Abstract
Unlike most other effector cells of the innate, as well as the adaptive immune systems, the neutrophil is a relatively undiscerning aggressor with scant regard for damage limitation. Although this highly combative, professional phagocyte has become increasingly implicated in the immunopathogenesis of many acute and chronic inflammatory disorders, of both infective and noninfective origin, effective pharmacological strategies to counter neutrophilaggression have remained elusive. Activation of neutrophils results in rapid mobilization of both stored and extracellular Ca2+, resulting in abrupt, usually transient increases in cytosolic Ca2+, which precede, and are a prerequisite for activation of the Ca2+-dependent pro-inflammatory activities of these cells. Mobilization of Ca2+ by, and restoration of Ca2+ homeostasis to activated neutrophils are multistep processes which present a number of potential targets, some well recognized and others noveland unconventional, for the pharmacological control of neutrophil-mediated inflammation. Uncovering these targets represents the primary focus of this review.
Introduction
Polymorphonuclear leukocytes, of which the neutrophil granulocyte is the most abundant, are key components of the innate immune system. These cells are activated and recruited to sites of infection by inflammatory stimuli generated by microbial pathogens, or by the interaction of these with pattern recognition molecules present on/in neighboring tissue cells such as epithelial cells, mast cells and tissue macrophages. Notwithstanding their abruptly mobilizable arsenal of antimicrobialagents, which include antimicrobial peptides/proteins, enzymes, bioactive lipids, and reactive oxidant species (ROS) (CitationAnderson et al 1998; CitationTheron et al 2002; CitationHatanaka et al 2004), these cells also have biosynthetic capability, albeit limited (CitationCassatella 1999; CitationWitko-Sarsat et al 2000), which enables them to produce chemokines/cytokines, especially interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNF-α). Acting in concert, TNF-α and IL-8 amplify neutrophil-mediated inflammatory responses by promoting extravasation and accumulation of neutrophils at sites of tissue injury and infection through induction and upregulation of expression of endothelialadhesion molecules and chemotaxis, respectively. Neutrophil migration and localization are further potentiated by leukotriene B4 (LTB4), an endogenously generated bioactive lipid with potent chemoattractant activity (CitationChen et al 2006).
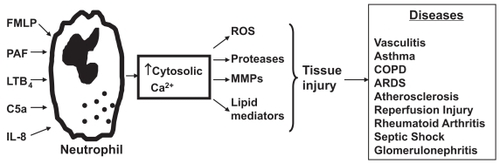
Although extremely effective in eradicating microbial pathogens, the neutrophil, with its arsenal of indiscriminate oxidants and proteases, poses a potential threat to bystander host cells and tissues in the vicinity of the inflammatory reaction. Accordingly, neutrophil influx must be effcient, protective and promptly downregulated. Nevertheless, there is increasing awareness of the involvement of inappropriate activation of neutrophils in the etiology of many acute/hyper acute and chronic inflammatory disorders of both infective and noninfective origin, important examples of which are shown in , with the airways and cardiovascular system being particularly vulnerable.
Figure 1 Activation of neutrophils by chemoattractants such as FMLP, PAF, C5a, and LTB4 increases cytosolic Ca2+ concentrations with resultant generation of toxic reactive oxygen species (ROS) and release of proteases, matrix metalloproteinases (MMPs) and lipid mediators. The tissue injury that may be associated with release of these harmful molecules into the vicinity of innocent bystander host tissues contributes to the pathogenesis of numerous diseases, including chronic obstructive pulmonary disease (COPD) and the acute respiratory distress syndrome (ARDS).
Responsiveness to corticosteroids
Few currently available therapeutic agents, including corticosteroids, effectively control the harmful pro-inflammatory activities of neutrophils. Indeed, insensitivity to corticosteroids appears to be a feature of those disorders in which the neutrophil is the predominant offender.
The apparent insensitivity of neutrophils is attributable to the coexistence of several different resistance mechanisms in these cells. Firstly, in contradistinction to other types of immune and inflammatory cells, glucocorticoids delay neutrophil apoptosis. This antiapoptotic effect of glucocorticoids in neutrophils is achieved by a mechanism which involves sustained expression of the antiapoptotic Bcl-2 family protein, Mcl-1L (CitationSivertson et al 2007). Secondly, neutrophils contain high levels of the functionally inactive beta isoform of the glucocorticoid receptor (GR), the synthesis of which is further upregulated on exposure of the cells to IL-8 (CitationStrickland et al 2001), rendering them even less sensitive to corticosteroids. Although not yet described in neutrophils, the activity of the enzyme histone deacetylase, which is recruited by activated GRs as a mechanism of repression of expression of multiple inflammatory genes, is decreased in macrophages and circulating mononuclear leukocytes of patients with COPD and severe asthma respectively (reviewed in CitationBarnes 2007). Thirdly, many of the proinflammatory activities of neutrophils, including generation of ROS, release of granule proteases, and generation of prostanoids, eicosanoids, and platelet-activating factor (PAF) occur within seconds of receptor-mediated activation of these cells and are independent of de novo protein synthesis.
Clearly the identification of novel targets for effective neutrophil-directed anti-inflammatory chemotherapy is a priority.
Calcium and neutrophils
Interaction of neutrophil membrane receptors with chemoattractants, opsonized particles, or endothelialadhesion molecules, results in abrupt, transient increases in cytosolic Ca2+ which precede, and are a prerequisite, for initiation of the proinflammatory activities of these cells. Ca2+-activatible inflammatory functions include generation of superoxide by the membrane-associated electron transporting NADPH oxidase, adhesion to vascular endothelium, degranulation, activation of cytosolic phospholipase A2 and 5-lipoxygenase, as well as synthesis of IL-8. Because of this critical dependence of activation of the proinflammatory activities of neutrophils on Ca2+, the mechanisms utilized by these cells to mobilize and dispose of the cation represent attractive, and in several cases, novel potential targets for neutrophil-directed anti-inflammatory chemotherapy.
Calcium handling by activated neutrophils
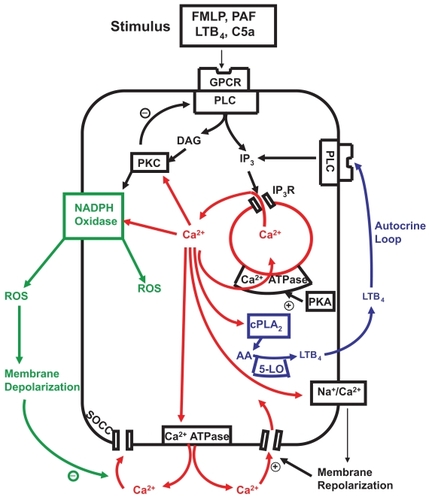
A model of calcium mobilization and restoration of calcium homeostasis in activated human neutrophils is presented in and discussed below. Release of Ca2+ from intracellular stores following receptor-mediated activation of neutrophils with various stimuli, including chemoattractants such as N-formylated peptides/polypeptides (FMLP), C5a, leukotriene B4 (LTB4), PAF, or IL-8, occurs rapidly, reaching peak values within several seconds of ligand-receptor binding which are 5–10-fold above the basal value of about 100 nM (CitationFavre et al 1996). The receptors for the aforementioned chemoattractants belong to the 7-transmembrane, G-protein-coupled family of receptors. Occupation of these receptors, which are regulated by various Gα and Gβγ subunits, results in activation of the β isoforms of phospholipase C (PLC), which in turn mediate production of inositol-1,4,5-triphosphate (IP3) by hydrolysis of phosphatidylinositol–4,5,-biphosphate (CitationAliet al 1998; CitationYue et al 1998). IP3 interacts with Ca2+-mobilizing receptors on intracellular storage vesicles, resulting in discharge of stored Ca2+ into the cytosol. Only modest increases in IP3 of around 15% of maximal are required to mobilize the total pool of stored Ca2+. The duration of the peak increase in cytosolic Ca2+ varies according to the type and concentration of the chemoattractant, but is usually brief, being followed by a progressive decline in the concentration of cytosolic Ca2+ with a return to basal values within several minutes. The duration of the peak cytosolic Ca2+ response, as wellas the rate of decline in the concentration of cytosolic Ca2+, are determined by at least 4 mechanisms. These are: i) shuttling of Ca2+ between the stores and the cytosol (ie, repetitive bouts of release from, and resequestration of Ca2+ into stores (CitationAnderson et al 2005); ii) activation of a secondary wave of Ca2+ influx due to endogenously-generated LTB4 by chemoattractant-activated neutrophils (CitationSteel et al 2007); iii) the efficiency of the systems which promote clearance of Ca2+ from the cytosol (CitationAnderson et al 1998; CitationSteeland Anderson 2002); and iv) the efficiency of the systems which regulate the time of onset, rate and magnitude of influx of extracellular cation (CitationTintinger et al 2001a).
Figure 2 Calcium-mobilizing stimuli interact with membrane G-protein coupled receptors (GPCR) to activate phospholipase C (PLC) generating inositol triphosphate (IP3) which interacts with IP3 receptors (IP3R) releasing Ca2+ from storage vesicles. Cytosolic Ca2+ phospholipase A2 (cPLA2) Which mobilizes arachidonic acid (AA) for the 5-lipoxygenase (5-LO) pathway. The AA metabolite leukotriene B4 (LTB4) is actively transported to the cell exterior where it binds to its receptor to activate PLC, completing a positive feedback autocrine loop. Ca2+ released into the cytosol is rapidly extruded from the cell by the plasma membrane Ca2+ ATPase and resequested into storage vesicles by the protein kinase A (PKA)-sensitive endomembrane Ca2+ ATPase. Protein kinase C (PKC) activated by Ca2+ and diacylglycerol (DAG) facilitates assembly and activation of NADPH oxidase on the outer membrane which generates reactive oxygen species (ROS) with concomitant membrane depolarization. The depolarized membrane potential delays Ca2+ entry through store operated channels (SOCCs) until the Ca2+-activatible Na+/Ca2+ exchanger, operating in reverse mode, mediates recovery of the membrane potential promoting Ca2+ reuptake via SOCCs. PKC down-regulates PLC as part of a negative feedback loop to terminate IP3 production
Restoration of Ca2+ homeostasis
Rapid and efficient removal of Ca2+ from the cytosol of activated neutrophils ensures that activation of the cells is brief, thereby avoiding Ca2+ overload and hyperreactivity. This is achieved primarily by removal of Ca2+ from the cytosol by two adenosine triphosphate (ATP)-driven Ca2+ pumps operating in unison. These are the plasma membrane Ca2+-ATPase and the endomembrane Ca2+-ATPase, which mediate Ca2+ efflux and resequestration respectively. These two Ca2+ pumps appear to contribute equally to the removal of Ca2+ from the cytosol of activated neutrophils (CitationAnderson and Goolam Mahomed 1997; Pettit and Hallett 2000).
The plasma membrane Ca2+-ATPase of neutrophils is upregulated by calmodulin, acidic phospholipids, and polyunsaturated fatty acids, all of which shift the pump to a higher affinity for Ca2+, resulting in enhanced maximal velocity (Carafoliet al 1992).
Apart from causing activation of PLC and release of stored Ca2+, activation of neutrophils with chemoattractants such as FMLP is also accompanied by transient activation of adenylate cyclase (CitationIannone et al 1989; CitationTheron et al 2002). This results from the interaction of adenosine, generated by dephosphorylation of adenylates, presumably by ecto-5′nucleotidase, with G-protein/adenylyl cyclase-coupled adenosine receptors (AR) of the A2A subtype on the neutrophil membrane (CitationIannone et al 1989; CitationTheron et al 2002), resulting in activation of adenosine 3′,5′-cyclic monophosphate (cAMP)-dependent protein kinase A (PKA). Phospholamban, a polypeptide regulator of the endomembrane Ca2+-ATPase, undergoes PKA-mediated phosphorylation which results in up-regulation of the Ca2+ sequestering/resequestering activity of the pump (CitationChu et al 2000).
Importantly, efficient clearance of Ca2+ by the plasma membrane and endomembrane Ca2+-ATPases is facilitated by the membrane depolarizing action of NADPH oxidase which restricts the influx of extracellular Ca2+. NADPH oxidase undergoes Ca2+-dependent activation on exposure of neutrophils to chemoattractants such as C5a, FMLP, and LTB4, but not PAF or IL-8 (CitationSteeland Anderson 2002; CitationGuichard et al 2007), as well as to opsonized antigens. Activation of NADPH oxidase is accompanied by an abrupt and steep decrease in membrane potential which results primarily from the electrogenic properties of the oxidase (CitationTintinger et al 2001a). When the cells are depolarized, the driving force for entry of Ca2+ is abolished because the electrical component of the electrochemical gradient promoting Ca2+ entry is markedly reduced. Consequently, NADPH oxidase-mediated membrane depolarization enables the plasma membrane and endomembrane Ca2+-ATPases to mediate clearance of Ca2+ from the cytosol of activated neutrophils, unhindered by influx of extracellular Ca2+.
Influx of Ca2+
Depletion of intracellular Ca2+ stores following receptor-mediated activation is followed by refilling of the stores, a process known as store-operated Ca2+ influx, or capacitative Ca2+ influx (CitationParekh and Penner 1997). In neutrophils, the time of onset and rate of store-operated Ca+ influx are determined primarily by the duration and intensity of activity of NADPH oxidase. In the case of cells activated with Ca2+-mobilizing stimuli which are inefficient activators of the oxidase, such as PAF (CitationSteeland Anderson 2002), influx of Ca2+occurs rapidly, overwhelming the Ca2+-ATPases, resulting in prolonged elevations in peak cytosolic Ca2+ concentrations. On the other hand, exposure of the cells to Ca2+-mobilizing activators of the oxidase, such as FMLP, is accompanied by efficient clearance of store-derived cytosolic Ca2+, in the setting of a gradual influx of Ca2+, the rate of which is super-imposable on that of membrane repolarization (CitationTintinger and Anderson 2004).
While the magnitude and duration of the maximal membrane depolarization responses of neutrophils activated with FMLP are determined by the intensity and duration of activation of NADPH oxidase, as wellas by the counteracting effects of an efflux of protons from the cells (CitationSchrenzel et al 1998; CitationBánfi et al 2000), until relatively recently less was known about the mechanisms which contribute to membrane repolarization. We have identified a role for the electrogenic Na+/Ca2+ exchanger operating in reverse mode in mediating membrane repolarization in activated neutrophils (CitationTintinger and Anderson 2004).
As opposed to being a major transporter of extracellular Ca2+ for store refilling, the primary role of the exchanger when operating in reverse mode is to mediate recovery of the membrane potential which is necessary to drive the influx of Ca2+ through store-operated Ca2+ channels (CitationTintinger and Anderson 2004). However, the Na+/Ca2+ exchanger is vulnerable to oxidative inactivation, being sensitive to the phagocyte-derived oxidants, hydrogen peroxide, and especially hypochlorous acid (CitationCoetzee et al 1994; CitationTintinger et al 2007). The efficiency of the exchanger in activated neutrophils is therefore dependent on its level of exposure to phagocyte-derived oxidants.
Store-operated Ca2+ channels
Until very recently, the precise molecular identity of the store-operated Ca2+ channels operative in human neutrophils and other cell types had not been conclusively established. One particular family of nonvoltage-activated Ca2+ channels which had attracted considerable attention and interest was the family of transient receptor potential (TRP) channels (CitationElliot 2001; CitationLiet al 2002). Although two members of this family, LTFP2 and TRP6, have been described in neutrophils (CitationLiet al 2002; CitationHeiner et al 2003), the characteristics of these channels are not entirely compatible with those of putative, prototype store-operated Ca2+ channels. However, in the past two years the identities of the major components of store-operated Ca2+ channels have been elucidated. These are the proteins Stim1 (and possibly Stim2), and Orai1 (and possibly Orai2 and 3), which function as the Ca2+ sensing and channel-forming proteins, respectively (CitationSpassova et al 2006). Stim1 is located in the endoplasmic reticulum where it binds reversibly to Ca2+. Following store depletion, dissociation of Ca2+ causes Stim1 to redistribute within the endoplasmic reticulum to areas which are in close proximity to Orai1 within the plasma membrane. Stim1 then activates the Ca2+-selective Oraichannels by a mechanism which remains to be elucidated (reviewed by CitationPutney 2007).
Neutrophil-directed anti-inflammatory strategies
Notwithstanding the increasing awareness of the anti-inflammatory potential of macrolide antimicrobialagents (CitationSimpson et al 2008), several other categories of anti-inflammatory agents have recently been described which have the potential to target neutrophils. These agents, described in detail elsewhere (CitationBarnes 2007), include: i) antagonists of receptors for chemoattractants such as C5a, IL-8, and LTB4; ii) antagonists of endothelialadhesion molecules; iii) inhibitors of pro-inflammatory enzymes and transcription factors such as phosphoinositide 3-kinase, 38 mitogen-activated protein kinase, and nuclear factor kappa B; and iv) activators of histone deacetylase 2 which is recruited by activated glucocorticoid receptors to switch off multiple proinflammatory genes.
In addition to these, recent insights into the mechanisms utilized by activated neutrophils to mobilize both intracellular and extracellular Ca2+, as wellas to restore Ca2+ homeostasis to the cells present a number of novel targets which are amenable to pharmacological control. In some cases target enhancement of activity improves the efficiency of clearance of Ca2+ from the cytosol, while in others this is achieved by target inhibition. Targets in the former category include endomembrane Ca2+ ATP-ases, NADPH oxidase, myeloperoxidase, and protein kinase C, while in the latter category, phospholipase C, the Na2+/Ca2+ exchanger, 5′-lipoxygenase, and store-operated Ca2+ channels represent attractive targets.
Ca2+ mobilization and restoration of Ca2+ homeostasis as targets for neutrophil-directed anti-inflammatory chemotherapy
Inhibitors of phospholipase C
The importance of PLC as a potential target is underscored by the critical involvement of IP3, not only in mediating the release of Ca2+ from intracellular stores, but also in sustaining elevations in cytosolic Ca2+ by promoting shuttling of the cation between the stores and the cytosol (CitationAnderson et al 2005) and possibly by initiating influx of Ca2+ (CitationMa 2000; CitationBolotina 2004). Furthermore, diacylglycerol (DAG) generated by PLC activates protein kinase C (PKC), which in turn promotes the assembly and activation of NADPH oxidase (CitationTauber 1987).
Although pharmacological inhibitors of PLC and antagonists of IP3 receptors such as U73122 and 2-aminoethoxydiphenyl borate respectively, are effective in experimental systems, no such inhibitors are available for clinicaluse. However, PLC activity is inhibited in various cell types such as vascular endothelial cells (CitationAvdonin and Ryan 2000) and platelets (CitationMurphy et al 1991) by a negative feedback loop involving PKC. This feedback inhibition on PLC by PKC appears to be operative in human neutrophils as we have demonstrated recently that in the presence of PKC inhibitors, such as GF109203X, IP3, and cytosolic Ca2+ concentrations in chemoattractant-activated neutrophils reach higher peaks and remain elevated longer than those measured in untreated cells (unpublished observations). This finding is important as it provides insight into the physiological mechanisms that down-regulate PLC in activated neutrophils and has implications for the design of pharmacological strategies targeting PKC, as inhibitors of this enzyme may paradoxically enhance the Ca2+-dependent proinflammatory activities of phagocytes. In contradistinction to the paucity of clinically useful PLC or PKC inhibitors, the endomembrane Ca2+ ATPase is considerably more amenable to pharmacologic interventions.
Upregulation of endomembrane Ca2+ ATP-ases
The activity of the Ca2+ resequestering endomembrane Ca2+ ATP-ases can be upregulated by the cAMP-sensitive enzyme, protein kinase A (PKA) (CitationChu et al 2000). PKA-mediated enhancement of the activity of the endomembrane Ca2+ ATP-ase markedly accelerates the clearance of cytosolic Ca2+ following release of the cation from storage vesicles (CitationAnderson et al 1998; CitationTintinger et al 2001b). The apparent sensitivity of PKA to cAMP has been exploited with the introduction of a wide range of pharmacologic agents, all of which share the ability to elevate intracellular cAMP concentrations. This can be achieved by several mechanisms, including activation of adenylate cyclase, or inhibition of phosphodiesterase enzymes (PDEs) responsible for the metabolism of cAMP (CitationMoore and Willoughby 1995). Adenylate cyclase is activated via G-protein-coupled receptors linked to membrane associated β-adrenergic, adenosine (A2A) and prostaglandin E2 receptors. PDE isoenzymes of various classes have been identified in neutrophils and PDE 4B2 is probably the most abundant of these (CitationWang et al 1999). The relative tissue specificity of PDE 4 isoenzymes for inflammatory cells has enabled some degree of pharmacological targeting of neutrophils by new generation agents such as roflimulast (CitationSanz et al 2007) and cilomilast (CitationBaumer et al 2007).
The validity of this pharmacologic approach has been confirmed in vitro as cAMP-elevating agents markedly suppress a range of Ca2+-dependent proinflammatory activities of neutrophils, including adhesion to vascular endothelium (CitationBloemen et al 1997), oxidant production (Tintinger et al 2000), as wellas release of proteases, eicosanoids, and cytokines (CitationMoore and Willoughby 1995; Tintinger et al 2000). Furthermore, numerous clinical trials are ongoing or have been completed in which the efficacy of these agents has been verified in vivo (CitationPauwels et al 1997; CitationSullivan et al 2001; CitationVignola 2004). Less well documented, however, is the role of cGMP during the restoration of Ca2+ homeostasis in activated neutrophils. Intracellular cGMP concentrations can be increased by activation of guanylate cyclase, or by inhibition of PDEs that metabolize cGMP, predominantly PDEs 3 and 5 (CitationTorphy 1998). The endomembrane Ca2+-ATPase has been identified as a putative target of cGMP acting via protein kinase G (CitationAy et al 2006), although other mechanisms may exist whereby cGMP accelerates the clearance of cytosolic Ca2+. In this regard, cGMP may elevate intracellular cAMP levels by effectively acting as a competitive antagonist of PDE 3-mediated hydrolysis of cAMP (PDE 3 hydrolyzes both cyclic nucleotides) (CitationBoswell-Smith et al 2006). The selective PDE 3 inhibitor, cilostazol, significantly attenuated FMLP-mediated adhesion of neutrophils to human umbilical endothelial cells in vitro (CitationYang et al 2006), and suppressed the cough threshold in elderly asthmatics (CitationIshiura et al 2005).
Inhibitors of 5-Lipoxygenase
Leukotrienes, such as LTB4, are produced by activated neutrophils consequent to upregulation of the 5-lipoxygenase (5-LO) pathway (CitationPeters-Golden and Henderson 2007). Phospholipase A2 hydrolyzes membrane phospholipids liberating arachidonic acid, the substrate for 5-LO, and in the presence of calcium and 5-lipoxygenase-activating protein (FLAP), activated 5-LO generates leukotriene A4, which in neutrophils is converted to LTB4. The diverse role of LTB4 in promoting inflammation includes recruitment of inflammatory cells (CitationDe Caterina and Zampolli 2004), activation of cytosolic PLA2 and degranulation (Boyce 2007), as well as the release of cytokines and matrix metalloproteinases, thus playing a role in immunoregulation (Rola-Pleszczynkiet al 1986; CitationFord-Hutchinson 1990; CitationLeppert et al 1995). In addition to the physiological responses above, LTB4 also participates in a positive feedback autocrine loop which activates phospholipase C and triggers sustained elevations of cytosolic Ca2+ in PAF-activated neutrophils (CitationMcDonald et al 1994; CitationSteel et al 2007). Intracellular LTB4 derived from 5-LO is actively transported to the cell exterior where it interacts with high affinity LTB4 receptors (BLT1) linked to PLC on the neutrophil plasma membrane. This interaction induces a secondary pulse of IP3 which mediates sustained release of Ca2+ into the cytosoland this in turn potentiates Ca2+-dependent pro-inflammatory responses (CitationSurette et al 1999; CitationSteel et al 2007). The validity of pharmacological anti-inflammatory strategies targeting LTB4 is underscored by recent evidence for the involvement of this leukotriene in the pathogenesis of bronchialasthma (CitationDe Caterina and Zampolli 2004), COPD (CitationMarian et al 2006), and inflammatory arthritis (CitationChen et al 2006). Importantly, the imidazole antimycotic, itraconazole, antagonizes the effects of LTB4 in PAF-activated neutrophils by inhibiting 5-LO (CitationSteel et al 2007). The concentrations of itraconazole required to inhibit 5-LO are similar to those achieved in the plasma of patients receiving this antimycotic (CitationKageyama et al 1999). Interestingly, other agents in this class, fluconazole and flutrimazole, may also possess anti-inflammatory properties as the former decreased mortality in critically ill patients by a mechanism unrelated to its antifungal properties (CitationJacobs et al 2003), while the latter inhibited arachidonic acid-induced ear edema in a murine model (CitationMerlos et al 1996). Other 5-LO inhibitors such as zileuton, which inhibits production of both LTB4 and the cysteinyl leukotrienes, LTC4 and LTD4, by activated eosinophils, have shown efficacy in clinical trials of patients with bronchialasthma (CitationPeters-Golden and Henderson 2007).
This is in keeping with the well established pathogenetic role of cysteinyl leukotrienes in inflammatory airway diseases (CitationArm 2004), and although neutrophils do not generate LTC4, or LTD4, receptors for cysteinyl leukotrienes, namely CysLT1 and CysLT2 are present on the plasma membrane of these cells. The physiological significance of these receptors is unknown, but we have found recently that cysteinyl leukotrienes act as receptor-mediated priming agents for human neutrophils in vitro. Pretreatment of neutrophils with these agents augments the subsequent responses to FMLP with marked increases in oxidant production and elastase release compared with cells activated with the chemoattractant alone (unpublished observations). This finding is likely to be of importance given the widespread use of CysLT1 receptor antagonists such as montelukast, pranlukast and zafirlukast, all of which may modulate cysteinyl leukotriene-mediated priming of neutrophils in a clinical setting.
NADPH oxidase
The membrane-associated NADPH oxidase complex is assembled on the plasma membrane and becomes incorporated into the phagocytic vacuole following phagocytosis of microbial pathogens. When activated, the oxidase generates highly ROS by transferring electrons from NADPH to molecular oxygen. This electrogenic transfer of electrons results in significant depolarization of the resting membrane potential. The oxygen radicals formed during this process include superoxide anions, hydroxyl radicals, hydrogen peroxide and hypochlorous acid (CitationNagata 2005), all of which may participate in the killing of microbial pathogens. However, these toxic molecules have also been implicated in the pathogenesis of numerous diseases as their release to the extracellular environment may damage innocent bystander host tissues (CitationMoraes et al 2006). Although inhibition of the oxidase may abolish the generation of toxic ROS, this also abrogates the membrane depolarization response and dissipates the electrical gradient required to restrain Ca2+ entry into leukocytes. Veritably, neutrophils lacking a functional oxidase such as those of patients with chronic granulomatous disease (CGD), are inherently predisposed to Ca2+ overload (CitationTintinger et al 2001a; CitationRada et al 2003) with consequent exaggeration of Ca2+-dependent proinflammatory activity, including increased release of proteolytic enzymes (CitationTintinger et al 2001a) and cytokines (CitationBylund et al 2007). NADPH oxidase thus fulfi lls dual roles as destroyer of microbial pathogens and in protecting the cell from Ca2+ flooding of the cytosol mediated by its important membrane depolarizing action. Therefore, somewhat paradoxically, enhancing the activity of the oxidase may uncover unforeseen anti-inflammatory properties (CitationHallett 2003; CitationHultqvist et al 2006).
In addition to mediating membrane depolarization, recent evidence suggests that the oxidase also indirectly modulates the rate of neutrophil membrane repolarization (CitationTintinger et al 2007). In this regard, myeloperoxidase (MPO) and hypochlorous acid have emerged as important regulators of the neutrophil membrane potential following activation of NADPH oxidase.
Myeloperoxidase and hypochlorous acid
Myeloperoxidase is stored in tertiary granules in the neutrophil cytosol and following the formation of phagocytic vacuoles is released into the phagosome, together with ROS generated from the concomitant activation of NADPH oxidase. Hydrogen peroxide thus formed acts as a substrate for MPO which catalyzes its conversion to the highly reactive oxidant, hypochlorous acid (HOCl). In the presence of MPO inhibitors such as sodium azide or 4-aminobenzoyl hydrazide (ABAH), the magnitude of neutrophil membrane depolarization in response to activating stimuli such as FMLP is not altered. However, the rate of recovery of the membrane potential is significantly accelerated (CitationTintinger et al 2007) and this is associated with an increased rate and magnitude of Ca2+ reuptake. Thus, inhibition of HOCl generation promotes rapid recovery of the membrane potentialand accelerates the rate of capacitative Ca2+ entry, underscoring the regulatory roles of MPO and HOCl during these events. This may paradoxically represent a physiologicalanti-inflammatory activity of HOCl given the calcium requirements of numerous pro-inflammatory neutrophil functions. Lending support to this contention, exaggerated inflammatory responses have been observed in the skin and lungs of MPO-deficient mice exposed to injurious ultraviolet light and irradiation, respectively (CitationMilla et al 2004; CitationKomatsu et al 2006).
Inhibition of store-operated calcium channels
The IP3-mediated release of Ca2+ from neutrophil storage vesicles is associated with activation of Ca2+ entry channels on the plasma membrane of these cells in order to facilitate store refilling. The mechanism of activation of these store-operated channels (SOCCs) remains controversial, although recent evidence suggests a role for proteins Stim 1 and Orai 1 (CitationSpassova et al 2006). Given the elusive nature of the structure and mechanism/s of activation of these channels, it has been difficult to design pharmacologic inhibitors of SOCCs. However, the recent advances in our understanding of these channels, notably Stim 1 and Orai 1 (CitationPutney 2007), may pave the way for the development of agents that antagonize this mechanism of Ca2+ reuptake. Regardless of the precise identity of, or coupling mechanisms that activate SOCCs, the rate of Ca2+ entry through these channels is regulated by the electrochemical gradient for Ca2+ across the plasma membrane (CitationGeiszt et al 1997). Accordingly, at depolarized potentials, Ca2+ reuptake is opposed by the electrical gradient and is only initiated during the recovery phase of the membrane potential. Crucially, the rate of membrane repolarization is modulated by the activity of the plasma membrane Na+/Ca2+ exchanger.
Inhibition of the Na+/Ca2+ exchanger
The reverse mode of the Na+/Ca2+ exchanger is activated by the rapid rise in cytosolic Ca2+ consequent to release of the cation from stores. When operating in reverse mode, the exchanger extrudes 3 Na+ ions for each Ca2+ ion entering the cell (CitationBlaustein and Lederer 1999), with a resultant net loss of positive charge. Although proton efflux from the cell is the primary charge compensating mechanism (CitationSchrenzel et al 1998; Bánfi et al 1999), the electrogenic Na+/Ca2+ exchanger contributes to recovery of the membrane potential (CitationTintinger and Anderson 2004) and represents a target amenable to pharmacologic intervention. Inhibitors of the reverse mode of the Na+/Ca2+ exchanger such as KB-R7943 markedly retard the rate of membrane repolarization with consequent attenuation of the rate and magnitude of Ca2+ reuptake (CitationTintinger and Anderson 2004). It is likely that delayed Ca2+ entry with incomplete refilling of Ca2+ storage vesicles mediated by inhibitors of the exchanger may down-regulate the proinflammatory activity of neutrophils. Agents that inhibit the reverse mode of the Na+/Ca2+ exchanger including KB-R7943 and SN-6 have shown promise in animal models of ischemia-reperfusion injury (Iwamato 2007), a condition in which activated neutrophils are considered to play a central pathogenetic role (CitationBuras and Reenstra 2007). Interestingly, the expression of Na+/Ca2+ exchanger genes is upregulated in leukocytes from patients with cardiac failure (CitationSeiler et al 2004).
Conclusions
Advances in our understanding of the physiologic mechanisms pertaining to calcium handling by activated neutrophils have facilitated recognition of novel pharmacologic strategies designed to suppress the pro-inflammatory activities of these cells. The distinct potential for agents such as imidazole anti-mycotics, antagonists of cysteinyl leukotriene receptors, inhibitors of the Na+/Ca2+ exchanger, long-acting beta-adrenergic receptor agonists and phosphodiesterase inhibitors to modulate neutrophil-mediated inflammation in vivo, creates exciting new opportunities for researchers in this important field.
Disclosure
The authors declare no conflicts of interest.
References
- AliHSozzaniSFisherI1998Differential regulation of formyl peptide and platelet-activating factor receptors: role of phospholipase Cβ, phosphorylation by protein kinase AJ Biol Chem27311012169556582
- AndersonRGoolam MahomedATheronAJ1998Effect of rolipram and dibutyryl cyclic AMP on resequestration of cytosolic calcium in FMLP-activated human neutrophilsBr J Pharmacol124547559647480
- AndersonRGoolam MahomedA1997Calcium efflux and influx in f-met-leu-phe (fMLP)-activated human neutrophils are chronologically distinct eventsClin Exp Immunol110132389353160
- AndersonRSteelHCTintingerGR2005Inositol 1,4,5-triphosphate-mediated shuttling between intracellular stores and the cytosol contributes to the sustained elevation in cytosolic calcium in FMLP-activated human neutrophilsBiochem Pharmacol6915677515896336
- ArmJP2004Leukotriene generation and clinical implicationsAllergy Asthma Proc25374215055561
- AvdoninPRyanUS2000Receptor-dependent regulation of [Ca2+]; and phospholipase C in vascular endothelial cellsJ Recept Signal Transduct Res202355411192020
- AyBIyanoyeASieckGC2006Cyclic nucleotide regulation of store-operated Ca2+ influx in airway smooth muscleAm J Physiol Cell Mol Physiol290L27883
- BánfiBMaturanaAJaconiS2000A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1Science2871384210615049
- BarnesPJ2007New molecular targets for the treatment of neutrophilic diseasesJ Allergy Clin Immunol11910556217353033
- BaumerWHoppmannJRundfeldtC2007Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasisInflamm Allergy Drug Targets6172617352685
- BlausteinMPLedererWJ1999Sodium/Calcium exchange: Its physiological implicationsPhysiol Rev7976385410390518
- BloemenPGMVan den TweelMCHendricksPAJ1997Increased cAMP levels in stimulated neutrophils inhibit their adhesion to human bronchial epithelial cellsAm J Physiol Lung Cell Mol Physiol16L5807
- BolotinaVM2004Store-operated channels: diversity and activation mechanisms [online]Sci STKE: Signal Transduction Knowledge Environment 200424pe34
- Boswell-SmithVSpinaDPageCP2006Phosphodiesterase inhibitorsBr J Pharmacol147S252716402111
- BurasJAReenstraWR2007Endothelial-neutrophil interactions during ischemia and reperfusion injury: basic mechanisms of hyperbaric oxygenNeurol Res291273117439696
- BylundJMacdonaldKLBrownKL2007Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-dependent activation of NF-kappa BEur J Immunol3710879617330823
- CarafolieEKesslerFFalchettoR1992The molecular basis of the modulation of the plasma membrane calcium pump by calmodulinAnn NY Acad Sci67158681337686
- CassatellaMA1999Neutrophil-derived proteins: selling cytokines by the poundAdv Immunol7336950910399011
- ChenMLamBKKanaokaY2006Neutrophil-derived leukotriene B4 is required for inflammatory arthirisJ Exp Med2038374216567388
- ChuGXLesterJWYoungKB2000A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta-agonistsJ Biol Chem275389384310988285
- CoetzeeWAIchikawaHHearseDJ1994Oxidant stress inhibits Na-Ca-exchange current in cardiac myocytes: mediation by sulphydryl groups?Am J Physiol Heart Circ Physiol266H90919
- De CaterinaRZampolliA2004From asthma to atherosclerosis – 5-lipoxygenase, leukotrienes, and inflammationN Engl J Med3504714702420
- ElliottAC2001Recent developments in non-excitble cell calcium entryCell Calcium30739311440466
- FavreCJNüsseOLewDP1996Store-operated calcium influx: what is the message from the stores to the membrane?J Lab Clin Med12819268759933
- Ford-HutchinsonAW1990Leukotriene B4 in inflammationCrit Rev Immunol101122155000
- GeisztMKapusANémetK1997Regulation of capacitative Ca2+ influx in human neutrophil granulocytes: alterations in chronic granulomatous diseaseJ Biol Chem2722647189334224
- GuichardCPedruzziEDewasC2007Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid raftsJ Biol Chem280370213216115878
- HallettMB2003Holding back neutrophilaggression; the oxidase has potentialClin Exp Immunol132181412699403
- HatanakaECarvalhoBTCCondino-NetoA2004Hyperresponsiveness of neutrophils from gp91phox deficient patients to lipopolysaccharide and serum amyloid AImmunol Lett9443615234534
- HeinerIEisfeldJHalaszovichCR2003Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes, evidence for currents through long TRP channel2 induced by ADP-ribose and NADBiochem J37110455312564954
- HultqvistMOlofssonPGeldermanKA2006A new arthritis therapy with oxidative burst inducersPLoS Med3162536
- IannoneMAWolbergGZimmermanTP1989Chemotactic peptide induces cAMP elevation in human neutrophils by amplification of the adenylate cyclase response to endogenously produced adenosineJ Biol Chem26420177802555342
- IshiuraYFujimuraMNobataK2005Phosphodiesterase 3 inhibition and cough in elderly asthmaticsCough (London, England)111
- IwamotoT2007Na+/Ca2+ exchange as a drug target – insights from molecular pharmacology and genetic engineeringAnn N Y Acad Sci10995162817446496
- JacobsSPrice EvansDATariqM2003Fluconazole improves survival in septic shock: a randomized double-blind prospective studyCrit Care Med3119384612847386
- KageyamaSMasuyaMTanakaI1999Plasma concentration of itraconazole and its antifungal prophylactic efficacy in patients with neutropenia after chemotherapy for acute leukemiaJ Infect Chemother52131611810520
- KomatsuJKoyamaHMaedaN2006Earlier onset of neutrophil-mediated inflammation in the ultraviolet-exposed skin of mice deficient in myeloperoxidase and NADPH oxidaseInflamm Res55200616830107
- LeppertDHausserSLKishiyamaJL1995Stimulation of matrix metalloproteinase-dependent migration of T-cells by eicosanoidsFASEB J91473817589989
- LiSWWestwickJPollCT2002Receptor-operated Ca2+ influx channels in leukocytes: a therapeutic target?Trends Pharmacol Sci23637011830262
- MaH-T2000Requirements of the inositol triphosphate receptor for activation of store-operated Ca2+ channelsScience28716475210698739
- MarianEBaraldoSVisentinA2006Up-regulated membrane and nuclear leukotriene B4 receptors in COPDChest12915233016778270
- McDonaldPPMcCollSRBraquetP1994Autocrine enhancement of leukotriene synthesis by endogenous leukotriene B4 and platelet-activating factor in human neutrophilsBr J Pharmacol111852608019762
- MerlosMVericatMLGarcia-RafanellJ1996Topical anti-inflammatory properties of flutrimazole, a new imidazole antifungalagentInflamm Res452058821774
- MillaCYangSCornfieldDN2004Myeloperoxidase deficiency enhances inflammation after allogeneic marrow transplantationAm J Physiol Lung Cell Mol Physiol287L7061415020295
- MooreARWilloughbyDA1995The role of cAMP regulation in controlling inflammationClin Exp Immunol10138797664483
- MoraesTJZurawskaJHDowneyGP2006Neutrophil granule contents in pathogenesis of lung injuryCurr Opin Hematol1321716319683
- MurphyCTElmoreMKellieS1991The relationship between cytosolic Ca2+, sn-1,2-diacylglyceroland inositol 1,4,5-triphosphate elevation in platelet-activating factor-stimulated rabbit platelets. Influence of protein kinase C on production of signal moleculesBiochem J278255611883334
- NagataM2005Inflammatory cells and oxygen radicalsCurr Drug Targets Inflamm Allergy4503416101529
- ParekhAPennerR1997Store depletion and calcium influxPhysiol Rev77901309354808
- PauwelsRALöfdahlC-GPostmaDS1997Effect of inhaled for-moteroland budesonide on exacerbations of asthmaN Engl J Med3371405119358137
- Peters-GoldenMHendersonWRJr2007Mechanisms of disease: LeukotrienesN Engl J Med35718415417978293
- PettitEJHalletMB1998Two distinct Ca2+ storage and release sites in human neutrophilsJ Leuk Biol6322532
- PutneyJWJr2007Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here)Cell Calcium421031017349691
- RadaBKGeisztMVan BruggenR2003Calcium signalling is altered in myeloid cells with a deficiency in NADPH oxidase activityClin Exp Immunol132536012653836
- Rola-PleszczynskiMChavaillazPALemaireI1986Stimulation of interleukin 2 and interferon gamma production by leukotriene B4 in human lymphocyte culturesProstaglandins Leukot Med23207103020587
- SanzMJCortijoJTahaMA2007Roflumilast inhibits leukocyte-endothelial cell interactions, expression of adhesion molecules and microvascular permeabilityBr J Pharmacol1524819217704822
- SchrenzelJSerranderLBànfiB1998Electron currents generated by the human phagocyte NADPH oxidaseNature392734379565037
- SeilerPUStypmannJBreidthardtG2004Real-time RT-PCT for gene expression profiling in blood of heart failure patients – A pilot studyBasic Res Cardiol99230815088108
- SimpsonJLPowellHBoyleMJ2008Clarithromycin targets neutrophilic airway inflammation in refractory asthmaAm J Respir Crit Care Med1771485517947611
- SivertsonKLSeedsMCLongDL2007The differential effect of dexamethasone on granulocyte apoptosis involves stabilization of Mcl-1L in neutrophils but not in eosinophilsCell Immunol246344517573055
- SpassovaMASoboloffJHeL-P2006STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channelsProc Natlacad Sci USA10340405
- SteelHCAndersonR2002Dissociation of the PAF-receptor from NADPH oxidase and adenylate cyclase in human neutrophils results in accelerated influx and delayed clearance of cytosolic calciumBr J Pharmacol13681911976271
- SteelHCTintingerGRTheronAJ2007Itraconazole-mediated inhibition of calium entry into platelet-activating factor-stimulated human neutrophils is due to interference with production of leukotriene B4Clin Exp Immunol1501445017683509
- StricklandIKisichKHaukPJ2001High constitutive glucocorticoid receptor β in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroidsJ Exp Med1935859311238589
- SullivanGWRiegerJMScheldWM2001Cyclic AMP-dependent inhibition of human neutrophil oxidative activity by substituted 2-propynylcyclohexyladenosine A2A receptor agonistsBr J Pharmacol13210172611226132
- SuretteMEKrumpEPicardS1999Activation of leukotriene synthesis in human neutrophils by exogenous arachidonic acid: inhibition by adenosine A(2a) receptor agonists and crucial role of autocrine activation by leukotriene B(4)Mol Pharmacol5610556210531413
- TauberAI1987Protein kinase C and the activation of human neutrophil NADPH-oxidaseBlood69711203545319
- TheronAJSteelHCTintingerGR2002Endogenous adenosine regulates neutrophil pro-inflammatory activities by cyclic AMP-dependent accelerated clearance of cytosolic calciumInflamm Res511911845995
- TintingerGRAndersonR2004Counteracting effects of NADPH oxidase and the Na+/Ca2+ exchanger on membrane repolarisation and store-operated uptake of Ca2+ by chemoattractant-activated human neutrophilsBiochem Pharmacol6722637115163557
- TintingerGRTheronAJAndersonR2001bThe anti-inflammatory interactions of epinephrine with human neutrophils in vitro are achieved by cyclic AMP-mediated accelerated resequestration of cytosolic calciumBiochem Pharmacol6113192811322936
- TintingerGRTheronAJPotjoM2007Reactive oxidants regulate membrane repolarization and store-operated uptake of calcium by formyl peptide-activated human neutrophilsFree Radic Biol Med421851717512464
- TintingerGRTheronAJSteelHC2001aAccelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous diseaseClin Exp Immunol1232546311207656
- TorphyTJ1998Phosphodiesterase enzymes: molecular targets for novelantiasthma agentsAm J Respir Crit Care Med157351709476844
- VignolaAM2004PDE4 inhibitors in COPD – a more selective approach to treatmentResp Med98495503
- WangPWuPOhlethKM1999Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophilsMol Pharmacol56170410385698
- Witko-SarsatVRieuPDescamps-LatschaB2000Biology of disease. Neutrophils: Molecules, functions and pathophysiologicalaspectsLab Invest56175310830774
- YangYLuoJKazumuraK2006Cilostazol suppresses adhesion of human neutrophils to HUVECs stimulated by FMLP and its mechanismsLife Sci706293616564549
- YueCDodgeKLWeberG1998Phosphorylation of serine 1105 by protein kinase A inhibits phospholipase Cβ, stimulation by GalphaqJ Biol Chem2731802379660757