Abstract
Obesity constitutes a critical risk factor for the development of many life threatening diseases, particularly insulin resistance and type 2 diabetes. Adipose tissue plays an important role in regulating whole body energy homeostatsis and obesity-related insulin resistance. Inflammation has been commonly linked to insulin resistance. Recent studies demonstrated that adipose tissue is an important source for producing inflammatory molecules in the obese state, primarily due to accumulation of macrophages. Animal models deficient in key inflammatory molecules or with reduced adipose macrophage infiltration are protected from development of obesity-related insulin resistance. Repression of adipose inflammation may be a useful approach to ameliorate obesity-associated metabolic disorders.
Correlation between obesity-related insulin resistance and inflammation
Approximately 30% of US adults and 20% of school-aged children are obese. Obesity has become an epidemic disease and its incidence is still rapidly increasing at an alarming rate (CitationHill et al 2003). The hallmark of obesity is manifested by massive expansion of adipose tissue, attributable to excessive food consumption and sedentary life style in modern society. Obesity has been identified as a major risk factor for the development of many life-threatening diseases, such as hypertension, atherosclerosis, dyslipidemia, and particularly type 2 diabetes (CitationKopelman 2000). Type 2 diabetes itself is also a risk factor for the development of heart disease and stroke. As a matter of fact, more than 65% of people with diabetes die from heart disease or stroke. The underlying molecular mechanism for obesity-related metabolic disorders is still not well understood. The correlation between obesity, diabetes and inflammation have been observed as early as several decades ago, when increased circulating levels of acute phase proteins, such as fibrinogen, haptoglobin, and α-1 antitripsin, were reported (CitationFearnley et al 1959; CitationOgston and McAndrew 1964; CitationGanrot et al 1967). In more recent studies, elevated serum concentrations of more acute phase proteins were shown, including sialic acid, α-1 acid glycoprotein, C-reactive protein (CRP) and serum amyloid A (CitationCrook et al 1993; CitationPickup et al 1997). In addition, inflammatory proteins such as IL-6 and plasminogen activator inhibitor 1 (PAI-1), as well as increased white cell count are also found to correlate with type 2 diabetes (CitationFesta et al 2002; CitationPradhan et al 2001; CitationVozarova et al 2002). However, findings from these human population studies alone were not sufficient to stimulate thoughts regarding whether inflammation could be a causative factor for the pathogenesis of obesity-related insulin resistance and type 2 diabetes.
Recent studies indicate that inflammatory event in the adipose tissue may play a critical role in the development of insulin resistance. Tumor necrosis factor-α (TNFα) is the first cytokine identified with elevated expression in adipose tissue from obese rodents and humans, resulting from both increased production and abnormal processing (CitationHotamisligil et al 1993, Citation1995; CitationXu et al 2002b). TNFα inhibits signaling from the insulin receptor and neutralization of TNFα by infusion with a tumor necrosis factor receptor-immunoglobin G (TNFR-IgG) chimeric protein improves insulin sensitivity (CitationFeinstein et al 1993; CitationHotamisligil et al 1993, Citation1994). TNFα can also cause dysregulated lipolysis in adipocytes, therefore potentially contributes to the elevation of circulating free fatty acid (FFA) concentration (CitationGreen et al 1994). FFA has been demonstrated to be a critical factor for causing systemic insulin resistance in multiple tissues, such as liver and muscle. The discovery that inflammatory cytokine TNFα impairs insulin signaling greatly stimulated the field and scientists started to view that inflammation may be a causative event for the pathogenesis of obesity-related insulin resistance. Subsequently, more cytokines, chemokines and other secretary proteins are found to increase in adipose tissue of obese rodents and/or humans, including leptin, interleukin-6 (IL-6), IL-8, monocyte chemotactic factor 1 (MCP-1), angiotensinogen, visfatin, resistin, retinol-binding protein-4, serum amyloid A (SAA). Among these molecules, IL-6, IL-8, and MCP-1 have been reported to impair insulin action (CitationRotter et al 2003; CitationSartipy and Loskutoff 2003). The reason that cytokine and chemokine production increases in response to triglyceride accumulation in adipose tissue is not clear. One possibility is that these factors might have designated roles to limit excessive fat mass expansion, which unfortunately is at the cost of inducing insulin resistance. Consistent with this hypothesis, TNFα, IL-8, and MCP-1 have been reported to inhibit adipocyte differentiation (CitationXu et al 1999; CitationGerhardt et al 2001; CitationXu and Hotamisligil 2001). Animals deficient in CCR2, the major receptor of MCP-1, have larger adipocytes but are more insulin sensitive (CitationWeisberg et al 2006). This hypothesis is further supported by two animal models: adipose tissue TNFα transgenic and fat-specific insulin receptor knockout mice. Both models demonstrated reduced adiposity but accompanied by severely impaired insulin signaling in adipose tissue (CitationBluher et al 2002; CitationXu et al 2002a). However, the systemic insulin sensitivity has been improved despite adipose insulin resistance, suggesting the importance of overall fat mass on metabolism.
Animal models deficient in key inflammatory molecules
Intensive investigations with various animal models deficient in key molecules in the inflammatory pathway further provided solid evidence for the causative role of inflammation in the development of obesity-related insulin resistance. The first proof of principle animal model is TNFα-deficient mice. In the absence of TNFα, mice are partially protected from development of hyperlipidymia, hyperglycemia and hyperinsulinemia without an effect on body weight in obesity models induced by high fat diet, deficiency of leptin, or goldthioglucose injection (CitationUysal et al 1997; CitationVentre et al 1997). Increased tyrosine phosphorylation of insulin receptor was observed in both adipose tissue and muscle in obese mice deficient in TNFα. In addition, KB-R7785, a synthetic compound that inhibits TNFα production, can improve insulin sensitivity in obese/diabetic KKAy mice (CitationMorimoto et al 1997). These in vivo data support a role of TNFα as a candidate mediator of obesity-induced insulin resistance but also indicate the involvement of other factors since the protection is partial. TNFα signals through two receptors, p55 and p75. The role of TNFα receptors in mediating the effect of TNFα on obesity-related insulin resistance is controversial in two independent studies, which might be attributable to differences in genetic backgrounds (CitationSchreyer et al 1998; CitationUysal et al 1998).
Recently, the c-Jun NH2-terminal kinase (JNK) has appeared as a vital regulator in obesity and insulin resistance. Both TNFα and FFA are potent stimulators for JNK activation. Indeed, JNK activity is elevated in liver, adipose tissue and muscle in both diet-induced obese mice and ob/ob mice (CitationHirosumi et al 2002). Interestingly, different JNK isoforms seem to play distinct roles. JNK1 deficient mice have decreased adiposity and adipocyte size, increased serum adiponection and lowered serum resistin concentrations, lowered glucose and insulin concentrations, enhanced insulin signaling in the liver and improved whole body insulin sensitivity upon high fat diet (CitationHirosumi et al 2002). Absence of JNK1 also partially protected ob/ob mice from developing hyperglycemia and hyperinsulinemia (CitationHirosumi et al 2002). In contrast, deficiency of JNK2 does not have any influence on diet-induced insulin resistance. These results demonstrate that JNK1 accounts for the majority of increased JNK activity in insulin target tissues and is an important regulator in obesity-related insulin resistance.
IKKβ, the master regulator of inflammation which can also be activated by TNFα and FFA, is another important serine kinase involved in the development of obesity-related insulin resistance. Homozygous IKKβ deficiency is lethal due to increased liver apoptosis but heterozygous IKKβ+/− mice appear normal. Lowered fasting glucose and insulin concentrations were observed in IKKβ+/− mice on both standard diet and high-fat diet compared to wild-type littermates (CitationYuan et al 2001). Furthermore, absence of one IKKβ allele is sufficient to lower blood glucose and plasma FFA levels as well as improve glucose tolerance in ob/ob mice (CitationYuan et al 2001). CitationArkan and colleagues (2005) further selectively deleted IKKβ in hepatocytes or myeloid cells and showed that IKKβ deficiency in hepatocytes locally protected liver from development of obesity-related insulin resistance, but fat and muscle still develop insulin resistance upon high fat diet. Consistent with these results, transgenic mice selectively expressing the constitutively active form of IKKβ in the liver have increased hepatic production of proinflammatory cytokines, which caused profound hepatic insulin resistance (CitationCai et al 2005). In addition, these proinflammatory cytokines also cross talk with other tissues and lead to hyperglycemia, and modest systemic insulin resistance (CitationCai et al 2005). Interestingly, specific deletion of IKKβ in myeloid cells of diet-induced obese mice or ob/ob mice is sufficient to improve systemic insulin sensitivity, which is attributable to decreased production of proinflammatory cytokines or chemokines that are known to inhibit insulin signaling in cultured cells (CitationArkan et al 2005). These data demonstrate that IKKβ plays an important role in the development of obesity-related insulin resistance by acting in multiple cell types, particularly myeloid cells.
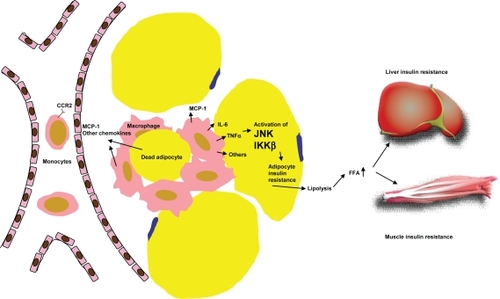
Figure 1 Mechanism of adipose macrophage infiltration and the role of adipose inflammation in systemic insulin resistance. Accumulation of triglycerides in adipocytes causes increased production of MCP-1 and the other monocyte chemotactic factors, which attract circulating monocytes to migrate into adipose tissue. These inflamed monocytes mature into classically activated macrophages and secrete a variety of proinflammatory factors that can impair insulin signaling in adipocytes. Dysregulated adipocyte lipolysis contributes to elevation of circulating FFA level which subsequently induces insulin resistance in other tissues such as liver and muscle.
TLR4, a family member of toll-like receptors (TLRs), plays a critical role in pattern recognition and activation of innate immunity (CitationMedzhitov et al 1997). Lipopolysaccharide (LPS) is the well characterized ligand for TLR4 (CitationPoltorak et al 1998). Several recent studies demonstrate that FFAs, whose levels are elevated in obesity, can also activate TLR4 (CitationLee et al 2001). Deficiency of TLR4 can prevent FFA-induced production of inflammatory cytokines in both macrophages and adipocytes. In lipid-infused animals, absence of TLR4 significantly improved insulin signaling in muscle and decreased production of inflammatory cytokines in adipose tissue (CitationShi et al 2006). Absence of TLR4 also modestly improved systemic insulin sensitivity in female mice upon high fat challenge, presumably through reduced production of proinflammatory cytokines in liver and adipose tissue. Similar phenotypes are also observed in diet-induced obese mice with a loss-of-function mutation in TLR4, confirming a role of TLR4 in linking innate immunity and lipid-induced insulin resistance (CitationTsukumo et al 2007).
Adipose tissue macrophage accumulation in obesity
Despite the substantive amount of data demonstrating the link between inflammation and obesity-related insulin resistance, the specific cell source for production of inflammatory molecules are unclear though it appears to be originated from adipose tissue. It was originally thought that adipocytes are the cell type that produces proinflammatory factors. It has been shown that adipocytes possess many features of immune cells and adipocyte precursors have phagocytic capacity and can be converted into macrophage-like cells in response to certain stimuli (CitationCousin et al 1999; CitationCharriere et al 2003). Two studies in 2003, which used transcription profiling approach to compare adipose tissue from lean and several rodent obesity models, or adipose tissue from obese animals before and after treatment with compounds of the thiazolidinedione class of anti-diabetic agents, have showed that the majority of genes significantly upregulated in obese adipose tissue are macrophage and inflammatory genes, which can be repressed by rosiglitazone (CitationWeisberg et al 2003; CitationXu et al 2003). By separating adipocytes from stromal-vascular cells and histological examination, these studies revealed massive macrophage accumulation in adipose tissue in the obese state. Bone marrow transplant study further demonstrated that these macrophages are predominantly infiltrated, rather than converted from local preadipocytes. Activated macrophages are known to secrete a variety of pro-inflammatory cytokines and chemokines. In these two studies, isolated stromal-vascular cells, the fraction containing infiltrated macrophages, are demonstrated to be the predominant source for producing proinflammatory molecules in comparison to adipocytes, including TNFα, MCP-1, IKKβ, IL-1β, and inducible nitric oxide synthase (iNOS). These results suggest that infiltrated macrophages in adipose tissue in state of obesity are likely an important source contributing to elevation of circulating inflammatory marker proteins. Blood mononuclear cells (MNCs) in the obese state are also reported to be in a pro-inflammatory state, as reflected by increased nuclear factor kappa-B (NF-κB) binding activity, decreased IκB content and increased transcription of NF-κB controlled proinflammatory genes, indicating that inflamed circulating MNCs may also contribute to elevation of circulating inflammatory marker proteins as well (CitationGhanim et al 2004).
The triggering factors for macrophage infiltration into adipose tissue and the roles of infiltrating macrophages are still not well understood. One speculation is that enlargement of fat cells, which commonly occurs in obese state at relatively early stage, may trigger the initial secretion of critical monocyte chemotactic factors. Once macrophages infiltrate into adipose tissue and get activated, they become the major source of inflammatory molecules and continue to attract more macrophages to further amplify the harmful cycle. The prototype monocyte chemotactic factor, MCP-1, has been under intensive investigation as such a candidate for recruiting macrophages into adipose tissue in obesity since its expression is upregulated in adipose tissue in early stage of diet-induced obesity. However, controversial results have been reported for the role of MCP-1. One publication showed that mice deficient in MCP-1 have partially reduced adipose tissue macrophage accumulation upon high fat diet and are accompanied with improved insulin sensitivity and hepatic steatosis (CitationKanda et al 2006). Acute expression of a dominant-negative MCP-1 mutant also ameliorated insulin resistance in both diet-induced obese mice and db/db mice (CitationKanda et al 2006). Another publication demonstrated no change of adipose macrophage content in MCP-1 knock out mice (CitationInouye et al 2007). These results suggest the roles of other monocyte chemotactic factors. Nevertheless, reports from two independent laboratories reported that mice overexpressing MCP-1 transgene in adipose tissue under the promoter of adipocyte fatty acid binding protein have increased macrophage accumulation in adipose tissue and are insulin resistant (CitationKamei et al 2006; CitationKanda et al 2006). The major receptor of MCP-1 is CCR2, which also mediates signaling of other monocyte chemotactic factors such as MCP-2 and MCP-3. Macrophages recruited into adipose tissue in obese state have increased expression of CCR2. Mice deficient in CCR2 have a lean phenotype when fed on a high fat diet due to reduced food intake. When high fat-diet fed CCR2 deficient mice and control mice are matched for adiposity, partially decreased adipose macrophage content was observed. These mice have improved inflammatory profile in adipose tissue, along with improved systemic insulin sensitivity and ameliorated hepatic steatosis (CitationWeisberg et al 2006). DIO mice treated with CCR2 antagonist have a 28% reduction of adipose macrophage content and have improved hyperglycemia (CitationWeisberg et al 2006). These data suggest that CCR2 plays a role in adipose tissue macrophage recruitment in obese state and also point out the potential role(s) of other chemokine receptor(s) since absence of CCR2 only partially blocked macrophage infiltration upon high fat diet.
Macrophages are phagocytic cells that clear pathogens, dead cells and cell debris in innate immunity and wound healing. It has been shown that adipocyte death increases significantly in obese humans and mice via the mechanism of necrosis. The majority of infiltrated macrophages in adipose tissue are localized around dead adipocytes to form crown-like structure (CLS) (CitationCinti et al 2005). This scenario highly resembles atherosclerosis, which is caused by macrophage infiltration into aorta to clean excessive cholesterol and subsequent formation of foam cells. Interestingly, frequency of adipocyte death in epidydimal fat pat correlates with the extent of macrophage infiltration (CitationStrissel et al 2007). These data indicate that infiltrated macrophages are involved in clearance of cell debris, leaked lipids and perhaps are important for adipose tissue remodeling. Macrophages are heterogeneous in function and their properties and activation state are dependent upon local environment factors. Two polarization states, M1 or “classically activated” and M2 or “alternatively activated”, have been defined for macrophage activation. M1 macrophages have high proinflammatory cytokine expression and produce reactive oxygen species; M2 macrophages express high levels of anti-inflammatory cytokines, such as IL-10. IL-10 can selectively inhibit transcription rates of inflammatory gene and activate STAT3 to attenuate inflammatory signals induced by TNFα. Macrophages infiltrated into adipose tissue in obese state possess features of M1 macrophages and more importantly, macrophages in CLS express protein of TNFα and IL-6 (CitationLumeng et al 2007; CitationStrissel et al 2007). In contrast, resident macrophages in adipose tissue of lean mice have many features of M2 macrophages, including expression of M2 macrophage characteristic genes such as Ym1, arginase 1, and IL-10. High fat diet is sufficient to promote macrophage phenotypic switch from anti-inflammatory M2 polarization to proinflammatory M1 polarization. Peroxisome proliferator activated receptor-γ (PPARγ), the molecular target for the anti-diabetic thiazolidinedione class of compounds, has been reported to play a key role in maturation of alternatively activated macrophages with anti-inflammatory properties (CitationBouhlel et al 2007). Mice deficient of PPARγ in myeloid cells have impaired activation of M2 macrophages, as demonstrated by decreased expression and activity of arginase, hallmarks of alternatively activated macrophages (CitationHevener et al 2007; CitationOdegaard et al 2007). These mice have lower content of alternatively activated macrophages, increased local inflammation in adipose tissue, impaired insulin signaling in adipose tissue, muscle, and liver on chow diet. These mice are also prone to the development of obesity-related insulin resistance. These results indicate that phenotypic switch from M2 to M1 macrophages are critical for impairing insulin sensitivity.
Transient neutrophil infiltration into adipose tissue in obesity is also reported, which occurs 3 and 7 days upon high fat diet in epididymal but not subcutaneous fat and disappears on all subsequent times points (CitationElgazar-Carmon et al 2008). A potential chemotactic factor for neutrophil infiltration has been proposed to be IL-8, which can be secreted by adipose tissue in obesity. Neutrophils have been demonstrated to directly adhere to adipocytes and this process can be influenced by cations and activation state of neutrophils. The time course of adipose tissue neutrophil infiltration precedes macrophage infiltration, which does not start increase till 8 weeks upon high fat diet (CitationStrissel et al 2007). Neutrophil infiltration reflects acute inflammation while macrophage infiltration symbols chronic inflammation. Activated neutrophils can release ROS and TNFα, whether infiltrated neutrophils contribute to adipose inflammation remains a question to be studied.
Targeting inflammation as a potential therapeutic approach for improving insulin sensitivity
The findings for a potential role of adipose inflammation in the development of insulin resistance from animal models can also be extended to humans. Macrophage accumulation has been observed in human adipose tissue and the extent has strong positive correlation with BMI and adipocyte area but negatively related to insulin sensitivity (CitationWeisberg et al 2003). The macrophage content is depot dependent. Omental fat of obese humans contains more macrophages than subcutaneous fat. Omental fat macrophage infiltration correlates to fasting glucose, insulin and hepatic fibroinflammatory lesions in obese patients (CitationCancello et al 2006). In addition, obese subjects with impaired glucose homeostasis have preferential omental adipose macrophage infiltration (CitationHarman-Boehm et al 2007). Weight loss in obese subjects either through low calorie diet or bariatric surgery have decreased expression of inflammatory factors and increased expression of anti-inflammatory molecules, accompanied with decreased macrophage number and improved metabolic profile (CitationClement et al 2004; CitationCancello et al 2005). These data suggest that anti-inflammatory reagents or blockage of macrophage infiltration into adipose tissue may be beneficial for improving systemic insulin resistance in obese patients.
Inflammation has been generally correlated with insulin resistance in many situations, such as trauma, burn and infection. Although it is a relatively recent finding that adipose inflammation is associated with obesity-related insulin resistance, the effect of anti-inflammation reagent sodium salicylate on improving symptoms of diabetes mellitus was demonstrated a century ago (CitationWilliamson 1901). Later on, high dose aspirin was discovered to not only improve arthritis in a diabetic patient, but also make daily insulin injection unnecessary for controlling blood glucose level (CitationReid et al 1957). Cessation of aspirin upon disappearance of joint symptoms caused recurrence of glucose intolerance. Subsequent clinical trial with high dose-aspirin reconfirmed the effect on lowering blood glucose levels (CitationHecht and Goldner 1959). The molecular target mediating the hypoglycemic effect of salicylates has now been identified, which is IKKβ (CitationYuan et al 2001). IKKβ activates NF-κB, which is a master transcription factor that activates transcription of numerous inflammatory genes. Potent small molecule inhibitor aginst IKKβ may be beneficial for improving insulin reistance in obese subjects.
Another line of evidence that repression of inflammation may be useful for controlling hyperglycemia in type 2 diabetic patients derives from the anti-inflammatory effect of the widely used insulin sensitizer. Thiazolidinediones (including rosiglitazone and pioglitazone), a new and highly efficacious class of insulin-sensitizing drugs that mainly targeting adipose tissue by activating PPARγ, have been reported to have anti-inflammatory effect. PPARγ is a ligand-dependent nuclear hormone receptor predominantly expressed in adipocyte and is a master regulator of adipocyte differentiation. Expression of PPARγ is significantly increased upon macrophage activation and treatment with thiazolidinediones inhibits production of monocyte inflammatory cytokines and expression of iNOS, gelatinase B and scavenger genes, partly through repressing the activities of transcription factors AP-1, STAT, and NF-κB. In animal models, PPARγ agonists have been reported to improve development of atherosclerosis (CitationLi et al 2000; CitationStojanovska et al 2007). These results indicate that thiazolidinediones can repress macrophage-mediated inflammation. Interestingly, atherosclerosis-prone apolipoprotein E-deficient mice transplanted with inflamed visceral fat developed significantly more atherosclerosis compared to sham-operated animals. Treatment with pioglitazone decreased macrophage content of the transplanted visceral fat tissue, reduced plasma MCP-1 and reduced atherosclerosis (CitationOhman et al 2008). Ten-week treatment with pioglitazone in subjects with impaired glucose tolerance reduced macrophage number in adipose tissue and decreased inflammatory cytokine production (CitationDi Gregorio et al 2005). Furthermore, thiazolidinediones are also reported to suppress NF-κB activity in inflamed circulating MNCs in obese subjects (CitationGhanim et al 2001, Citation2006; CitationMohanty et al 2004). These results suggest that the insulin sensitizing effect of thiazolidinediones could be partially achieved through repression of adipose tissue macrophage content and subsequent reduction of inflammatory factor production.
In summary, substantive evidence derived from both animal models and human studies supports an important role of adipose inflammation in the development of obesity-related insulin resistance. Development of new therapeutic agents targeting improving adipose inflammation, such as blocking macrophage infiltration and preventing phenotypic switch of M2 to M1 macrophages, may be effective for attenuating insulin resistance and hyperglycemia associated with obesity.
Disclosure
The authors report no conflicts of interest in this work.
References
- ArkanMCHevenerALGretenFR2005IKK-beta links inflammation to obesity-induced insulin resistanceNat Med11191815685170
- BluherMMichaelMDPeroniOD2002Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intoleranceDev Cell3253812110165
- BouhlelMADerudasBRigamontiE2007PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory propertiesCell Metab61374317681149
- CaiDYuanMFrantzDF2005Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaBNat Med111839015685173
- CancelloRHenegarCViguerieN2005Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight lossDiabetes5422778616046292
- CancelloRTordjmanJPoitouC2006Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesityDiabetes5515546116731817
- CharriereGCousinBArnaudE2003Preadipocyte conversion to macrophage. Evidence of plasticityJ Biol Chem2789850512519759
- CintiSMitchellGBarbatelliG2005Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humansJ Lipid Res4623475516150820
- ClementKViguerieNPoitouC2004Weight loss regulates inflammation-related genes in white adipose tissue of obese subjectsFASEB J1816576915522911
- CousinBMunozOAndreM1999A role for preadipocytes as macrophage-like cellsFASEB J13305129973318
- CrookMATuttPSimpsonH1993Serum sialic acid and acute phase proteins in type 1 and type 2 diabetes mellitusClin Chim Acta21913187508342
- Di GregorioGBYao-BorengasserARasouliN2005Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazoneDiabetes5423051316046295
- Elgazar-CarmonVRudichAHadadN2008Neutrophils transiently infiltrate intra-abdominal fat early in the course of high fat feedingJ Lipid Res49189490318503031
- FearnleyGRVincentCTChakrabartiR1959Reduction of blood fibrinolytic activity in diabetes mellitus by insulinLancet2106713821819
- FeinsteinRKanetyHPapaMZ1993Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substratesJ Biol Chem2682605588253716
- FestaAD’AgostinoRJrTracyRP2002Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis studyDiabetes511131711916936
- GanrotPOGydellKEkelundH1967Serum concentration of alpha-2-macroglobulin, haptoglobin and alpha-1-antitrypsin in diabetes mellitusActa Endocrinol (Copenh)55537444166389
- GerhardtCCRomeroIACancelloR2001Chemokines control fat accumulation and leptin secretion by cultured human adipocytesMol Cell Endocrinol175819211325518
- GhanimHAljadaAHofmeyerD2004Circulating mononuclear cells in the obese are in a proinflammatory stateCirculation11015647115364812
- GhanimHDhindsaSAljadaA2006Low-dose rosiglitazone exerts an antiinflammatory effect with an increase in adiponectin independently of free fatty acid fall and insulin sensitization in obese type 2 diabeticsJ Clin Endocrinol Metab913553816804037
- GhanimHGargRAljadaA2001Suppression of nuclear factor-kappaB and stimulation of inhibitor kappaB by troglitazone: evidence for an anti-inflammatory effect and a potential antiatherosclerotic effect in the obeseJ Clin Endocrinol Metab8613061211238525
- GreenADobiasSBWaltersDJ1994Tumor necrosis factor increases the rate of lipolysis in primary cultures of adipocytes without altering levels of hormone-sensitive lipaseEndocrinology134258188194485
- Harman-BoehmIBluherMRedelH2007Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesityJ Clin Endocrinol Metab922240717374712
- HechtAGoldnerMG1959Reappraisal of the hypoglycemic action of acetylsalicylateMetabolism84182813666386
- HevenerALOlefskyJMReichartD2007Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinedionesJ Clin Invest11716586917525798
- HillJOWyattHRReedGW2003Obesity and the environment: where do we go from here?Science299853512574618
- HirosumiJTuncmanGChangL2002A central role for JNK in obesity and insulin resistanceNature420333612447443
- HotamisligilGSArnerPCaroJF1995Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistanceJ Clin Invest952409157738205
- HotamisligilGSBudavariAMurrayD1994Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alphaJ Clin Invest94154397523453
- HotamisligilGSShargillNSSpiegelmanBM1993Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistanceScience25987917678183
- InouyeKEShiHHowardJK2007Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissueDiabetes5622425017473219
- KameiNTobeKSuzukiR2006Overexpression of monocyte che-moattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistanceJ Biol Chem281266021416809344
- KandaHTateyaSTamoriY2006MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesityJ Clin Invest116149450516691291
- KopelmanPG2000Obesity as a medical problemNature4046354310766250
- LeeJYSohnKHRheeSH2001Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4J Biol Chem27616683911278967
- LiACBrownKKSilvestreMJ2000Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient miceJ Clin Invest1065233110953027
- LumengCNBodzinJLSaltielAR2007Obesity induces a phenotypic switch in adipose tissue macrophage polarizationJ Clin Invest1171758417200717
- MedzhitovRPreston-HurlburtPJanewayCAJr1997A human homologue of the Drosophila Toll protein signals activation of adaptive immunityNature38839479237759
- MohantyPAljadaAGhanimH2004Evidence for a potent antiinflammatory effect of rosiglitazoneJ Clin Endocrinol Metab8927283515181049
- MorimotoYNishikawaKOhashiM1997KB-R7785, a novel matrix metalloproteinase inhibitor, exerts its antidiabetic effect by inhibiting tumor necrosis factor-alpha productionLife Sci617958039275009
- OdegaardJIRicardo-GonzalezRRGoforthMH2007Macrophage-specific PPARgamma controls alternative activation and improves insulin resistanceNature44711162017515919
- OgstonDMcAndrewGM1964Fibrinolysis in obesityLancet21205714215560
- OhmanMKShenYObimbaCI2008Visceral adipose tissue inflammation accelerates atherosclerosis in apolipoprotein E-deficient miceCirculation11779880518212290
- PickupJCMattockMBChusneyGD1997NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome XDiabetologia401286929389420
- PoltorakAHeXSmirnovaI1998Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 geneScience282208589851930
- PradhanADMansonJERifaiN2001C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitusJAMA2863273411466099
- ReidJMacDougallAIAndrewsMM1957Aspirin and diabetes mellitusBr Med J21071413472052
- RotterVNagaevISmithU2003Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjectsJ Biol Chem278457778412952969
- SartipyPLoskutoffDJ2003Monocyte chemoattractant protein 1 in obesity and insulin resistanceProc Natl Acad Sci U S A10072657012756299
- SchreyerSAChuaSCJrLeBoeufRC1998Obesity and diabetes in TNF-alpha receptor- deficient miceJ Clin Invest102402119664082
- ShiHKokoevaMVInouyeK2006TLR4 links innate immunity and fatty acid-induced insulin resistanceJ Clin Invest11630152517053832
- StojanovskaLHonisettSYKomesaroffPA2007The anti-atherogenic effects of thiazolidinedionesCurr Diabetes Rev3677418220657
- StrisselKJStanchevaZMiyoshiH2007Adipocyte death, adipose tissue remodeling, and obesity complicationsDiabetes562910817848624
- TsukumoDMCarvalho-FilhoMACarvalheiraJB2007Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistanceDiabetes5619869817519423
- UysalKTWiesbrockSMHotamisligilGS1998Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesityEndocrinology139483289832419
- UysalKTWiesbrockSMMarinoMW1997Protection from obesity-induced insulin resistance in mice lacking TNF-alpha functionNature38961049335502
- VentreJDoebberTWuM1997Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and non-obese miceDiabetes461526319287059
- VozarovaBWeyerCLindsayRS2002High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetesDiabetes514556111812755
- WeisbergSPHunterDHuberR2006CCR2 modulates inflammatory and metabolic effects of high-fat feedingJ Clin Invest1161152416341265
- WeisbergSPMcCannDDesaiM2003Obesity is associated with macrophage accumulation in adipose tissueJ Clin Invest112179680814679176
- WilliamsonRT1901On the treatment of glycosuria and diabetes mellitus with sodium salicylateBr Med J1760220759517
- XuHBarnesGTYangQ2003Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistanceJ Clin Invest11218213014679177
- XuHHirosumiJUysalKT2002aExclusive action of transmembrane TNF alpha in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistanceEndocrinology14315021111897709
- XuHHotamisligilGS2001Signaling pathways utilized by tumor necrosis factor receptor 1 in adipocytes to suppress differentiationFEBS Lett5069710211591379
- XuHSethiJKHotamisligilGS1999Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1J Biol Chem274262879510473584
- XuHUysalKTBechererJD2002bAltered tumor necrosis factor-alpha (TNF-alpha) processing in adipocytes and increased expression of transmembrane TNF-alpha in obesityDiabetes5118768312031976
- YuanMKonstantopoulosNLeeJ2001Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IkkbetaScience2931673711533494