Abstract
This article examines the role of pegvisomant in the treatment of acromegaly. This syndrome, caused by excessive growth hormone (GH) secretion by a pituitary adenoma, is associated with a doubled mortality rate and poor quality of life. Pituitary microsurgery has long been the first choice of treatment since it cures many patients, especially those with localized tumors. Adjuvant irradiation was given if insulin-like growth factor-I (IGF-I) or GH did not normalize. The introduction of long-acting slow- release somatostatin analogs was a breakthrough for adjuvant treatment, although not always effective. Rather, targeting excessive GH production, muting the GH signal at its receptor, was a totally different approach. The development of GH antagonists (by mutation of glycine at position 120) and other modifications to enhance receptor binding, and subsequent pegylation of the molecule led to the development of B2036. After pegylation of B2036 at 5 positions the distribution volume is restricted and its serum half-life considerably increased. In short-term clinical studies performed in selected, mostly pretreated, acromegalic patients, IGF-I normalized in the majority of cases. Combination therapy with long-acting somatostatin analogs and weekly rather than daily pegvisomant injections appears to be successful in one clinical study and might limit the high cost of pegvisomant. Long-term efficacy and safety has to be demonstrated. The drug does not cross the blood–brain barrier, and whether it distributes freely into the extracellular space of other organs than the liver has not been investigated, which might have implications for persistent local IGF-I production under unrestrained GH concentrations.
Introduction
The most obvious effect of growth hormone (GH) is promoting height in prepubertal and pubertal subjects by stimulating cell division and differentiation of chondroblasts in the growth plates of bones. In adult life, however, many other effects can be attributed to GH. These actions include stimulation of protein synthesis, increased lipolysis and inhibition of protein catabolism. GH is also involved in bone remodeling, muscle growth, insulin antagonism, renal sodium retention, and immuno-modulation. Most of these mentioned effects, however, are not the result of the direct action of GH, but they are mediated via insulin-like growth factor-I (IGF-I), a peptide synthesized and secreted after GH-signaling, which acts locally in a paracrine or autocrine fashion or distantly as a hormone, when secreted by the liver (CitationLe Roith et al 2001). The latter organ is responsible for 70%–80% of the circulating IGF-I (CitationSjogren et al 1999; CitationSjogren et al 2002). The importance of IGF-I for body growth is underscored by observations of severely stunted growth in animals and the human with inactivating mutations IGF-I or its receptor (CitationWoods et al 1996; CitationWalenkamp et al 2005). Acquired GH deficiency in the human leads to a characteristic syndrome, which can be treated successfully with physiological replacement therapy (CitationJanssen et al 1997). In contrast, chronic excessive secretion of GH by a pituitary adenoma leads to acromegaly, a disfiguring and debilitating condition, causing premature death (CitationEzzat et al 1994).
The purpose of this review is to discuss the role of the nanomedicine, pegvisomant in the treatment of acromegaly. Before discussing its role, we shall first review briefly essential aspects of GH physiology, GH molecular structure and signaling, the acromegaly syndrome and the currently used therapeutical approaches in the treatment of this disease.
Growth hormone physiology
Growth hormone is a single chain polypeptide hormone that is synthesized, stored and secreted by the somatotropic cells of the pituitary gland. The 22 kDa GH isoform represents 90% of plasma GH (CitationBaumann 1991). The other circulating GH molecule is the 20 kDa isoform, which is a post-translational RNA splicing variant form. In plasma, GH is mainly bound to the GH-binding protein (GHBP), which is derived from the extracellular domain of the GH receptor by proteolytic cleavage (CitationSotiropoulos et al 1993). This binding protein may enhance or limit tissue actions of GH and reduces the GH clearance rate from 2–5 minutes (free GH) to 16–19 minutes (bound GH). Growth hormone is cleared via renal and hepatic mechanisms.
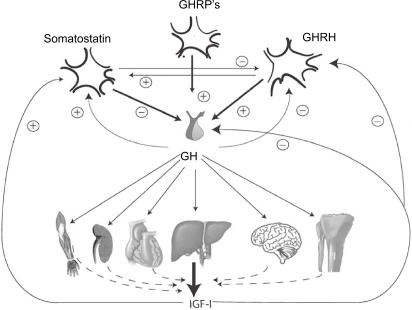
Control of GH secretion is regulated at the hypothalamic and the pituitary level by stimulatory and inhibitory hormones resulting in a diurnal pulsatile secretion pattern by which the majority of GH is released during sleep (CitationGiustina and Veldhuis 1998). The hypothalamic stimulatory growth hormone-releasing hormone (GHRH) stimulates GH gene transcription, GH-cell proliferation, and GH release. The hypothalamic inhibitory hormone somatostatin acts via binding of the somatostatin receptors and inhibits GH release from the secretory granules in the somatotropes, but also attenuates GHRH release as illustrated in . Other negative feedback signals regulating GH secretion are GH itself at the hypothalamic level and IGF-I at the hypothalamic and pituitary level. The physiological role of the GH-stimulatory hormone ghrelin, the native substrate for the GH- releasing peptide (GHRP) receptor, is not fully elucidated, but large GH responses are induced by GHRP or ghrelin infusion and this hormone acts synergistically with GHRH (CitationKojima et al 1999).
Figure 1 Schematic outline of the principal components of GH regulation, ie, somatostatin, GHRH, GHRPs (including ghrelin) and IGF-I. Circulating IGF-I is derived mainly from the liver and from other organs to a lesser extend. Positive stimulation and feedback are indicated by the + symbol, and inhibition or negative feedback by the – symbol.
Abbreviations: GHRH, hypothalamic stimulatory growth hormone-releasing hormone; GHRPs, GH- releasing peptides; IGF-1, insulin-like growth factor-I.
GH stimulates the production of IGF-I in many organs. IGF-I is a polypeptide belonging to the same family of growth factors as insulin. Most circulating IGF-I is bound to one of the six IGF binding proteins (IGFBPs), mainly to IGF-BP3 in a ternary complex with acid labile subunit (ALS). The serum IGF-I concentration reflects the 24-hour GH secretion and is increased or decreased, when GH production is elevated or decreased, respectively. An important determinant for the IGF-I concentration is age. Increasing age is associated with a decreasing GH production and IGF-I concentration. Sex steroids, especially estrogens, inhibit GH-mediated IGF-I production. Malnutrition and anorexia nervosa are associated with high GH levels, but with low serum IGF-I concentrations. On the other hand, obesity, especially visceral obesity is associated with a low serum GH concentrations, but with a normal serum IGF-I concentration (CitationGiustina and Veldhuis 1998; CitationPijl et al 2001).
Acromegaly
Acromegaly is a rare pituitary disorder, in which a pituitary adenoma hyper secretes GH. The incidence of acromegaly is 3–4 per one million per year and the prevalence is 60–70 per one million as was found in the UK, Spain, and Sweden, without geographical and sex differences (CitationAlexander et al 1980; CitationBengtsson et al 1988; CitationRitchie et al 1990; CitationMestron et al 2004). The disease is likely under-diagnosed and it is possible that incidence and prevalence rates are therefore higher than previously reported. Acromegaly is associated with increased incidence of vascular disease, cardiomyopathy, and an increased prevalence of cardiac valvular abnormalities and malignancies, especially of the gastrointestinal tract (CitationColao 2004). Patients with active acromegaly have a two- to three-fold increased mortality risk due to cardiovascular and respiratory diseases and possibly cancer (CitationWright et al 1970).
Assessment of body composition in active acromegaly shows increased body weight and height, increased total body water and extracellular water, and reduced body fat (CitationBengtsson et al 1989). Due to hyperprolactinemia patients may have galactorrhea, amenorrhea, hirsutism, impotence, or infertility. Local tumoral effects include headache, visual field defects with typical hemianopsia, and sporadically cerebral nerve dysfunction, especially of the trigeminal, trochlear, or abducens nerves. Hypopituitarism may occur in a small percentage of patients (~10%) especially those with large tumors leading to symptoms related to hypothyroidism, hypogonadism and hypocortisolism.
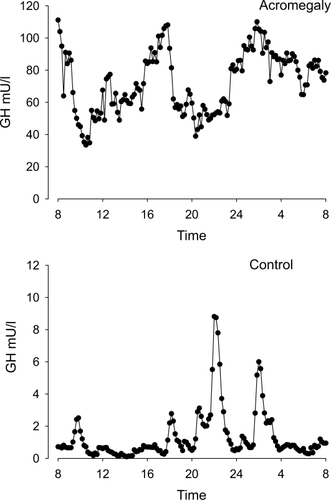
Detailed studies of GH secretion in acromegaly have demonstrated increased basal GH production, increased pulse frequency, disturbed diurnal secretion and decreased regularity () (CitationHo and Weisberger 1994; Citationvan den Berg et al 1994).
Figure 2 Serum GH concentrations obtained by 10 min blood sampling for 24 hours in a female patient with acromegaly and an age- and gender-matched healthy control subject. Note the difference in y-scale. In the patient, GH secretion is characterized by the increased non-pulsatile component, increased pulsatile secretion, and burst frequency.
Abbreviation: GH, growth hormone.
Biochemical criteria for diagnosis and remission largely depend on the GH assay used. Therefore, reference values should be determined in each laboratory for all biochemical parameters used in the diagnosis and follow-up of patients with acromegaly. In healthy controls, after oral glucose load of 75 g the serum GH is suppressed to low levels. In contrast, in active acromegaly, the serum GH concentration is insufficiently suppressed after glucose loading. The glucose tolerance test (GTT) therefore provides a reproducible standardized test, although there are interpretative difficulties in overt diabetes mellitus and renal and hepatic disease. In addition, the test has not been validated for evaluation of the GH suppression during medical therapy (CitationGiustina et al 2000; CitationFreda 2003). No data are available on the relation between glucose-suppressed GH concentrations and morbidity and mortality.
Serum IGF-I, a marker that reflects the mean serum GH concentration, is elevated in all patients with active acromegaly and may be decreased in the GH-deficient state (CitationHoffman et al 1994; Citationvan der Lely et al 1997). As IGF-I concentration decreases with age, values should be interpreted after adjustment for age. In addition, gender, sex hormone status and body mass index may influence the serum IGF-I (and GH) concentrations. Importantly, normalized IGF-I after pituitary surgery leads to normal survival (CitationSwaeringen et al 1998; CitationBiermasz et al 2004).
Treatment of acromegaly
Treatment options in acromegaly are surgery, radiotherapy, and medical therapy or combinations of these and the various modalities are reviewed below.
Surgery
In most centers pituitary surgery is performed via the trans-sphenoidal route with very low mortality and low morbidity, although transcranial surgery may be required in very large suprasellar tumors. After selective and complete adenoma removal, GH secretion is reported to normalize completely (Citationvan den Berg et al 1998). Microadenoma (ie, adenoma smaller than 10mm) removal is successful in most cases (80%–90%), but with increasing size and expansion of the adenoma total tumor removal is more difficult and only 30%–40% of the patients with a large macroadenoma can be cured (CitationFreda et al 1998; CitationBiermasz et al 2000c). Second surgical procedures are generally safe, but less successful than primary surgery (CitationLong et al 1996). The experience of the neurosurgeon is crucial for the success rate (CitationAhmed et al 1999).
Radiotherapy
Conventional radiotherapy is administered by a linear accelerator (4–8 MeV) in a total dose of 40–50 Gy fractionated in at least 20 sessions. A rotational field, laterally opposed fields, or 3 fields have been used. A decline of about 50% in serum GH levels is observed in the first 2 years after radiotherapy and after 5 years a 75% decline (CitationBiermasz et al 2000a, Citation2000b; CitationWass 2003). Whether GH and IGF-I levels normalize in the follow-up mainly depends on the pre-irradiation serum GH concentration. Remission rates of radiotherapy are thus affected by surgical intervention (debulking) prior to radiotherapy. The incidence of hypopituitarism increases with the duration after radiotherapy and about 50% of the patients require replacement therapy following radiotherapy (CitationBiermasz et al 2000b).
Other radiation techniques include proton beam irradiation and stereotactic radiosurgery (gamma knife) (CitationMahmoud-Ahmed et al 2001). With radiosurgery a high single dose is administered at the stereotactically mapped region so that the tumor is ablated precisely while the surrounding tissue receives a low radiation dose. At present there is no convincing evidence available that radiosurgery is superior to conventional radiotherapy in terms of GH control, time needed for reaching safe GH levels, and incidence of hypopituitarism (CitationLandolt et al 1998; CitationAttanasio et al 2003; CitationBiermasz et al 2006).
Medical treatment
Medical therapy for acromegaly started with the use of estrogens and later with chlorpromazine, but the important break-through came with the discovery that dopamine and bromocriptine could decrease GH levels in many acromegalic patients, although not in all. A further major improvement in treatment was the introduction of somatostatin analogs. The development of GH receptor antagonists is the latest in this line.
Dopamine agonists
Bromocriptine, a dopamine agonist, effectively reduces GH secretion in only a minority of GH-secreting adenoma (CitationRoelfsema et al 1979; CitationJaffe and Barkan 1992). Cabergoline, a new more potent dopamine agonist with prolonged duration of action, normalized GH in 35% and IGF-I in 44% of 46 patients with a purely GH-secreting adenoma in a dose of 1–1.75 mg/week (CitationAbs et al 1998), but this result has not been confirmed by others. The efficacy was somewhat larger in patients co-secreting prolactin. Quinagolide, another dopamine agonist, normalized IGF-I in 28% of the patients (CitationFreda 2003). Therefore it is not surprising that the addition of the dopamine agonist cabergoline to chronic somatostatin analog treatment in patients who fail to normalize GH secretion under monotherapy is still able to decrease serum IGF-I concentrations into the normal range in 30%–40% of patients, irrespective of the prolactin concentration (CitationCozzi et al 2004). Side-effects of cabergoline are rare.
Somatostatin analogs
Somatostatin analog treatment has been the most important medical therapy for more than a decade. Compared with dopamine agonists this class of drugs is much more efficient in suppressing GH in acromegaly. The currently used analogs, octreotide and lanreotide inhibit GH secretion via the somatostatin receptor subtypes 2 and 5 (CitationHofland and Lamberts 2003). The plasma half-life is about 20 times longer than that of native somatostatin. Although the most important effect of somatostatin analogs is the inhibition of tumor-derived GH and the subsequent fall in circulating liver-derived IGF-I, part of the peripheral effects are caused by a direct inhibition of IGF-I gene transcription via activation after binding to the somatostatin receptor (CitationSerri et al 1992; CitationLe Roith et al 2001; CitationMurray et al 2004). The magnitude of this latter effect in the various organs is not precisely known. The introduction of long-acting repeatable (LAR) forms using monthly intramuscularly injectable microspheres of octreotide (octreotide-LAR, Sandostatin® LAR®, Novartis Pharmaceutical, Basel, Switzerland), the 1- to 2-weekly injections with lanreotide prolonged release (Somatuline® PR, Ipsen SA, Paris, France), and the more recent subcutaneously monthly injection formula lanreotide Autogel® (Somatuline® Autogel®, Ipsen SA, Paris, France) has improved further the treatment results and facilitated the use of these agents (CitationFreda 2003) Several multicenter studies have shown that disease activity is controlled in 40%–60% of the patients (CitationLancranjan et al 1999; CitationCozzi et al 2003; CitationCozzi et al 2006). Other somatostatin analogs currently investigated in Phase 2 and 3 studies were recently reviewed (CitationRoelfsema et al 2005).
Tumor volume reduction of GH adenoma with a weighted mean of 19.4% has been reported to occur in 62% of acromegalic patients during primary therapy with somatostatin analogs (CitationMelmed et al 2005). Medical pretreatment before surgery of especially macroadenoma, however, does not clearly improve outcome (CitationBiermasz et al 1999; CitationBen Shlomo and Melmed 2003). Clinically important is the observation that the decrease of GH below 2.2 μg/L during an intravenous or subcutaneous test with octreotide predicts the long-term GH suppression with octreotide LAR to safe levels, thus avoiding non-successful treatment in some patients, who will be better off with pegvisomant or combined treatment (see below) (CitationBiermasz et al 2005; CitationGilbert et al 2005; CitationKaravitaki et al 2005).
GH antagonists
Above we have outlined the various therapeutical approaches to cure or to control excessive GH secretion. Rather than diminishing or normalizing GH secretion at the pituitary level, it is also possible to neutralize or diminish the GH signal at the peripheral cellular level. Theoretically, this could be accomplished by the construction of GH receptor antagonists and by RNA antisense techniques, thus preventing the synthesis of the GH receptor. Indeed, the recently developed antisense oligonucleotide, ATL 227446, effectively inhibited hepatic GH receptor expression and IGF-I production and decreased body weight gain in mice (CitationTachas et al 2006). Below we shall review the molecular structure of GH and its receptor and physiology which all are keystones for the development of GH receptor antagonists.
GH and GH receptor structure
GH is synthesized and secreted by the somatotroph cells of the anterior pituitary gland. The GH gene consists of 5 exons and 4 introns encoding a 217-amino acid precursor protein. By proteolysis the amino-terminal signal peptide is removed, yielding the mature single chain 191-amino acids polypeptide, with a molecular mass of 22 kDa. The 3-dimensional structure of human (h) GH and of GH from other mammalian species was established by X-ray crystallography (CitationUltsch et al 1991, Citation1993; Citationde Vos et al 1992). The protein consists of four α-helices, with 20–30 residues bound together by stretches of non-helical chains which are packed together in an antiparallel bundle (CitationUltsch et al 1994).
The GH receptor belongs to the cytokine-hemopoietine receptor superfamily (CitationBazan 1990). This transmembrane protein contains 620 amino acids. The receptor lacks intrinsic kinase activity, but GH binding to its extracellular domain induces the recruitment and activation of the tyrosine kinase, Janus kinase 2 (JAK2). Subsequently, the activated JAK2 phosphorylates the receptor and STATs (signal transducer and activator of transcription), and dimers of the latter are translocated to the nucleus to promote gene transcription (CitationCarter-Su and Smit 1998).
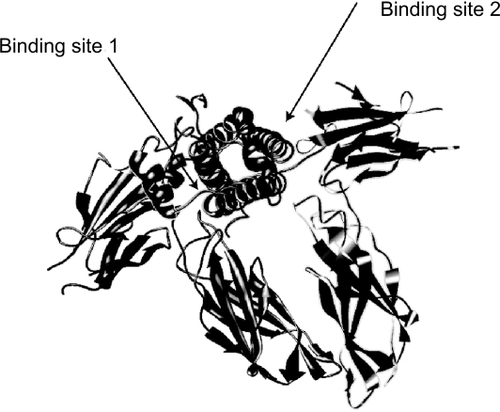
X-ray crystallography studies of the GH-GH-receptor complex and of GH with the extracellular domain of the receptor have demonstrated that each GH molecule is bound by two receptor molecules, suggesting dimerization of the receptor (). A former, well-accepted model was that the binding site 1 of the GH molecule binds to the first GH receptor, followed by the binding of site two of the same GH molecule to the second GH receptor, thus leading to dimerization of the receptor (CitationFuh et al 1992; CitationBehncken and Waters 1999). Recent data, however, suggest that the mature unbound glycosylated GH receptor is already present as a dimer on the cell surface, and moreover that GH receptor precursors in the endoplasmatic reticulum already form dimers, indicating that dimerization and binding are independent processes (CitationGent et al 2003). Binding of GH to the extracellular domains of the receptor dimer causes a relative shift and rotation of the subunits (CitationBrown et al 2005). The conformational change in the GH receptor-dimer results in the recruitment and cross-activation of two JAK2 tyrosine kinase molecules, leading to the activation of the receptor and this step is followed by a cascade of intracellular phosphorylation and further activations of other pathways, including the important STAT-5 pathway, but also those of the MAPK, IRS, and/or PKC pathways and followed by gene transcription (CitationPelekanos and Waters 2006). Mutations of the extracellular domain of the receptor prevent GH binding and lead to the so called Laron syndrome, characterized by severely stunted growth, GH insensitivity, and increased serum GH concentration (CitationLaron et al 1966). Later it was demonstrated that patients with this syndrome had low IGF-I and IGFBP3 concentrations. More than 40 different mutations in the human have been described, but mutations of the intracellular domain, preventing activation but not GH binding, are rare (CitationRosenbloom 2000).
Figure 3 Crystal structure of growth hormone complexed to the extracellular domains of its receptor dimer. The binding sites are indicated by arrows (crystal structure determined by Citationde Vos et al 1992, Protein Database code 3HHR, downloaded graph modified in Chimera UCSF).
The number of GH receptors on the cell surface is a strictly regulated process, and an important physiological determinant of the sensitivity to GH. The GH receptor is synthesized and degraded continuously with a half-life of less than 60 minutes. The number of receptors at the cell surface is mainly regulated by the ubiquitin system (CitationAlves dos Santos et al 2001; Citationvan Kerkhof et al 2002). An example of receptor down-regulation is starvation, which leads to high GH and low IGF-I serum concentrations and is caused by increased GH receptor degradation (CitationStrous et al 2004).
Growth hormone antagonists
In the search for GH molecules with increased biological activity the group of Kopchick substituted amino acids in the third α-helix of GH. This site was considered as essential for the biological activity of GH (CitationChen et al 1990).
They generated an altered bovine bGH in which glutamate-117 was changed into leucine, glycine-119 into arginine, and alanine-122 into aspartate and thus generated an idealized amphiphilic α-helix. Surprisingly, transgenic mice that expressed the mutated hormone were smaller than the wild type littermates. Nevertheless, the altered GH had the same binding affinity to mouse liver membrane preparations as the wild type bGH, indicating that binding and function could be dissociated and that the mutated hormone acted as an antagonist. Interestingly, the same group of scientists found that substitution of glycine for glutamate at position 126 of bGH increased growth 1.6-fold in transgenic mice (CitationChen et al 1991).
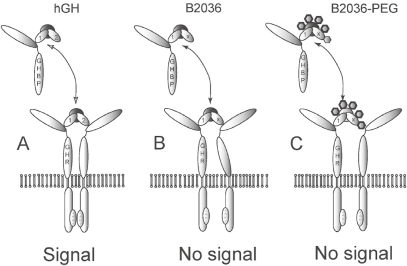
Substitution of glycine at position 120 of the third α-helix of human (h) GH with arginine (G120R) or with leucine (G120K) decreased growth and serum IGF-I concentration, but with full binding to the receptor (CitationChen et al 1994). Originally, it was thought that the antagonistic effect of G120R (or any long amino acid substitution) prevented receptor dimerization, because of the mutation at binding site 2. Indirect evidence from several studies, however, showed that the mutated molecule is bound by the receptor dimer as illustrated in (CitationHarding et al 1996; CitationRoss et al 2001). The mutated hormone-receptor complex is rapidly internalized to the same extent and rate as the native GH-GHR complex (CitationHarding 1996; CitationMaamra et al 1999). In the human the GH antagonists are specific for the GH receptor and they do not bind to the prolactin receptor, in contrast to hGH itself (CitationGoffin et al 1999).
Figure 4 Interaction of GH, B2036, and B2036/PEG (pegvisomant) with the GH receptor. A: GH binds to GH binding protein (GHBP). Binding to its cell surface receptor through sites 1 and 2 induces a change in conformation of the receptor leading to activation and subsequent signaling. B: the site 2 mutation (x) prevents the conformational change needed for activation. C: B2036-PEG binds to GHBP via site 1, which is protected from pegylation by mutation of lysines. High dose of B2036-PEG is required to overcome the steric hindrance by pegylation. Modified after CitationRoss RJ, Leung KC, Maamra M, et al 2001. Binding and functional studies with the growth hormone receptor antagonist B2056-PEG (pegvisomant) reveal effects of pegylation and evidence that it binds to a receptor dimmer. J Clin Endocrinol Metab, 86:1716–23. Copyright © 2001, with permission from the Endocrine Society.
The G120K mutation of hGH was further explored for the treatment of acromegaly. Unbound GH has a very short half-life of only a few minutes and bound GH (to GH-binding protein) of 16–18 minutes, so that treating GH excess with the native mutated GH is not practical. Therefore, two strategies were used, first to increase the binding affinity of site 1 and second the pegylation of the molecule by increasing its molecular weight in order to increase its half-life in the circulation. The affinity of site 1 for GHBP can be increased by mutating residues (CitationLowman and Wells 1993). In B2036, the G120K mutation of hGH, 8 other mutations were introduced at binding site 1, thereby increasing affinity for recombinant GHBP by 4–5-fold compared with GH. However, binding affinity to the cell surface receptor is equivalent between B2036 and GH (CitationRoss et al 2001).
Pegylation was developed in the 1970s by Davis and colleagues (CitationDavis et al 1978). It is the covalent attachment of polyethylene glycol moieties to therapeutic compounds, including proteins, such as interleukins, interferon, asparagines, and GH. Pegylation increases the size and molecular weight of the compound and alters the physicochemical properties, including the conformation, spatial hindrance, and electrostatic binding. The physico-chemical changes lead to decreased systemic clearance by different mechanisms, including decrease in renal clearance, proteolysis, and macrophage uptake. In addition, the pegylated molecule is heavily hydrated, so that the effective molecular weight is much greater than the apparent molecular weight, which is important for the passage of the drug across blood–tissue barriers. Furthermore, the pegylated moieties and associated hydration water shield the protein from immunogenic recognition and increase the resistance to proteolytic enzymes (CitationHarris et al 2001).
In earlier studies long-acting hGH was constructed by pegylation of recombinant hGH and the biological effects and pharmacokinetics studied in intact and hypophysectomized rats (CitationClark et al 1996). Depending on the level of polyethylene glycol (PEG) modification the affinity for the receptor was decreased and the clearance rate was inversely proportional to the effective molecular weight. The efficacy for the PEG-analogs in vitro and in vivo was optimal for hGH conjugated with 5 equivalents of PEG5000 and the potency was increased by about 10-fold compared with unmodified hGH.
Based on these findings and mutational studies of the binding site 1 of hGH which increase binding to the receptor, the pegylated B2036 molecule contained three alterations: (1) the amino acid substitution in the third alpha helix of binding site 2 (Glyc120Lys); (2) 4–6 PEG moieties that increase the in vivo half-life of the antagonist; and (3) 8 other amino acid alterations at site 1 that increase the affinity to the GHR (see and ). Nevertheless, this molecule has a 4–5-fold reduced binding to GHBP, but a much greater reduction of around 20-fold when binding is studied at the cell surface receptor and presumably explained by steric hindrance (CitationPradhananga et al 2002). The construction of the drug explains the very long half-life, but also the very high dose required for effective receptor blockage in clinical practice. This compound has been named pegvisomant and is marketed for the treatment of acromegaly Pfizer (Pfizer Inc, New York, USA) as Somavert®.
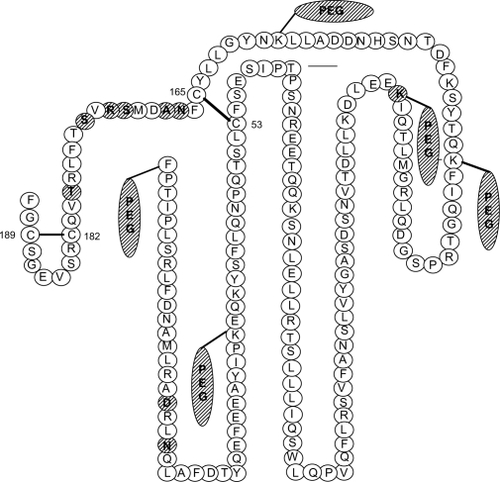
Figure 5 Schematic representation of pegvisomant and the attached PEG polymers. The amino acid substitutions were introduced at positions G120R (binding site 2) and at H18D, H21N, R167N, K168A, D171S, K172R, E174S, and I179T (binding site 1). The mutated amino acids are hatched. Two disulfide bridges between amino acids 53 and 165 and between 182 and 189 are shown by solid lines.
Pharmacokinetic and pharmacodynamic studies with pegvisomant
Most of the pharmacokinetic data on pegvisomant were published in two abstracts and as attachment on the internet site of the European Medicines Agency (http://www.emea.eu.int/humandocs/Humans/EPar/Somavert/somavert.htm). In a double-blind, placebo-controlled, single rising dose study the safety, pharmacokinetics, and pharmacodynamics of pegvisomant were investigated in 36 healthy male subjects (CitationRodvold et al 1997). The drug was tolerated in doses from 0.03 up to 1 mg/kg s.c. Maximal serum concentration was dose-dependent and reached values of 9600 ng/ml for the highest dose. The time of Cmax was also dose-dependent ranging from 15±10 hours at the lowest dose (0.03 mg/kg) to 60±18.6 hours for 1.0 mg/kg. The β-component of the serum half-life of pegvisomant was dose-independent and was about 75 hours. Serum IGF-I was suppressed by the 0.3 and 1.0 mg/kg doses. Comparable results were obtained in 6 patients with acromegaly (CitationRodvold and van der Lely 1999). The bioavailability after s.c injection was 56.7% (range 48.6%–64.7%). The distribution volume amounted to 23.3 L for s.c. administration and 12.4 L for i.v. injection. Only a very small fraction of unchanged pegvisomant is excreted via the urine (0.0015 after s.c. administration vs 0.0041 (comment: fractions are dimensionless) after i.v. injection). The mean systemic clearance was estimated to be 28 mL/hour for s.c. administered pegvisomant in doses of 10–20 mg. The clearance increased by 0.6 mL/hour for each kg of body weight greater than the average weight of 94 kg.
Thorner’s group investigated endogenous GH and pegvisomant concentrations in 36 healthy young male adults, within 15% of ideal body weight. The hormones were measured with specific assays. Although IGF-I decreased in the higher dose groups (1 mg/kg) no increase in endogenous GH occurred, suggesting to the authors that pegvisomant did not interfere with hypothalamic feedback (CitationThorner et al 1999). In contrast, the group of Veldhuis convincingly demonstrated with the aid of intensive venous sampling the increase in GH secretion after 1 mg/kg in 12 healthy volunteers (CitationVeldhuis et al 2001). Specifically, they found a linear relation between the decrease in IGF-I and the increase in GH burst amplitude. In patients with acromegaly GH concentrations increase while on GH receptor blockade, similar to that demonstrated in healthy subjects. However, it is not known whether diminished feedback via circulating IGF-I at the pituitary and/or hypothalamic level is solely responsible for the increased GH secretion. Direct intracerebroventricular administration of the unpegylated GH antagonist B2036 in rats leads to increased GH release, pointing to the presence of a short feedback loop (CitationNass et al 2000). At present it is not well known whether pegvisomant can penetrate into the hypothalamus, but it is stated that the radio-labeled drug does not cross the blood–brain barrier (product information EMEA), thus favoring an indirect effect via IGF-I.
Comparable questions may be asked for effects observed in organs other than the liver, ie, are eventual changes in organ responses the result of diminished liver IGF-I synthesis and subsequent decrease in circulating IGF-I or are effects also (or mainly) mediated via local blocking of the GH receptor? Unfortunately, this issue was never specifically addressed, but below we summarize some in vivo and in vitro studies in which peripheral effects were investigated. In mice, pegvisomant increased, in a dose-dependent way, liver mRNA of the GH receptor and the binding protein and decreased mRNA of IGF-I, but no corresponding changes were found in the kidneys (CitationVan Neck et al 2000). In another study, high protein feeding in mice caused renal hypertrophy associated with increased circulating IGF-I (CitationVan Neck et al 2002). However, pegvisomant treatment did not prevent renal hypertrophy and the authors concluded that this effect is GH-independent, a conclusion which is certainly acceptable only if pegvisomant was blocking effectively the renal GH receptors. Indeed, both studies did not provide any evidence for blocking of the renal GHR.
In another study, human meningiomas were xeno-grafted in nude mice. Tumors in pegvisomant-treated mice showed a mean volume decrease of 31.8% while the placebo- treated group had a 23.2% increase after 8 weeks. Serum IGF-I was moderately decreased in the pegvisomant group (257 vs 319 μg/L), but IGF-I was not detectable in the tumors (CitationMcCutcheon et al 2001), suggesting that the drug acted directly on these tumors. Pegvisomant administration to the rhesus monkey (slowly) decreased bone turnover, and the inhibitory effect could be abolished quickly by the simultaneous treatment with IGF-I (CitationWilson 2000). However, this study does not allow a distinction between endocrine and auto- or paracrine mechanisms of IGF-I.
Nevertheless, pegvisomant had a clear direct cellular effect in vitro in the following two studies. Growth hormone, both the 20 kDa and the 22 kDa isoforms, amplify GHR and GHBP gene transcription in human mesangial cells in vitro and this effect was blocked specifically by pegvisomant (CitationMeinhardt et al 2003). That pegvisomant could block the GH receptor in vitro was also demonstrated in an elegant series of experiments on phosphorylation and transcriptional effects of autocrine hGH in human mammary carcinoma cell lines (CitationKaulsay et al 2001).
Clinical studies with pegvisomant
The first study reporting efficacy of the drug was a multicenter, double-blind study comprising 112 patients (). The majority of the patients had undergone previous treatment by surgery, radiation therapy, or a combination. Because of active disease 81 patients were on treatment with somatostatin analogs and/or dopamine agonists. Nine patients were treated only with medication and 4 patients had never been treated before. As expected, GH concentration was rather low after withholding the drugs (8.1±10.6 ng/mL). After a loading dose of 80mg pegvisomant (or placebo) patients received placebo, 10, 15, or 20 mg pegvisomant per day for 6 weeks. IGF-I normalized in 0%, 38%, 75%, and 82% of the patients, respectively. Soft tissue swelling, perspiration, and fatigue all improved in the 15- and 20-mg treated groups. No serious side-effects were noted, except for 1 patient who developed liver function abnormalities on 2 occasions when exposed to the drug. Liver function normalized after withholding the drug (CitationTrainer et al 2000). The second large study enrolled 160 patients who were treated for a mean duration of 425 days (Citationvan der Lely et al 2001). These patients had undergone previous treatments: surgery 86%, pituitary irradiation 56%, somatostatin analog medication 74%, and dopamine agonists 48%. Patients received daily s.c. injections, with a starting dose of 10mg and increases of 5 mg until a normal IGF-I was obtained or the maximal dose of 40 mg/day was reached. The total number of dropouts for various reasons was 30 patients, including 5 patients in whom therapy was withheld because of lack of efficacy. The entry criterion for the study was a 1.3-fold increased IGF-I level, corrected for age after discontinuation of GH-suppressive medicines. The mean GH concentration at entry was only moderately increased to 10.9 μg/L. The mean GH increase after 18 months was 33.8 μg/L. Normal IGF-I was obtained in 87 of 90 patients at 12 months, and of all post-basal blood samples normal IGF-I was measured in 91.7%. Fasting insulin concentration decreased to 12.2 mU/L, but HbA1c values did not change. Residual tumor mass did not increase (2.41±0.31 to 2.37±0.31mL) but in 2 patients the size of the adenoma increased, requiring surgery in on patient and radiotherapy in the other. Two patients showed a 10-fold reversible increase in alanine amino transferase and aspartate amino transferase. A drawback of these two studies is that they may be biased by patient selection, and therefore the conclusion that the drug is excellent might only be applicable for patients who had persisting disease activity after surgery and/or pituitary radiotherapy.
Table 1 Effect of pegvisomant treatment on IGF-I normalization in acromegaly
Other GH-dependent parameters were generally studied in small groups of patients, being part of larger studies (). Trainer and colleagues found during pegvisomant treatment in 7 patients a shift from cortisone to cortisol metabolism, indicating the reversal of the well-known inhibition of GH on 11-β-hydroxysteroid dehydrogenase (CitationTrainer et al 2001). In a case-controlled study of cardiovascular risk factors in acromegaly, the normalization of the very low serum C-reactive protein concentration was described, but other risk factors including cholesterol, interleukin-6, homocystein, and insulin concentrations remained unchanged during the 3-month treatment period. The clinical significance of this finding is presently unknown (CitationSesmilo et al 2002). In a study by Parkinson in 16 patients increased leptin levels were found during pegvisomant treatment (CitationParkinson et al 2003a). Several studies showed improved glucose tolerance during treatment with pegvisomant (CitationRose and Clemmons 2002). Three other studies with more patients showed insulin sensitivity and glucose tolerance in patients converted from depot octreotide to pegvisomant (CitationParkinson et al 2002; CitationDrake et al 2003; CitationBarkan et al 2005). Increased bone turnover is a well-known feature of untreated acromegaly. In two studies the increased serum markers for bone formation and bone resorption decreased in parallel with normalization of IGF-I during pegvisomant therapy, while in one study PTH increased with concomitant decrease of 1, 25-dihydroxy-vitamin D3 (CitationFairfield et al 2002; CitationParkinson 2003b). These results on bone metabolism, but also on insulin and leptin, are not unexpected since they are under GH-IGF-I control. The unsolved question, however, is whether these effects are caused exclusively by diminished liver-derived IGF-I or whether the eventually locally decreased GH signal and paracrine effect of IGF-I contributes to peripheral (non-liver-associated) effects.
Table 2 Effect of pegvisomant treatment on metabolic parameters in acromegaly
Three other studies have explored either different dosing interval of pegvisomant or combining pegvisomant with soma-tostatin analogs on efficacy. In an elegant and well-designed study in 11 patients by Jørgensen and co-workers, treatment with low-dose pegvisomant (15 mg/day) and octreotide–LAR was clearly superior in suppressing IGF-I. Notwithstanding the well-known suppressive effect of somatostatin analogs on insulin secretion the combined treatment had similar effects on glucose and insulin secretion as pegvisomant alone. An interesting, unexplained observation in this study was the 20% increased circulating pegvisomant concentrations during combined treatment with octreotide (CitationJørgensen et al 2005). In the study by Jehle and colleagues, comprising only 10 patients, alternate day pegvisomant therapy was equally effective as daily injections in 5 patients (CitationJehle et al 2005). Finally, in a single-center, 42-week, open-label prospective dose finding study, 26 acromegalic patients (of whom 14 were previously untreated) received monthly injections with 30 mg Sandostatin LAR or 120 mg lanreotide Autogel and increasing weekly doses of pegvisomant. Under somatostatin analog mono-treatment all patients had elevated IGF-I levels, but after combined treatment with a median weekly dose of 60 mg pegvisomant 95% of the patients had normalized their IGF-I levels. Mild increases in liver enzymes were found in 38% of the patients during combined treatment. These studies together suggest that a longer dosing interval than daily injections is feasible in patients and that a relatively low dose of pegvisomant combined with slow-release somatostatin analogs is able to suppress effectively the GH-IGF-I axis, even in untreated patients (CitationFeenstra et al 2005). The results of the last two studies fully concur with the available data on plasma half-life and plasma residence time of the drug in man.
Clinical practice
Pegvisomant (Pfizer Inc, New York, USA) is supplied as a lyophilized powder and the vial must be reconstituted with 1 mL of the diluent (sterile water, USP). After reconstitution each vial contains 10, 15, or 20 mg of the pegvisomant protein in 1 mL of solution. The company advises a subcutaneous loading dose of 40 mg under the supervision of a physician and then daily s.c. injections with 10 mg or more (dose increases of 5 mg in 6-weekly intervals) until a normal age-adjusted IGF-I level is reached. The maximal daily dose should not exceed 30 mg. However, the pegvisomant dose may be tailored and less frequent injections may be sufficient in selected patients, as reviewed above. Less frequent dosing seems logical because of the known half-life, although weekly dosing might become critical at lower doses in many patients, as was observed in unpublished trials. Although not recommended by the Pfizer, patients might benefit from the combination of a slow-release somatostatin analog and weekly pegvisomant injection (CitationFeenstra et al 2005).
The most important side-effects of pegvisomant are disturbance of liver function tests and, in rare cases, growth of the pituitary adenoma. Therefore, regular control of liver function tests should be performed (CitationBiering et al 2006). In addition, before starting treatment with pegvisomant, MRI studies of the adenoma should be carried out and repeated at yearly intervals. In selected cases, especially in patients with suprasellar extension of the adenoma, tests of the visual acuity and visual fields should be performed on a regular basis. Most other side-effects are mild and generally restricted to local irritation at the injection site. The dose of hypoglycemic drugs in diabetic patients might be reduced, because of improved insulin sensitivity, and careful monitoring of glucose levels is advised.
Neutralizing, low titer GH antibodies are present in about 17% of patients, but no tachyphylaxis has occurred. Pituitary-derived GH concentration rises during pegvisomant treatment until a stable IGF-I level is obtained. The GH increase is dose-dependent, but can be demonstrated only with specially adapted assays.
Unsolved questions
All studies with pegvisomant are short-term studies and the long-term efficacy is not known. Clarification is needed whether in man the drug is mainly acting via its IGF-I-suppressive effect on the liver or whether the GH receptor is blocked effectively in all organs. Even if the latter is true then the brain still escapes from this effect, since pegvisomant does not cross the blood–brain barrier and its long-term consequence on brain function is unknown.
Long-term studies are required to evaluate the safety on pituitary adenoma size in patients primarily treated with pegvisomant only, since these tumors have a slow growth rate.
No studies have compared the efficacy of slow-release somatostatin analogs with that of pegvisomant. Such a (long-term) study is badly needed and should primarily focus on normalization of IGF-I, but should also document parameters which are more dependent on local GH-IGF-I effects and less on circulating IGF-I.
No study with primary medical treatment has investigated the influence on the quality of life and on mortality in acromegaly, in contrast to what is known presently from surgical patient series.
Place of pegvisomant in the treatment of acromegaly
For the initial treatment of acromegaly, both trans-sphenoidal surgery and treatment with somatostatin analogs can be attempted. The choice between the two modalities depends on several factors, including the available surgical experience of the center, the preference of the patient and comorbidity, resulting in contraindications for either treatment option. In addition, the size of the adenoma, the presence of optic chiasm compression, and the level of GH should be taken into account. Octreotide is able to achieve disease control, especially in patients with documented good sensitivity for octreotide and relatively low GH levels. Primary radiosurgery could be applied in experienced centers in patients harboring tumors invading the cavernous sinus, not related to optic structures, and that have a low a priori chance for surgical cure. Pegvisomant seems to be an effective drug as supplement to long-acting slow-release somatostatin analogs (ie, Sandostatin LAR or lanreotide Autogel), when IGF-I levels remain elevated. Primary treatment with pegvisomant alone should be done at present only as part of long-term studies which are necessary to define its precise role. In case of equal potency the choice should be dictated by the costs of life-long GH-suppressive therapy.
References
- AbsRVerhelstJMaiterD1998Cabergoline in the treatment of acromegaly:a study in 64 patientsJ Clin Endocrinol Metab8337489467544
- AhmedSElsheikhMStrattonIM1999Outcome of transsphenoidal surgery for acromegaly and its relationship to surgical experienceClin Endocrinol (Oxf)50561710468920
- AlexanderLAppletonDHallR1980Epidemiology of acromegaly in the Newcastle regionClin Endocrinol (Oxf)127197379316
- Alves dos SantosCMten BroekeTStrousGJ2001Growth hormone receptor ubiquitination, endocytosis, and degradation are independent of signal transduction via Janus kinase 2J Biol Chem276326354111418602
- AttanasioREpaminondaPMottiE2003Gamma-knife radiosurgery in acromegaly: A 4-Year follow-up studyJ Clin Endocrinol Metab8831051212843150
- BaumannG1991Growth hormone heterogeneity: genes, isohormones, variants, and binding proteinsEndocr Rev12424491760996
- BarkanALBurmanPClemmonsDR2005Glucose homeostasis and safety in patients with acromegaly converted from long-acting octreotide to pegvisomantJ Clin Endocrinol Metab9056849116076947
- BazanJF1990Structural design and molecular evolution of a cytokine receptor superfamilyProc Natl Acad Sci U S A87639348
- BehnckenSNWatersMJ1999Molecular recognition events involved in the activation of the growth hormone receptor by growth hormoneJ Mol Recognit123556210611645
- Ben ShlomoAMelmedS2003The role of pharmacotherapy in peri-operative management of patients with acromegalyJ Clin Endocrinol Metab88963812629068
- BengtssonBAEdenSErnestI1988Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984Acta Med Scand223327353369313
- BengtssonBABrummerRJEdenS1989Body composition in acromegalyClin Endocrinol (Oxf)30121302612014
- BieringHSallerBBauditzJ2006Elevated transaminases during medical treatment of acromegaly: a review of the German pegvisomant surveillance experience and a report of a patient with histologically proven chronic mild active hepatitisEur J Endocrinol1542132016452533
- BiermaszNRDekkerFWPereiraAM2004Determinants of survival in treated acromegaly in a single center: predictive value of serial insulin-like growth factor-I measurementsJ Clin Endocrinol Metab8927899615181059
- BiermaszNRvan DulkenHRoelfsemaF1999Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotideJ Clin Endocrinol Metab843551510522994
- BiermaszNRvan DulkenHRoelfsemaF2000aPostoperative radio-therapy in acromegaly is effective in reducing GH concentration to safe levelsClin Endocrinol (Oxf)533212710971449
- BiermaszNRvan DulkenHRoelfsemaF2000bLong-term follow-up results of postoperative radiotherapy in 36 patients with acromegalyJ Clin Endocrinol Metab8524768210902796
- BiermaszNRvan DulkenHRoelfsemaF2000cTen-year follow-up results of transsphenoidal microsurgery in acromegalyJ Clin Endocrinol Metab85459660211134114
- BiermaszNRPereiraAMNeelisKJ2006The role of radiotherapy in the management of acromegalyExpert Rev Endocrinol MetabIn press.
- BiermaszNRPereiraAMSmitJW2005Intravenous octreotide test predicts the long term outcome of treatment with octreotide-long-acting repeatable in active acromegalyGrowth Horm IGF Res15200615935982
- BrownRJAdamsJJPelekanosRA2005Model for growth hormone receptor activation based on subunit rotation within a receptor dimerNat Struct Mol Biol128142116116438
- Carter-SuCSmitLS1998Signaling via JAK tyrosine kinases: growth hormone receptor as a model systemRecent Prog Horm Res5361829769703
- ChenWYWightDCWagnerTE1990aExpression of a mutated bovine growth hormone gene suppresses growth in transgenic miceProc Natl Acad Sci U S A875061652367524
- ChenWYWightDCChenNY1991Mutations in the third α-helix of bovine growth hormone dramatically affect its intracellular distribution in vitro and growth enhancement in transgenic miceJ Biol Chem266225281989980
- ChenWYChenNYYunJ1994In vitro and in vivo studies of antagonistic effects of human growth hormone analogsJ Biol Chem2691589278195244
- ClarkROlsonKFuhG1996Long-acting growth hormones produced by conjugation with polyethylene glycolJ Biol Chem27121969778703002
- ColaoAFeroneDMarzulloP2004Systemic complications of acromegaly: epidemiology, pathogenesis, and managementEndocr Rev251025214769829
- CozziRAttanasioRMontiniM2003Four-year treatment with octreotide-long-acting repeatable in 110 acromegalic patients: predictive value of short-term results?J Clin Endocrinol Metab883090812843148
- CozziRAttanasioRLodriniS2004Cabergoline addition to depot somatostatin analogues in resistant acromegalic patients: efficacy and lack of predictive value of prolactin statusClin Endocrinol (Oxf)612091515272916
- CozziRMontiniMAttanasioR2006Primary treatment of acromegaly with octreotide LAR: A long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkageJ Clin Endocrinol Metab91139740316449332
- DavisFFAbuchowskiAvan EsT1978Enzyme-polyethylene glycol adducts: modified enzymes with unique propertiesEnzyme Eng416973
- de VosAMUltschMKossiakoffAA1992Human growth hormone and extracellular domain of its receptor: crystal structure of the complexScience255306121549776
- DrakeWMRowlesSVRobertsME2003Insulin sensitivity and glucose tolerance improve in patients with acromegaly converted from depot octreotide to pegvisomantEur J Endocrinol149521714640992
- EzzatSForsterMJBerchtoldP1994Acromegaly. Clinical and biochemical features in 500 patientsMedicine (Baltimore)73233407934807
- FairfieldWPSesmiloGKatznelsonL2002Effects of a growth hormone receptor antagonist on bone markers in acromegalyClin Endocrinol5738590
- FeenstraJde HerderWWten HaveSMTH2005Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegalyThe Lancet36516446
- FredaPUWardlawSLPostKD1998Long-term endocrinological follow-up evaluation in 115 patients who underwent transsphenoidal surgery for acromegalyJ Neurosurg8935389724106
- FredaPU2003Current concepts in the biochemical assessment of the patient with acromegalyGrowth Horm IGF Res131718412914750
- FuhGCunninghamBCFukunagaR1992Rational design of potent antagonists to the human growth hormone receptorScience2561677801535167
- GentJVan den EijndenMvan KerkhofP2003Dimerization and signal transduction of the growth hormone receptorMol Endocrinol179677512576487
- GilbertJAMiellJPChambersSM2005The nadir growth hormone after an octreotide test dose predicts the long-term efficacy of somatostatin analogue therapy in acromegalyClin Endocrinol (Oxford)622828
- GiustinaAVeldhuisJD1998Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the humanEndocr Rev19717979861545
- GiustinaABarkanACasanuevaFF2000Criteria for cure of acromegaly: a consensus statementJ Clin Endocrinol Metab85526910690849
- GoffinVBernichteinSCarriereO1999The growth hormone antagonist B2036 does not interact with the prolactin receptorEndocrinology1403853610433247
- HardingPAWangXOkadaS1996Growth hormone (GH) and a GH antagonist promote GH receptor dimerization and internalizationJ Biol Chem2716708128636090
- HarrisJMMartinNEModiM2001Pegylation: a novel process for modifying pharmacokineticsClin Pharmacokinet405395111510630
- HoKYWeisbergerAJ1994Characterization of 24-hour growth hormone secretion in acromegaly: implications for diagnosis and therapyClin Endocrinol (Oxford)417583
- HoflandLJLambertsSWJ2003The pathophysiological consequences of somatostatin receptor internalization and resistanceEndocr Rev24284712588807
- HoffmanDMO’SullivanAJBaxterRC1994Diagnosis of growth hormone deficiency in adultsLancet343106487512681
- JaffeCABarkanAL1992Treatment of acromegaly with dopamine agonistsEndocrinol Metab Clin North Am21713351355728
- JanssenYJHFrolichMRoelfsemaF1997A low starting dose of recombinant human growth hormone (GH) in adults with GH deficiencyJ Clin Endocrinol Metab82129358989246
- JehleSReyesCMSundeenRE2005Alternate-day administration of pegvisomant maintains normal serum insulin-like growth factor-I levels in patients with acromegalyJ Clin Endocrinol Metab9015889315585549
- JørgensenJOLFeldt-RasmusenUFrystykJ2005Cotreatment of acromegaly with a somatostatin analog and a growth hormone receptor antagonistJ Clin Endocrinol Metab9056273116046586
- KaravitakiNBotusanIRadianS2005The value of an acute octreotide suppression test in predicting long-term responses to depot somatostatin analogues in patients with active acromegalyClin Endocrinol (Oxf)62282815730408
- KaulsayKKZhuTBennettWF2001The effects of autocrine human growth hormone (hGH) on human mammary carcinoma cell behavior are mediated via the hGH receptorEndocrinology1427677711159849
- KojimaMHosodaHDateY1999Ghrelin is a growth-hormone-releasing acylated peptide from stomachNature4026566010604470
- LancranjanIAtkinsonABSandostatin LAR Group1999Results of a European multicentre study with Sandostatin LAR in acromegalic patientsPituitary11051411081188
- LandoltAMHallerDLomaxN1998Stereotactic radiosurgery for recurrent surgically treated acromegaly: comparison with fractionated radiotherapyJ Neurosurg88100289609294
- LaronZPertzelanAMannheimerS1966Genetic pituitary dwarfism with high serum concentrations of growth hormone – a new inborn error of metabolism?Isr J Med Sci215255916640
- Le RoithDBondyCYakarS2001The somatomedin hypothesisEndocr Rev22537411159816
- LongHBeauregardHSommaM1996Surgical outcome after repeated transsphenoidal surgery in acromegalyJ Neurosurg85239478755752
- LowmanHBWellsJA1993Affinity maturation of human growth hormone by monovalent phage displayJ Mol Biol234564788254660
- MaamraMFinidoriJvon LaueS1999Studies with a growth hormone antagonist and dual-fluorescent confocal microscopy demonstrate that the full-length human growth hormone receptor, but not the truncated isoform, is very rapidly internalized independent of Jak2-Stat5 signalingJ Biol Chem27414791810329677
- McCutcheonIEFlyvbjergAHillH2001Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude miceJ Neurosurg944879211235955
- Mahmoud-AhmedASSuhJHMaybergMR2001Gamma knife radio-surgery in the management of patients with acromegaly: a reviewPituitary42233012501972
- MeinhardtUEbleABessonA2003Regulation of growth-hormone-receptor gene expression by growth hormone and pegvisomant in human mesangial cellsKidney Int644213012846737
- MelmedSSternbergRCookD2005A critical analysis of pituitary tumor shrinkage during primary medical therapy in acromegalyJ Clin Endocrinol MetabDOI 10.1210/jc.2004–2466.
- MestronAWebbSMAstorgaR2004Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA)Eur J Endocrinol1514394615476442
- MurrayRDKimKRenS-G2004Central and peripheral actions of somatostatin on the growth hormone-IGF-I axisJ Clin Invest1143495615286801
- NassRToogoodAAHellmannP2000Intracerebroventricular administration of the rat growth hormone (GH) receptor antagonist G118R stimulates GH secretion: evidence for the existence of short loop negative feedback of GHJ Neuroendocrinol121194911106977
- ParkinsonCDrakeWMRobertsME2002aA comparison of the effects of pegvisomant and octreotide on glucose, insulin, gastrin, cholecystokinin, and pancreatic polypeptide responses to oral glucose and a standard mixed mealJ Clin Endocrinol Metab87179780411932320
- ParkinsonCDrakeWMWieringaW2002bSerum lipoprotein changes following IGF-I normalization using a growth hormone receptor antagonist in acromegalyClin Endocrinol5630311
- ParkinsonCWhatmoreAJYatesAP2003aThe effect of pegvisomant-induced serum IGF-I normalization on serum leptin levels in patients with acromegalyClin Endocrinol5916874
- ParkinsonCKassemMHeickendorffL2003bPegvisomant-induced serum insulin-like growth factor-I normalization in patients with acromegaly returns elevated markers of bone turnover to normalJ Clin Endocrinol Metab885650514671148
- PelekanosRAWatersMJ2006Activation of the growth hormone receptorExpert Rev Endocrinol Metab118998
- PijlHLangendonkJGBurggraafJ2001Altered neuroregulation of GH secretion in viscerally obese premenopausal womenJ Clin Endocrinol Metab865509551511701729
- PradhanangaSWilkinsonIRossRJM2002Pegvisomant: structure and functionJ Mol Endocrinol2911412200225
- RitchieCMAtkinsonABKennedyAL1990Ascertainment and natural history of treated acromegaly in Northern IrelandUlster Med J5955622349750
- RodvoldKABennettWFZibKA1997Single-dose safety and pharmacokinetics of B2036-PEG (Somavert) after subcutaneous administration in healthy volunteers [abstract]J Clin Pharmacol37869
- RodvoldKAvan der LelyAJ1999Pharmacokinetics and pharmacodynamics of B2036-PEG, a novel growth hormone receptor antagonist, in acromegalic subjects [abstract]Proceedings 81th annual meeting of the Endocrine Society1049
- RoelfsemaFGoslingsBMFrolichM1979The influence of bromocriptin on serum levels of growth hormone and other pituitary hormones and its metabolic effects in active acromegalyClin Endocrinol (Oxf)1123544114344
- RoelfsemaFBiermaszNRRomijnJA2005Treatment strategies for acromegalyExpert Opin Emerging Drugs1087590
- RoseDRClemmonsDR2002Growth hormone receptor antagonist improves insulin resistance in acromegalyGrowth Horm IGF Res124182412423627
- RosenbloomAL2000Physiology and disorders of the growth hormone receptor (GHR) and GH-GHR signal transductionEndocrine121071910905371
- RossRJLeungKCMaamraM2001Binding and functional studies with the growth hormone receptor antagonist B2056-PEG (pegvisomant) reveal effects of pegylation and evidence that it binds to a receptor dimmerJ Clin Endocrinol Metab8617162311297608
- SerriOBrazeauPKachraZ1992Octreotide inhibits insulin-like growth factor-I hepatic gene expression in hypophysectomized rat: evidence for a direct and indirect mechanism of actionEndocrinology1301816211547711
- SesmiloGFairfieldWPKatznelsonL2002Cardiovascular risk factors in acromegaly before and after normalization of serum IGF-I levels with the GH antagonist pegvisomantJ Clin Endocrinol Metab871692911932303
- SjogrenKLiuJLBladK1999Liver-derived insulin-like growth factor I (IGF-I) is the principle source of IGF-I in blood but it is not required for postnatal body growth in miceProc Natl Acad Sci U S A9670889210359843
- SjogrenKJanssonJOIsakssonOG2002A model for tissue-specific inducible insulin-like growth factor (IGF-I) inactivation to determine the physiological role of liver-derived IGF-IEndocrine192495612624424
- SotiropoulosAGoujonLSimoninG1993Ece for generation of the growth hormone-binding protein through proteolysis of the growth hormone membrane receptorEndocrinology132186358462483
- StrousGJAlves dos SantosCGentJ2004Ubiquitin system –dependent regulation of growth hormone receptor signal transductionCurr Top Microbiol Immunol2868111815645711
- SwaeringenBBarkerFGKatznelsonL1998Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegalyJ Clin Endocrinol Metab83341189768640
- TachasGLofthouseSWraight2006A GH receptor antisense oligonucleotide inhibits hepatic GH receptor expression, IGF-I production and body weight gain in normal miceJ Endocrinol1891475416614389
- ThornerMOStrasburgerCJWuZ1999Growth hormone (GH) receptor blockade with a PEG-modified GH (B2036-PEG) lowers serum insulin-like growth factor-I but does not acutely stimulate serum GHJ Clin Endocrinol Metab84209810310372717
- TrainerPJDrakeWMKatznelsonL2000Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomantN Engl J Med3421171710770982
- TrainerPJDrakeWMPerryLA2001Modulation of cortisol metabolism by the growth hormone receptor antagonist pegvisomant in patients with acromegalyJ Clin Endocrinol Metab8629899211443156
- UltschMde VosAMKossiakoffAA1991Crystals of the complex between human growth hormone and the extracellular domain of its receptorJ Mol Biol22286581762154
- UltschMde VosAM1993Crystals of growth hormone –receptor complexes. Extracellular domains of the growth hormone and prolactin receptors and a hormone mutant designed to prevent receptor dimerizationJ Mol Biol2311133368515471
- UltschMHSomersWKossiakoffAA1994The crystal structure of affinity-matured growth hormone at 2 Å resolutionJ Mol Biol236286998107110
- Van den BergGFrolichMVeldhuisJD1994Growth hormone secretion in recently operated acromegalic patientsJ Clin Endocrinol Metab791706157989479
- van den BergGPincusSMFrolichM1998Reduced disorderliness of growth hormone release in biochemically inactive acromegaly after pituitary surgeryEur J Endocrinol13816499506860
- van der LelyAJde HerderWWJanssenJA1997Acromegaly:the significance of serum total and free IGF-I and IGF- binding protein-3 in diagnosisJ Endocrinol155Suppl 1S913discussion S15–6: S9–13.9389990
- van der LelyAJHutsonRKTrainerPJ2001Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonistLancet3581754911734231
- van KerkhofPSmeetsMStrousGJ2002The ubiquitin-proteasome pathway regulates the availability of the GH receptorEndocrinol143124352
- van NeckJWDitsNFJCingelV2000Dose-response effects of a new growth hormone receptor antagonist (B2036-PEG) on circulating, hepatic and renal expression of the growth hormone/insulin-like growth factor system in adult miceJ Endocrinol16729530311054644
- van NeckJWCingelVvan VlietAK2002High-protein induced renal enlargement is growth hormone independentKidney Int6211879512234289
- VeldhuisJDBidlingmaierMAndersonSM2001Lowering total plasma insulin-like growth factor I concentrations by way of a novel, potent, and selective growth hormone (GH) receptor antagonist, pegvisomant (B2036-Peg), augments the amplitude of GH secretory bursts and elevates basal/nonpulsatile GH release in healthy women and menJ Clin Endocrinol Metab8633041011443205
- WalenkampMJEKarperienMPereiraAM2005Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutationJ Clin Endocrinol Metab9028556415769976
- WassJA2003Radiotherapy in acromegaly: a protagonist’s viewpointClin Endocrinol (Oxf)581283112580924
- WilsonME2000Insulin-like growth factor-I (IGF-I) replacement during growth hormone receptor antagonism normalizes serum IGF-binding protein-3 and markers of bone formation in ovariectomized rhesus monkeysJ Clin Endocrinol Metab8515576210770197
- WoodsKACamacho-HubnerCSavageMO1996Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I geneN Eng J Med33513637
- WrightADHillDMLowyC1970Mortality in acromegalyQ J Med391165427331