMZhengXWitschiH2002The role of interleukin-6 in pulmonary inflammation and injury induced by exposure to environmental air pollutantsToxicological Sciences684889712151646
- YuQStamenkovicI2000Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesisGenes and Development141637610652271
- ZalacainRSobradilloVAmilibiaJ1999Predisposing factors to bacterial colonization in chronic obstructive pulmonary diseaseEuropean Respiratory Journal13343810065679
- ZhangXShanPJiangG2006Toll-like receptor 4 deficiency causes pulmonary emphysemaJ Clin Invest1163050917053835
- ZhuGWarrenLAponteJ2007The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populationsAmerican Journal of Respiratory and Critical Care Medicine1761677317446335
- ZhuJQiuYMajumdarS2001Exacerbations of bronchitis. Bronchial eosinophilia and gene expression for interleukin-4, inter-leukin-5, and eosinophil chemoattractantsAm J Respir Crit Care Med1641091611435248
- ZiegenhagenMBennerUZisselG1997Sarcoidosis: TNF-alpha release from alveolar macrophages and serum level of sIL-2R are prognostic markersAmerican Journal of Respiratory and Critical Care Medicine1561586929372680