Abstract
Glutathione S-transferase (GST) is a superfamily of detoxification enzymes, represented by GSTα, GSTμ, GSTπ, etc. GSTα is the predominant isoform of GST in human liver, playing important roles for our well being. GSTπ is overexpressed in many forms of cancer, thus presenting an opportunity for selective targeting of cancer cells. Our structure-based design of prodrugs intended to release cytotoxic levels of nitric oxide in GSTπ-overexpressing cancer cells yielded PABA/NO, which exhibited anticancer activity both in vitro and in vivo with a potency similar to that of cisplatin. Here, we present the details on structural modification, molecular modeling, and enzymatic characterization for the design of PABA/NO. The design was efficient because it was on the basis of the reaction mechanism and the structures of related GST isozymes at both the ground state and the transition state. The ground-state structures outlined the shape and property of the substrate-binding site in different isozymes, and the structural information at the transition-state indicated distinct conformations of the Meisenheimer complex of prodrugs in the active site of different isozymes, providing guidance for the modifications of the molecular structure of the prodrug molecules. Two key alterations of a GSTα-selective compound led to the GSTπ-selective PABA/NO.
Introduction
Glutathione S-transferases (GSTs, EC 2.5.1.18) are a superfamily of enzymes derived from distinct gene classes that have been designated α, μ, π, etc. GSTs catalyze the conjugation of the sulfur atom of glutathione (GSH) to an electrophilic center of endogenous and exogenous compounds, thereby increasing their aqueous solubility for subsequent excretion. Being ubiquitous and quite abundant in mammalian tissues, they initiate the metabolism of a broad range of alkylating agents and therefore play a central role in the detoxification of many carcinogens as well as anticancer chemotherapeutic agents (CitationJakoby and Habig 1980; CitationMannervik 1985; CitationPickett and Lu 1989; CitationArmstrong 1991, Citation1994, Citation1997; CitationHayes and Pulford 1995; CitationSheehan et al 2001; CitationDixon et al 2002). Of the superfamily, GSTπ is especially important in cancer therapy because it is often expressed at significantly higher levels in preneoplastic and neoplastic cells (CitationSato et al 1984; CitationSugioka et al 1985; CitationSuguoka et al 1985; CitationSato 1988, Citation1989; CitationMuramatsu et al 1995). It has also been shown that elevated levels of total GST and overexpression of GSTπ often accompany the development of drug resistance in tumors of patients undergoing chemotherapy (CitationMorgan et al 1996; CitationO’Brien and Tew 1996; CitationTownsend and Tew 2003). Such factors have stimulated recent efforts to target GSTs as a primary objective in the discovery of anticancer agents (CitationFlatgaard et al 1993; CitationLyttle et al 1994; CitationKauvar 1996; CitationRosario et al 2000; CitationTownsend et al 2002).
We have been trying to turn the GSTπ-overexpression to the tumor’s disadvantage by developing PABA/NO [O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]phenyl} 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate] that releases the established cytolytic agent nitric oxide (NO) upon metabolism by GSTπ (CitationFindlay et al 2004). PABA/NO belongs to a new family of anticancer prodrugs, the O2-aryl diazeniumdiolates (O2ADs), electrophilic species shown to transfer their aryl groups to attacking nucleophiles with cogeneration of ions that spontaneously release NO at physiological pH (CitationSaavedra et al 2001). The GST-catalyzed GSH addition of PABA/NO proceeds with the formation of a Meisenheimer-complex intermediate (), and subsequently the leaving group of the reaction releases two moles of NO (). Therefore, within the GST-overexpressing cancer cells, the intracellular GSH is irreversibly consumed, and the NO thus generated could contribute to chemotherapy by inhibiting DNA synthesis, forming toxic reactive nitrogen/oxygen intermediates, and inhibiting enzymes capable of preventing or repairing cellular damage. PABA/NO produces antitumor effects comparable with cisplatin in a human ovarian cancer model grown in SCID mice and is also potent against proliferation of the OVCAR-3 cell line (CitationFindlay et al 2004; CitationSaavedra et al 2006). PABA/NO is GSTπ-selective, ie, it is more efficiently metabolized by GSTπ It was designed on the basis of the mechanism of GST-catalyzed reaction and the structures of GSTs, especially the structural information of GSTs at the transition state.
Figure 1 The Meisenheimer complex. (a) The mechanism of GST-catalyzed reaction of GSH with PABA/NO and with CDNB, showing the formation of the Meisenheimer complex as the reaction intermediate. (b) The diazeniumdiolate ion releases two moles of NO at neutral pH. (c) A relatively stable model for the Meisenheimer complex is provided by the GSTCD− ion. (d) The GSTμ•GSTCD− structure (PDB entry 4GST). (e) The GSTπ•GSTCD− structure (PDB entry 1AQX). In panels d and e, the protein is shown as a molecular surface while the ligand as a stick model in atomic color scheme (carbon in gray, nitrogen in blue, oxygen in red, and sulfur in orange).
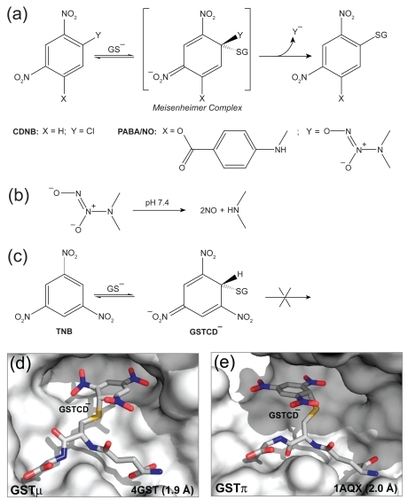
The attack of GSH on 1-chloro-2,4-dinitrobenzene (CDNB) is the basis for the most widely used spectrophotometric assay for measurement of GST activity (CitationHabig et al 1974). As shown in , the reaction proceeds in solution via the formation of the Meisenheimer-complex intermediate (CitationMiller 1968). Therefore, GST would stabilize the Meisenheimer-complex intermediate, at least to the extent that the intermediate resembles the transition state for its formation. However, the instability of the Meisenheimer complex has thus far precluded its structural elucidation. In contrast, the reversible reaction of GSH with 1,3,5-trinitrobenzene (TNB) to form the 1-(glutathion-S-yl)-2,4,6-trinitrocyclohexadienate anion (GSTCD−), a potent inhibitor of GST, provides a relatively stable model for this intermediate (CitationClark and Sinclair 1988; CitationGraminski et al 1989) as shown in . To date, crystal structures of the GSTμ•GSTCD− complex (PDB entry 4GST; CitationJi et al 1993) shown in and the GSTπ•GSTCD− complex (PDB entry 1AQX; CitationPrade et al 1997) shown in have been determined, which have guided us to model the GSTα•GSTCD− complex. Together, the structures and the model played an essential role in our structure-based development of PABA/NO.
Structure-based drug design is a growing field in which remarkable advances have been made in recent years (CitationAnderson 2003; CitationScapin 2006). Although the development of PABA/NO was briefly mentioned previously (CitationFindlay et al 2004), the details, involving structural modification, molecular modeling, and enzymatic characterization of two key intermediate compounds, have not been reported. Besides, the structure of PABA/NO in the previous report was not correct (CitationSaavedra et al 2006). Here, we present the structure-based design of PABA/NO in detail, during which the structures of different classes of GSTs, especially the structural information for the transition state of GST-catalyzed reaction of GSH and CDNB, played an essential role. With this unique example, we demonstrate again the importance of structure-based approach in drug development and the value of structural information for the transition state of an enzyme-driven reaction in structure-based drug design.
Materials and methods
Structural modifications of O2AD molecules
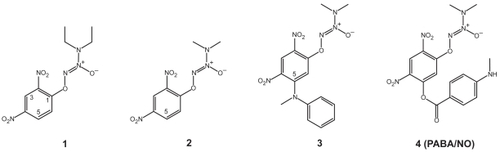
depicts our structural modifications starting from GSTα-selective compounds 1 and resulting in GSTπ-selective compound 4 (PABA/NO). Compounds 1, 2, and 3 were prepared as previously described (CitationSaavedra et al 2001), as was compound 4 (CitationFindlay et al 2004).
Figure 2 Structure-based design of PABA/NO. Structural modifications of the GSTα-selective compounds 1 and 2 have led to the GSTπ-selective compound 4 (PABA/NO).
Modeling the GSTα•GSTCD− complex
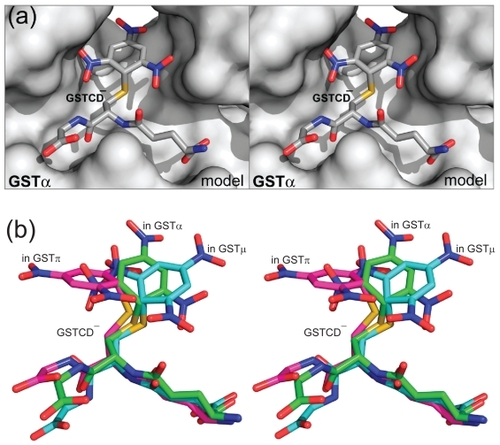
Molecular modeling studies were carried out on an SGI Fuel workstation with program packages CNS and O (CitationJones and Kjeldgaard 1997; CitationBrünger et al 1998). The initial model of GSTCD− bound to GSTα was built on the basis of the crystal structures of the GSTCD− found in the active sites of GSTμ (PDB entry 4GST), and GSTπ (PDB entry 1AQX), and then it was docked into the active site of GSTα in complex with the GSH adduct of ethacrynic acid (PDB entry 1GSE; CitationCameron et al 1995). The model complex, built in dimeric form because GSH interacts with the side chains from both subunits, was subject to geometry optimization using the conjugate gradient method by Powell embedded in CNS (CitationPowell 1977; CitationBrünger et al 1998). The Engh and Huber geometric parameters were used as the basis of the force field (CitationEngh and Huber 1991). The coordinates of the GSTα•GSTCD− model () are available from the corresponding author upon request.
Figure 3 The GSTα•GSTCD− model. (a) Stereoview showing the GSTα•GSTCD− model, constructed on the basis of the crystal structure of GSTα in complex with the GSH conjugate of ethacrynic acid (PDB entry 1GSE), the GSTμ•GSTCD− structure (PDB entry 4GST), and the GSTπ•GSTCD− structure (PDB entry 1AQX). The protein is shown as a molecular surface while the ligand as a stick model in atomic color scheme (carbon in gray, nitrogen in blue, oxygen in red, and sulfur in orange). (b) Stereoview showing the alignment of the GSTCD− ions in the GSTα•GSTCD− model, in the GSTμ•GSTCD− structure (PDB entry 4GST), and in the GSTπ•GSTCD− structure (PDB entry 1AQX). The GSTCD− ion is shown as a stick model in atomic color scheme (nitrogen in blue, oxygen in red, and sulfur in orange; carbon in green when bound in GSTα, cyan in GSTμ, and magenta in GSTπ).
Modeling GST with bound Meisenheimer complex of O2AD
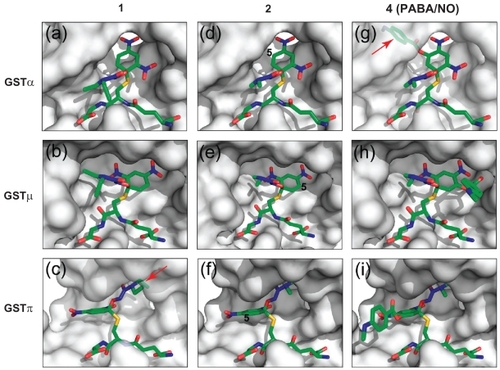
The initial models of the Meisenheimer complex of compounds 1 and 4 (GS1− and GS4−) bound to the three GST isozymes were built on the basis of the crystal structures of compounds 1 and 4 (CitationSaavedra et al 2001, Citation2006) and the GSTCD− structures in the GSTα•GSTCD− model, the GSTμ•GSTCD− structure (PDB entry 4GST), and the GSTπ•GSTCD− structure (PDB entry 1AQX). The initial models of GS2− were derived from those of GS4− by removing the 5-position bulky group, and then they were docked into the active sites of the corresponding GSTs. The dimeric model complexes, except for GSTπ•GS1− () and GSTα•GS4− (), were subject to geometry optimization in the same manner as for the GSTα•GSTCD− complex.
Figure 4 Models of the three GST isozymes with bound Meisenheimer complex of compound 1, 2, and 4. The GST active sites are illustrated as non-transparent surfaces, except for a transparent surface in panels c and g to show the unfavorable interaction between the ligand and the protein (indicated with red arrows), and the ligands as stick models in atomic color scheme (carbon in green, nitrogen in blue, oxygen in red, and sulfur in orange).
Kinetic studies
The recombinant GST isoenzymes were either purchased from Panvera or purified as described previously (CitationZimniak et al 1994). The reaction mixture, in a final volume of 1 mL, contained 0.1 M phosphate buffer (pH 6.5–7.0), 1–5 μg/mL of the desired GST protein, 1–2 mM GSH and varying concentrations of compound 1 or compound 2 (20–200 μM). Change in absorbance was monitored spectrophotometrically at 380 nm. The enzyme activity was calculated using an extinction coefficient of 3.58 mM−1 cm−1. Kinetic parameters were calculated using the Michaelis-Menten equation by software KiNET (Kinexus, Vancouver, Canada). The experiments were repeated three times and the results were averaged.
The same set of kinetic studies was carried out in two independent laboratory settings and the results were consistent.
Illustrations
The schematic illustrations for the crystal structures and the models were generated with PyMOL (CitationDeLano 2002).
Results and discussion
Structure of a transition-state analog in complex with GSTα
GSTα is the predominant isoform of GST in human liver, whereas GSTπ is most abundant in extrahepatic tissues and is overexpressed in many types of tumors. In order to target tumor cells while avoiding toxicity to the liver, structural information is needed for GSTα in complex with the transition-state analog GSTCD−. On the basis of available structural information, we have built a model of the GSTα•GSTCD− complex (). Together, the GSTμ•GSTCD− structure (PDB entry 4GST), the GSTπ•GSTCD− structure (PDB entry 1AQX), and the GSTα•GSTCD− model have shown that the conformation of the GSTCD− ion exhibits such dramatic differences when it is bound in the active site of different GSTs that the aromatic ring of the GSTCD− ion stretches out in distinct directions (), providing the guidance for our structure-based modification of O2AD molecules toward the GSTπ-selective PABA/NO.
Compound 1 is GSTα-selective
Compound 1 () is the first O2AD molecule we have ever synthesized (CitationSaavedra et al 2001). All three classes of GSTs catalyze the NO release of 1. However, it is GSTα-selective, 2-fold over GSTμ and 145-fold over GSTπ (). To understand the structural basis for the different catalytic efficiencies, we have constructed model complexes of the three GSTs with GS1−, the Meisenheimer complex of 1. The models of GSTα•GS1− () and GSTμ•GS1− () suggest favorable interactions between the protein and the reaction intermediate. The GSTπ•GS1− model, however, suggests unfavorable interaction between the diethyl group of the diazeniumdiolate moiety and the protein (). Therefore, the first modification of 1 was to replace the diethylamino group of the diazeniumdiolate moiety with a dimethyl-amino group, which was predicted to improve the fit of the reaction intermediate derived from the resulting compound to GSTπ.
Table 1 Kinetic data for human GST isoenzymes with Compound 1 as the variable substrate
Compound 2 indicates that our design is on the right track
As expected, compound 2 () is indeed a better substrate for GSTπ than compound 1. The catalytic efficiency of GSTπ toward 2 exhibits a noticeable increase (), which is likely due to the reduced steric interactions between the diazeniumdiolate moiety of the reaction intermediate and the protein (). Moreover, the two other GSTs are less active toward 2, with a 4-fold reduction in the catalytic efficiency for GSTα and a 3-fold reduction for GSTμ (). Although it is not as obvious, the reduction of the hydrophobic interactions between the protein and the diazeniumdiolate moiety of GS2−, the Meisenheimer complex of 2, may have played a role. Although 2 is still GSTα-selective, 1.5-fold over GSTμ and 24-fold over GSTπ, the improvement has indicated that our structure-based design is on the right track. The next modification is to further reduce the GSTα selectivity while increasing the GSTπ selectively.
Table 2 Kinetic data for human GST isoenzymes with Compound 2 as the variable substrate
We have constructed model complexes of GS2− with GSTα (), GSTμ (), and GSTπ (). The models suggest that the addition of a sterically demanding constituent at position 5 of the aryl ring should increase its accommodation in the active site of GSTπ () while decreasing its accommodation in the active site of GSTα (). Thus, compound 3 was designed and synthesized (). Unfortunately, the solubility of 3 was too low, rendering its enzymatic characterization impossible. Further modification aimed to improve the solubility has led to the synthesis of compound 4, ie, the PABA/NO ().
PABA/NO (Compound 4) is GSTπ-selective
As expected, PABA/NO (compound 4) is a better substrate for GSTπ compared to GSTα (CitationFindlay et al 2004). The model of GSTα in complex with the Meisenheimer complex of 4 (GSTα•GS4−) suggests unfavorable interaction between the bulky group at position 5 of the aryl ring system (). In contrast, both GSTμ and GSTπ can accommodate this group (, panels h and i). Thus, the structure-based modifications of 1 have reversed the GST isoenzyme selectively of O2AD.
GSTμ activity toward O2AD
The expression of GSTμ is null in 50% of the human population due to a gene deletion (CitationRowe et al 1997). Therefore, our current effort has been focused on GSTα and GSTπ while keeping GSTμ in the scope of our investigation for better understanding the reaction mechanism. Strategies have been developed to avoid GSTμ for the development of next-generation O2AD anticancer prodrugs.
The Meisenheimer complex of O2AD
We have previously shown that the transient Meisenheimer complex is generated in the reaction of compound 1 (CitationSaavedra et al 2001). On the basis of this mechanism, during which the Meisenheimer complex of O2AD is formed (), we have used the GSTμ•GSTCD− structure (), the GSTπ•GSTCD− structure (), and the GSTα•GSTCD− model () to guide the construction of the three GSTs in complex with the Meisenheimer complex of O2ADs ().
The model complexes of GST with the Meisenheimer complexes of O2AD have played a crucial role in guiding the modification of compounds 1 and 2, resulting in the development of compound 4, the GSTπ-selective PABA/NO as an anticancer drug lead. The key factors in this design include (1) understanding the difference in the shape and property of the substrate-binding site of different GSTs and (2) using the structures of GSTCD− in different GST complexes to guide the molecular modeling. For example, the substrate-binding site is broad in both GSTμ and GSTπ ( panels d and e). However, it is virtually a hydrophobic cavity in GSTμ, but is approximately half hydrophobic and half hydrophilic in GSTπ (CitationJi et al 1997). It is mainly the property of the binding site that dictates the orientation of the trinitrophenyl ring system of GSTCD− in these two isoenzymes. In support of this notion, the ring system of the GSH adduct of phenanthrene 9,10-oxide also points into opposite directions in these two isoenzymes (CitationJi et al 1994, Citation1997). Unlike GSTμ and GSTπ, GSTα has a narrower substrate-binding site, which limits the conformational freedom of the trinitrophenyl moiety of GSTCD− ().
Conclusions
This paper describes the details on structural modification, molecular modeling, and enzymatic characterization for structure-based design of PABA/NO. The design was efficient because it was on the basis of the reaction mechanism and the structures of related GST isozymes at both the ground state and the transition state. The ground-state structures outlined the shape and property of the substrate-binding site in different isozymes, and the known transition-state structures helped us build a model of GSTα at the transition state of which the structural information was not available. The structural information thus derived for the transition state, showing distinct conformations of the Meisenheimer complex of O2ADs in the active site of different isozymes, guided the modifications of the molecular structure of O2AD molecules. Two key alterations of a GSTα-selective compound, the reduction of the size of the amino group and the introduction of the bulky group, led to the GSTπ-selective PABA/NO.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute (NCI), Center for Cancer Research, and by federal funds from the NCI under contract N01-CO-12400. The content of this publication does not reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
References
- AndersonAC2003The process of structure-based drug designChem Biol107879714522049
- ArmstrongRN1991Glutathione S-transferases: reaction mechanism, structure, and functionChem Res Toxicol4131401782341
- ArmstrongRN1994Glutathione S-transferases: structure and mechanism of an archetypical detoxication enzymeAdv Enzymol Relat Areas Mol Biol691447817866
- ArmstrongRN1997Structure, catalytic mechanism, and evolution of the glutathione transferasesChem Res Toxicol102189074797
- BrüngerATAdamsPDCloreGM1998Crystallography and NMR system: A new software suite for macromolecular structure determinationActa Crystallogr D54905219757107
- CameronADSinningIL’HermiteG1995Structural analysis of human alpha-class glutathione transferase A1-1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugateStructure3717278591048
- ClarkAGSinclairM1988The Meisenheimer complex of glutathione and trinitrobenzene. A potent inhibitor of the glutathione S-transferase from Galleria mellonellaBiochem Pharmacol37259633342082
- DeLanoWL2002The PyMOL Molecular Graphics SystemSan Carlos, CADelano Scientific
- DixonDPLapthornAEdwardsR2002Plant glutathione transferasesGenome Biol3REVIEWS300411897031
- EnghRAHuberR1991Accurate bond and angle parameters for X-ray protein structure refinementActa Crystallogr A47392400
- FindlayVJTownsendDMSaavedraJE2004Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrugMol Pharmacol651070915102935
- FlatgaardJEBauerKEKauvarLM1993Isozyme specificity of novel glutathione-S-transferase inhibitorsCancer Chemother Pharmacol3363708269591
- GraminskiGFZhangPHSesayMA1989Formation of the 1-(S-glutathionyl)-2,4,6-trinitrocyclohexadienate anion at the active site of glutathione S-transferase: evidence for enzymic stabilization of sigma-complex intermediates in nucleophilic aromatic substitution reactionsBiochemistry28625282789999
- HabigWHPabstMJJakobyWB1974Glutathione S-transferases. The first enzymatic step in mercapturic acid formationJ Biol Chem249713094436300
- HayesJDPulfordDJ1995The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistanceCrit Rev Biochem Mol Biol304456008770536
- JakobyWGHabigWH1980Enzymatic Basis of DetoxicationNew YorkAcademic Press
- JiXArmstrongRNGillilandGL1993Snapshots along the reaction coordinate of an SNAr reaction catalyzed by glutathione transferaseBiochemistry3212949548241147
- JiXJohnsonWWSesayMA1994Structure and function of the xenobiotic substrate-binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthreneBiochemistry331043528110735
- JiXTordovaMO’DonnellR1997Structure and function of the xenobiotic substrate-binding site and location of a potential non-substrate-binding site in a class pi glutathione S-transferaseBiochemistry3696907029245401
- JonesTAKjeldgaardM1997Electron-density map interpretationMethods Enzymol27717320818488310
- KauvarLM1996Peptide mimetic drugs: a comment on progress and prospectsNat Biotechnol147099630974
- LyttleMHHockerMDHuiHC1994Isozyme-specific glutathione-S-transferase inhibitors: design and synthesisJ Med Chem37189948289195
- MannervikB1985The isoenzymes of glutathione transferaseAdv Enzymol Relat Areas Mol Biol573574173898742
- MillerJ1968Reaction Mechanisms in Organic ChemistryNew YorkElsevier
- MorganASCiaccioPJTewKD1996Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell linesCancer Chemother Pharmacol37363708548883
- MuramatsuMSuzukiTHisatakeKVermeulenNPEMulderGJNieuwenhuyseH1995Regulation of glutathione transferase P-expression during hepatocarcinogenesis of the RatGlutathione S-Transferease – Structure, Function and Clinical ImplicationsLondonTaylor and Francis8595
- O’BrienMLTewKD1996Glutathione and related enzymes in multidrug resistanceEur J Cancer32A967788763337
- PickettCBLuAY1989Glutathione S-transferases: gene structure, regulation, and biological functionAnnu Rev Biochem58743642673020
- PowellMJD1977Restart procedures for the conjugate gradient methodMath Prog1224154
- PradeLHuberRManoharanTH1997Structures of class pi glutathione S-transferase from human placenta in complex with substrate, transition-state analogue and inhibitorStructure51287959351803
- RosarioLAO’BrienMLHendersonCJ2000Cellular response to a glutathione S-transferase P1-1 activated prodrugMol Pharmacol581677410860939
- RoweJDNievesEListowskyI1997Subunit diversity and tissue distribution of human glutathione S-transferases: interpretations based on electrospray ionization-MS and peptide sequence-specific antiseraBiochem J325Part 248169230131
- SaavedraJESrinivasanABonifantCL2001The secondary amine/as nucleophile and leaving nitric oxide complex ion R2N[N(O)NO]− group in SNAr reactionsJ Org Chem663090811325274
- SaavedraJESrinivasanABuzardGS2006PABA/NO as an anticancer lead: analogue synthesis, structure revision, solution chemistry, reactivity toward glutathione, and in vitro activityJ Med Chem4911576416451080
- SatoK1988Glutathione S-transferases and hepatocarcinogenesisJpn J Cancer Res79556723136107
- SatoK1989Glutathione transferases as markers of preneoplasia and neoplasiaAdv Cancer Res52205552662713
- SatoKKitaharaASatohK1984The placental form of glutathione S-transferase as a new marker protein for preneoplasia in rat chemical hepatocarcinogenesisJpn J Cancer Res75199202
- ScapinG2006Structural biology and drug discoveryCurr Pharm Des1220879716796557
- SheehanDMeadeGFoleyVM2001Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamilyBiochem J36011611695986
- SugiokaYFujii-KuriyamaYKitagawaT1985Changes in polypeptide pattern of rat liver cells during chemical hepatocarcinogenesisCancer Res45365783965145
- SuguokaYKanoTOkudaA1985Cloning and the nucleotide sequence of rat glutathione S-transferase P cDNANucleic Acids Res136049572995915
- TownsendDTewK2003Cancer drugs, genetic variation and the glutathione-S-transferase gene familyAm J Pharmacogenomics31577212814324
- TownsendDMShenHStarosAL2002Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer cellsMol Cancer Ther110899512481432
- ZimniakPNanduriBPikulaS1994Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic propertiesEur J Biochem22489397925413