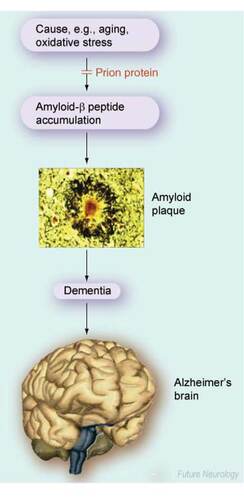
With the rise in life expectancy, Alzheimer‘s disease (AD) has become of increasingly significant concern to healthcare professionals and the population as a whole. As the most prevalent mental health issue affecting our aging population, with over 24 million sufferers worldwide and an estimated cost that runs into billions of dollars Citation[101], scientific interest remains prioritized on novel therapeutic strategies. Currently, there remains no adequate treatment to cure or prevent disease progression and only limited options to alleviate some of the symptoms Citation[1]. AD is characterized pathologically by extracellular senile plaques and intracellular neurofibrillary tangles. The major component of the senile plaque is the amyloid β (Aβ) peptide, which is derived from the proteolysis of the large transmembrane amyloid precursor protein (APP). Cleavage of APP by β-secretase (β-site APP-cleaving enzyme [BACE]1) is the first step in releasing neurotoxic Aβ peptides; Aβ1-40 is the most prevalent form, whilst Aβ1-42 is more fibrillogenic and is the major species found in the senile plaques Citation[2]. Rapid advances are being made to further understand the pathological development of AD, particularly relating to the roles of cellular proteins in the production, aggregation and metabolism of Aβ Citation[1].
The prion protein (PrP) is the causative agent of the transmissible spongiform encephalopathies (TSEs), which include scrapie in sheep, bovine spongiform encephalopathy or ‘mad cow disease‘ in cattle and Creutzfeldt–Jakob disease, Gerstmann–Straussler–Scheinker disease and fatal familial insomnia in humans Citation[3]. In TSEs, the normal cellular isoform of PrP (PrPC) undergoes a post-translational conformational change to create the infectious, disease-causing isoform PrPScCitation[4]. Whilst being an absolute prerequisite for disease propagation by providing the substrate for conversion, a precise physiological role for PrPC remains uncertain, especially as knockout mice in which the gene encoding PrP has been ablated are without gross anatomical defect or overt pathology. However, growing evidence indicates that PrPC may have a role in neuronal defence against oxidative stress, either by direct chelation of redox-active metal, as a sacrificial molecule or as a copper-sensitive stress sensor Citation[4].
Here, we summarize recent data demonstrating a direct role for PrPC in regulating the production of the Aβ peptide and, potentially, the progression of AD. As oxidative stress is an early event in the pathogenesis of AD and as PrPC can protect cells against oxidative stress, we highlight this link, and speculate whether molecules capable of mimicking the antiamyloidogenic and antioxidative functions of PrPC may provide a novel approach in the treatment of AD.
Protective role for PrPC in Alzheimer‘s disease
Recently, we reported that increasing the amount of PrPC within neuronal cells dramatically inhibited the formation of both Aβ1-40 and Aβ1-42Citation[5]. Conversely, reducing the amount of PrPC in cultured neuronal cells through the use of small interfering RNAs resulted in an increase in Aβ production, while the brains from PrP-knockout mice had increased Aβ levels compared with mice with normal amounts of PrPCCitation[5].
Together, these observations indicate that PrPC has a role in regulating the production of the neurotoxic Aβ peptide and, hence, protecting against AD . Further analyses of the metabolism of APP revealed that PrPC was regulating the production of Aβ at the first step in the proteolytic processing of APP, which is carried out by BACE1. However, PrPC did not alter the amount of BACE1 in the cells nor did it directly inhibit its catalytic activity towards a short synthetic peptide substrate. Coimmunoprecipitation studies indicated that PrPC interacted directly with BACE1, and the use of mutants of PrPC revealed that this interaction was glycosaminoglycan-dependent and required PrPC to be localized in cholesterol-rich domains of the cell membrane Citation[5]. Further investigation of the mechanism by which PrPC inhibits the cleavage of APP by BACE1 may provide the opportunity for the design of novel PrPC agonists that mimic this effect and that would have therapeutic potential for AD.
This novel role for PrPC in inhibiting the cleavage of APP by BACE1, thereby regulating the production of the neurotoxic Aβ peptide , raises the possibility that a small reduction in the level of functional PrPC in individuals may increase the amyloidogenic processing of APP in a subtle way over decades to affect long-term Aβ production, which in turn could accelerate the deposition of amyloid in the brain and the onset of AD. Further studies are required to address this hypothesis. However, if true, one would predict that complete absence of PrPC in humans would lead to the onset of AD at a relatively early age as there would be no PrPC to regulate the BACE1 cleavage of APP, with the result that the production of Aβ would be higher throughout life. Support for this hypothesis comes from two familial prion diseases that are due to mutations (Y145stop and Q160stop) in PrPC, which result in truncated proteins that are rapidly metabolized by the cell Citation[6,7]. In both cases, the onset of clinical disease occurred in the fourth decade of life and, in the Y145stop case, a diagnosis of AD was initially made Citation[6]. Thus, these natural mutations in PrPC provide compelling evidence that the loss of functional PrPC in humans may lead to the early onset of AD.
Oxidative stress in Alzheimer‘s disease
In addition to the direct effect of PrPC on the production of Aβ outlined above, it is also important to consider that AD is a disease of aging, a phenomena associated with an increase in reactive oxygen species (ROS) Citation[8]. Oxidative stress occurs when the rate at which a cell produces ROS is outweighed by the inherent ability of the cell to quench them. A substantial body of evidence exists to demonstrate that oxidative stress is an early event in the pathogenesis of AD and is present even before the hallmark features of AD are detectable Citation[9].
Oxidative modification in susceptible neurons of AD has been found in almost all classes of cellular macromolecules, and includes DNA-strand breaks and fragmentation, protein nitration and carbonyl formation and lipid peroxidation Citation[10,11]. Interestingly, oxidative damage decreases with disease progression, such that markers that were elevated during the earlier stages of disease decrease as the disease progresses Citation[12]. This would indicate that while ROS not only cause damage to the cellular infrastructure, they may also provoke physiological responses, such as complementary upregulation of antioxidant defences Citation[12]. Furthermore, the deposition of Aβ may be an attempt at functional adaptation to the pro-oxidative environment Citation[13,14]. The chelation of redox-active metals within senile plaques and intraneuronal inclusions, along with the demonstration that the neurons containing such inclusions are protected from cell death, indicate that Aβ deposition may have a beneficial effect.
As ROS-mediated damage appears to be a key factor in AD pathogenesis, measures to reduce the oxidant burden within the cell could contribute towards reducing disease etiology. A growing body of evidence indicates a role for PrPC in the protection of neuronal cells against oxidative stress Citation[15]. PrPC appears to provide crucial early detection of ROS in the local environment by undergoing cleavage following exposure to radical species Citation[16]. Cells in which this cleavage of PrPC occurred were able to cope better under oxidative stress.
However, if the protein was unable to undergo cleavage, for example owing to disease-associated mutation, the cells remained compromised Citation[16]. This substantiates a normal physiological function for PrPC in upregulating the antioxidant defence of neuronal cells. Interestingly, the expression level of PrPC increases with age Citation[17], suggesting that a physiological mechanism may be in place to reduce the consequence of age-related adventitious ROS production. Furthermore, aging mice showed impairment of both short- and long-term memory retention when there was an absence of PrPCCitation[18].
Conclusion
Taken together, these data suggest that the beneficial effect of PrPC may be at least twofold; as a direct cellular inhibitor of the amyloidogenic processing of APP to produce the neurotoxic Aβ and as a key component of the cellular protection against oxidative stress. As both Aβ and oxidative stress are intimately associated with the development and progression of AD, then it follows that PrPC may have a fundamental role to play in protecting against this devastating neurodegenerative disease. In the future, the identification of molecules able to mimic or recapitulate these antiamyloidogenic and antioxidative functions of PrPC may provide a new avenue for the development of AD therapeutics.
Future perspective
The discovery of a role for PrPC in regulating the production of the neurotoxic Aβ peptide, and thus in directly protecting against AD, as well as its antioxidant role, opens up new avenues for research in coming years. As we learn more about the mechanisms underlying both of these cellular roles of PrPC, it may be possible within 5–10 years to identify and/or develop small molecules that mimic these protective functions. Further ahead, such novel therapeutics could be used alongside drugs already in development and in clinical trials in our battle against the most prevalent and devastating neurodegenerative disease of old age.
Introduction
Alzheimer‘s disease (AD) is the major neurodegenerative disease of old age for which there is currently no cure.
AD is characterized by the build up of the neurotoxic amyloid-β (Aβ) peptide, which is deposited in senile plaques in the brain.
Cellular prion protein (PrPC), which causes the transmissible spongiform encephalopathies, is involved in the protection of neurons against oxidative damage.
Protective role for the prion protein in Alzheimer‘s disease
In neuronal cells PrPC dramatically inhibits the formation of Aβ through inhibiting the action of the β-secretase β-site amyloid precursor protein-cleaving enzyme 1.
In mice lacking PrPC, the amount of Aβ in the brain is increased, while in humans with mutations in PrPC that lead to lack of functional protein an early diagnosis of AD was made.
Oxidative stress in Alzheimer‘s disease
Aging is associated with an increase in reactive oxygen species and oxidative stress is an early event in the pathogenesis of AD.
PrPC protects cells against oxidative stress and the level of PrPC in the brain increases with age.
Conclusions
PrPC has beneficial effects, as a direct inhibitor of the production of Aβ and in the protection against the oxidative environment known to promote AD progression.
Identification of molecules able to mimic or recapitulate the beneficial functions of PrPC may provide a new avenue for AD therapeutics.
Future perspective
Such therapeutics could be used alongside drugs already in development and in clinical trials in our battle against the most prevalent and devastating neurodegenerative disease of old age.
Financial & competing interests disclosure
We thank the Medical Research Council of Great Britain for financial support of our work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
- Vardy ER , HussainI, HooperNM: Emerging therapeutics for Alzheimer‘s disease.Expert Rev. Neurother.6(5), 695–704 (2006).
- Mattson MP : Pathways towards and away from Alzheimer‘s disease.Nature430(7000), 631–639 (2004).
- Watts JC , BalachandranA, WestawayD: The expanding universe of prion diseases.PLoS Pathog.2(3), e26 (2006).
- Westergard L , ChristensenHM, HarrisDA: The cellular prion protein (PrPC): its physiological function and role in disease.Biochim. Biophys. Acta1772(6), 629–644 (2007).
- Parkin ET , WattNT, HussainIet al.: Cellular prion protein regulates β-secretase cleavage of the Alzheimer‘s amyloid precursor protein.Proc. Natl Acad. Sci. USA104(26), 11062–11067 (2007).
- Kitamoto T , IizukaR, TateishiJ: An amber mutation of prion protein in Gerstmann–Straussler syndrome with mutant PrP plaques.Biochem. Biophys. Res. Commun.192(2), 525–531 (1993).
- Finckh U , Muller-ThomsenT, MannUet al.: High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am. J. Hum. Genet.66(1), 110–117 (2000).
- Halliwell B : Oxidative stress and neurodegeneration: where are we now?J. Neurochem.97(6), 1634–1658 (2006).
- Lee HG , PerryG, MoreiraPIet al.: Tau phosphorylation in Alzheimer‘s disease: pathogen or protector?Trends Mol. Med.11(4), 164–169 (2005).
- Butterfield DA , ReedT, NewmanSF, Sultana R: Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer‘s disease and mild cognitive impairment. Free Radic. Biol. Med.43(5), 658–677 (2007).
- Zhu X , SuB, WangX, SmithMA, PerryG: Causes of oxidative stress in Alzheimer disease.Cell. Mol. Life Sci.64(17), 2202–2210 (2007).
- Nunomura A , CastellaniRJ, ZhuXet al.: Involvement of oxidative stress in Alzheimer disease.J. Neuropathol. Exp. Neurol.65(7), 631–641 (2006).
- Petersen RB , SiedlakSL, LeeHGet al.: Redox metals and oxidative abnormalities in human prion diseases.Acta Neuropathol. (Berl.)110(3), 232–238 (2005).
- Castellani RJ , LeeHG, PerryG, SmithMA: Antioxidant protection and neurodegenerative disease: the role of amyloid-β and tau.Am. J. Alzheimers Dis. Other Demen.21(2), 126–130 (2006).
- Vassallo N , HermsJ: Cellular prion protein function in copper homeostasis and redox signalling at the synapse.J. Neurochem.86(3), 538–544 (2003).
- Watt NT , TaylorDR, GillottAet al.: Reactive oxygen species-mediated β-cleavage of the prion protein in the cellular response to oxidative stress.J. Biol. Chem.280(43), 35914–35921 (2005).
- Williams WM , StadtmanER, MoskovitzJ: Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrP in mouse cerebral microvessels and brain parenchyma.Neuropathol. Appl. Neurobiol.30(2), 161–168 (2004).
- Coitinho AS , RoeslerR, MartinsVR, BrentaniRR, IzquierdoI: Cellular prion protein ablation impairs behavior as a function of age.Neuroreport14(10), 1375–1379 (2003).
Website
- Alzheimer‘s Research Trust www.alzheimers-research.org.uk