
Abstract
Oxidative stress has a significant impact on the development and progression of common human pathologies, including cancer, diabetes, hypertension and neurodegenerative diseases. Increasing evidence suggests that oxidative stress globally influences chromatin structure, DNA methylation, enzymatic and non-enzymatic post-translational modifications of histones and DNA-binding proteins. The effects of oxidative stress on these chromatin alterations mediate a number of cellular changes, including modulation of gene expression, cell death, cell survival and mutagenesis, which are disease-driving mechanisms in human pathologies. Targeting oxidative stress-dependent pathways is thus a promising strategy for the prevention and treatment of these diseases. We summarize recent research developments connecting oxidative stress and chromatin regulation.
Many pathogenic factors and pathophysiological processes lead to an increase in cytoplasmic and nuclear ROS. ROS can be detoxified, function as second messengers or damaging agents, altering gene expression, inducing mutagenesis or cell death.
Stars indicate macromolecular damage.
3-DG: 3-deoxyglucosone; Ang II: Angiotensin II; CAT: Catalase; COX: Cyclooxygenase; ER: Endoplasmic reticulum; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; GSH: Glutathione; LOX: Lipoxygenase; NO: Nitric oxide; NOS: Nitric oxide synthase; NOX: NADPH oxidase; PRDX: Peroxiredoxin; ROS: Reactive oxygen species; SOD: Superoxide dismutase; SOD*: Mutated SOD; TF: Transcription factor; TRX: Thioredoxin.
(A) Different oxidative histone modifications lead to alterations of chromatin structure. (B) Oxidation of deoxyguanine directly influences gene expression.
GS: Reduced glutathione; GSH: Glutathione; GSSG: Glutathione disulfide; NO: Nitric oxide; ROS: Reactive oxygen species; TF: Transcription factor.
Chromatin modifying enzymes can be inhibited under oxidative stress conditions by different mechanisms, including SAM depletion, oxidation, carbonylation or nitration. Activation of DNA methylation is mediated by oxidative DNA damage and the SIRT1 HDAC can be stimulated by LDH.
DNMT: DNA methyltransferase; GSH: Glutathione; HDAC: Histone deacetylase; HMT: Histone methyltransferase; LDH: Lactate dehydrogenase; MAT: Methionine adenosyltransferase; MS: Methionine synthase; NAD+: Nicotinamide adenine dinucleotide; NO: Nitric oxide; ROS: Reactive oxygen species; SAM: S-adenosylmethionine; TET: Ten-Eleven-Translocation DNA demethylase.
(A) Regulation of TRIM28 by oxidative stress. (B) Influence of oxidative stress on functions of HMGB1. (C) Regulation of chromatin proteins by PI(5)P.
PI(5)P: Phosphatidylinositol-5-phosphate; ROS: Reactive oxygen species; TF: Transcription factor.
Oxidative stress is involved in the pathogenesis of several diseases, like Alzheimer’s and Parkinson’s disease [Citation1,Citation2], amyotrophic lateral sclerosis [Citation3], diabetes [Citation4,Citation5], hypertension [Citation6,Citation7], autoimmune disorders [Citation8] or cancer [Citation9,Citation10]. Many of the pathogenic effects of oxidative stress are mediated by affecting chromatin.
Chromatin is a highly regulated signal integration platform, receiving inputs from extracellular and intracellular messengers and translating these into functional outputs such as activation or repression of transcription, DNA replication, chromatin condensation or DNA repair [Citation11]. Among others, these processes are regulated by covalent modifications of chromatin, including DNA methylation (for a review see [Citation12]) and modifications of histones, like methylation, acetylation, phosphorylation, ubiquitination, SUMOylation or poly-ADP-ribosylation (for a review see [Citation13]). These epigenetic marks as well as specific DNA sequences or DNA damage are recognized by effector proteins, which mediate the functional output.
Oxidative stress has been described to widely affect chromatin modifications as well as chromatin reader and repair proteins. These effects are thought to drive the physiological response to oxidative stress, but to also facilitate initiation and progression of several diseases. Here, we review how oxidative stress impacts on chromatin, how this is translated into a cellular outcome and how this is involved in common human diseases. Our analysis of the current data is focused on mechanisms directly linking oxidation or oxidative stress-mediated modification to chromatin and chromatin modifying enzymes. Effects of oxidative stress on upstream signal transduction pathways that indirectly influence chromatin have been omitted from our discussion.
Oxidative stress
In 2007, oxidative stress was redefined by Sies and Jones as ‘an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage’ [Citation14]. Physiologically, most cellular compartments are reducing environments [Citation15,Citation16], which are rich in antioxidants, like glutathione (GSH), ascorbic acid (vitamin C) or α-tocopherol (vitamin E) and antioxidant enzymes like peroxiredoxins, thioredoxins, superoxide dismutases (SODs) and catalase [Citation17,Citation18]. Consequently, most cysteines in intracellular proteins are in the reduced state under normal conditions to facilitate proper protein function [Citation16,Citation19]. Further, oxidative damage to DNA, RNA and lipids is prevented in such an environment because the antioxidant systems normally scavenge oxidants, like reactive oxygen or nitrogen species (ROS, RNS). ROS are produced when single electrons are transferred to oxygen, leading to the generation of the relatively inert superoxide radical (O2•–) [Citation20–23], the more reactive hydrogen peroxide (H2O2) and the highly reactive and damaging hydroxyl radical (•OH) [Citation24]. Damaging RNS result from the reaction of nitric oxide (NO•) with O2•–, generating the highly reactive peroxynitrite (ONOO-) [Citation25].
ROS & RNS as second messengers
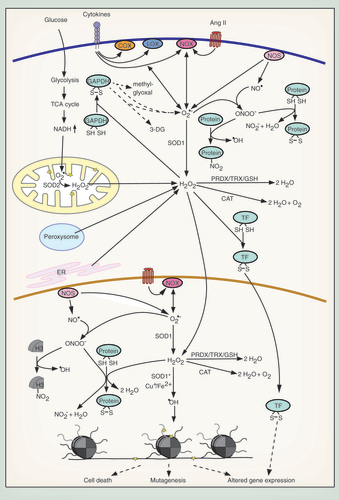
Due to their intermediate reactivity, H2O2 and NO• are important physiological second messengers. Several signal transduction pathways activate NADPH oxidases (NOX) in the cytosol and the nucleus and their sole function is to produce O2•–, which is then dismutated to H2O2 () [Citation24,Citation26,Citation27]. H2O2 has limited toxicity because it selectively oxidizes cysteine residues in specialized protein environments, whose pKa is particularly low, so that they exist as thiolate anions (S-) at physiological pH [Citation28,Citation29]. Oxidized cysteines often form disulfide bonds, altering protein folding and function and thus serving as ROS sensors () [Citation29]. This mechanism of redox regulation has been shown for a number of transcription factors, kinases, protein phosphatases and matrix metalloproteinases [Citation30–33] (see [Citation34,Citation35] for reviews).
NO• is generated in the cytosol and the nucleus through the action of nitric oxide synthases (NOS) () [Citation36,Citation37]. NOS are dependent on Ca2+/calmodulin and the cofactor tetrahydrobiopterin (BH4). They catalyze the production of NO• from O2, arginine and NADPH [Citation38]. Similar to H2O2, NO• is not highly reactive toward macromolecules and is mainly implicated in physiological cellular signaling [Citation39].
Pathways leading to increased ROS/RNS production
Although some ROS/RNS are important signaling molecules, overproduction of these molecules or pathological depletion of antioxidants can lead to oxidative stress. In eukaryotes, unintended ROS production is mainly a consequence of the oxidative metabolism. While oxygen itself has a very low reactivity [Citation40], many cellular enzymes are capable of inadvertently transferring a single electron to O2, leading to generation of O2•– [Citation20–23], which can damage biomolecules by either oxidizing or reducing them [Citation24]. Physiologically, O2•– is rapidly detoxified by SOD in the mitochondria, the cytosol and the nucleus to yield H2O2 [Citation20,Citation41]. H2O2 is also produced by a number of metabolic enzymes, like the peroxisomal Acyl-CoA oxidase, or through protein oxidation in the ER () [Citation42–44]. In contrast to O2•–, H2O2 can diffuse through aquaporins to cross organelle membranes and enter the cytosol and the nucleus [Citation45]. H2O2 is disproportionated by catalase or removed through the peroxiredoxin/thioredoxin and GSH systems () [Citation46,Citation47]. If H2O2 is not detoxified, it can be converted into •OH via the Cu+- or Fe2+-catalyzed Fenton reaction. •OH is highly toxic and rapidly oxidizes and damages DNA, lipids or proteins () [Citation29]. Similarly, ONOO- damages proteins through tyrosine nitration and disruption of iron–sulfur clusters. Also, its potential to oxidize perceptible cytosines is much higher than that of H2O2 () [Citation25].
One of the main causes of oxidative stress is mitochondrial dysfunction, resulting from damaged mitochondria [Citation1,Citation2,Citation6,Citation10] or from hyperglycemia-induced alterations in the cellular metabolism in diabetes () [Citation4,Citation5]. Other major contributors to oxidative stress are cytosolic and nuclear enzymes. Enhanced production of ROS by hyperactivated NOX, ‘uncoupled’ NOS or other enzymes, like lipoxygenase (LOX), cyclooxygenase (COX) or xanthine oxidase, can be the cause of oxidative stress [Citation6,Citation7,Citation9]. Among others, the activation of NOX, COX and LOX can be driven by acute or chronic inflammation through the exposure of cells to cytokines [Citation48,Citation49] or by hormones like angiotensin II in hypertension () [Citation50]. NOS can be uncoupled and release O2•– instead of NO• when cofactors are depleted or inhibited, when specific threonine residues are dephosphorylated or critical cysteines are oxidized [Citation51–54].
Defects in the ROS detoxifying machinery can also significantly contribute to the generation of oxidative stress. In some cases of familial amyotrophic lateral sclerosis, mutations of SOD1 enhance its ability to dismutate H2O2 to •OH and to catalyze tyrosine nitration () [Citation55,Citation56]. Decreased expression of or mutations in catalase lead to oxidative stress in cancer and diabetes [Citation57–59]. Apart from these enzymatic mechanisms for increased ROS production, non-enzymatic pathways are known. In Alzheimer’s and Parkinson’s disease intracellular accumulation of metals, like copper, iron and manganese leads to enhanced oxidative stress by catalyzing the Fenton reaction or by oxidizing dopamine, which results in ROS production [Citation60]. Furthermore, exposure to toxic chemicals, such as cigarette smoke, can induce oxidative stress.
Importantly, there is extensive cross-talk between the different ROS-generating pathways, driving a vicious cycle of ever increasing cellular oxidative stress. Mitochondrial ROS can, for example, stimulate NOX-mediated ROS production via a PKC-dependent pathway and vice versa [Citation61,Citation62]. This interplay can potentially drive nuclear ROS production in the context of mitochondrial dysfunction. Moreover, increased mitochondrial ROS inhibit GAPDH through cysteine oxidation [Citation63], causing an increase in all upstream glycolytic products. Under conditions of hyperglycemia (diabetes), this activates alternative arms of glucose metabolism, including the polyol and hexosamine pathways, which contribute to increased ROS formation () [Citation4]. Also, COX is activated by ROS and is itself a source of ROS () [Citation64].
Pathophysiological consequences of oxidative stress
An important pathogenic mechanism of oxidative stress in neurodegenerative diseases and diabetes is the induction of cell death [Citation65–68]. ROS can participate in cancer development and progression by inducing DNA damage, which leads to mutagenesis, and by affecting the expression of oncogenes [Citation69–71]. In diabetes, oxidative stress causes alterations in the cellular metabolism, contributing to the development of hyperglycemia-associated complications [Citation4,Citation5,Citation63]. Moreover, ROS/RNS-protein and -DNA adducts can function as autoantigens in autoimmune disorders like systemic lupus erythematosus (SLE) [Citation72,Citation73]. In hypertension, reaction of ROS with NO• is a major pathogenic mechanism inhibiting vasodilation by NO• and generating the damaging ONOO- [Citation74]. Major aspects of the ROS-dependent pathogenic mechanisms are mediated by affecting chromatin components.
Effects of oxidative stress on global chromatin structure
The global structure of chromatin is strictly organized within a cell. Based on different transcriptional activities, specific sets of chromatin modifications, associated proteins and distinct replication timings in S phase, at least four different chromatin states can be distinguished. These are transcriptionally active euchromatin and three distinct heterochromatic states: transcriptionally repressed chromatin, silent ‘null’ chromatin, which is not associated with specific proteins or histone marks, and HP1-associated heterochromatin [Citation75]. Maintaining appropriate levels of the latter is important to facilitate genome stability [Citation76,Citation77], proper transcription [Citation78,Citation79], mitosis [Citation80] and meiosis [Citation81]. Oxidative stress can stimulate global heterochromatin loss and thus potentially affect these processes [Citation82].
In this context, it has recently been shown that overexpression of tau, one of the pathogenic factors in Alzheimer’s disease, causes oxidative stress-induced heterochromatin loss and neurotoxicity in a Drosophila model [Citation83]. Mechanistically, it was hypothesized that oxidative stress-induced DNA damage promotes chromatin relaxation and thus a global reduction in heterochromatin in neurons. Heterochromatin loss then facilitates ectopic expression of normally silenced genes to induce cell cycle entry of postmitotic neurons [Citation83], which is known to be neurotoxic [Citation84]. In line with these findings, heterochromatin was reduced in neurons isolated from human postmortem Alzheimer’s brains and expression of heterochromatic, pluripotency-associated genes was increased [Citation83].
Different mechanisms are in place to protect cells from heterochromatin loss and enhance genome stability upon exposure to oxidative stress. 293F cells, for example, show an increase in the stability of pericentromeric heterochromatin upon exposure to hydrogen peroxide. Pericentromeric heterochromatin is characterized by high levels of the histone H3K9 methyltransferase SUV39H1, the SIRT1 deacetylase and HP1 proteins. Hydrogen peroxide upregulates the expression of SIRT1, which increases SUV39H1 stability by inhibiting its ubiquitination and proteasomal degradation. Increased SUV39H1 stability is directly linked to its enhanced turnover at pericentromeric heterochromatin, which decreases DNA damage in response to stress [Citation85]. This mechanism might play a role in the enhanced resistance of cancer cells to oxidative stress and it could possibly be exploited to inhibit heterochromatin loss in neuronal cells, ultimately preventing cell death.
Direct effects of oxidative stress on histones
Histones are extensively modified in an ROS- and RNS-dependent way and are glutathionylated in a redox-sensitive manner, which affects their folding and stability, as well as their ability to be post-translationally modified. Because histones are the most common chromatin proteins, any change in their abundance, structure or post-translational modifications (PTMs) will have a severe impact on the global structure of chromatin, influencing gene expression, genome stability and replication.
Modification of histones by peroxynitrite
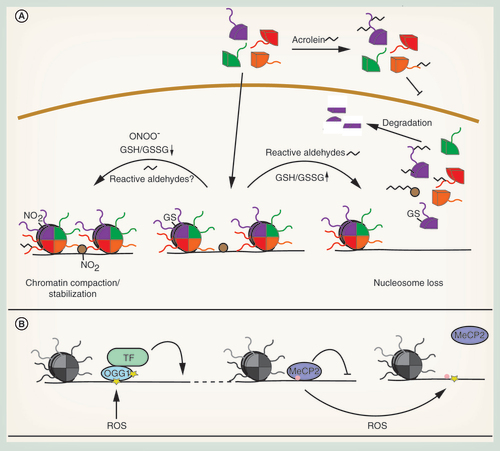
Histones can be directly modified by peroxynitrite. In vitro exposure of recombinant histones H1 and H3 to peroxynitrite leads to tyrosine nitration. Nitrated histones show an increase in structured domains, specifically β-sheet structures, and increased thermostability. This observed increased thermostability might contribute to protecting DNA from oxidative damage during oxidative stress (A) [Citation86,Citation87]. Further, nitrated H1 is a potent autoantigen in SLE and thus contributes to the pathogenesis of this disease [Citation87].
Modification of histones by reactive aldehydes
Reactive aldehydes are generated intracellularly in an ROS-dependent way. They readily react with proteins to form carbonyl adducts. Lipids can be enzymatically or non-enzymatically peroxidized to yield highly reactive α,β-unsaturated aldehydes, such as glyoxal, malondialdehyde, acrolein, 4-hydroxy-2-nonenal (4-HNE) or 4-oxo-2-nonenal (4-ONE) [Citation88]. Furthermore, ROS promote the production of the glucose metabolites methylglyoxal and 3-deoxyglucosone [Citation89,Citation90]. These reactive carbonyl species form adducts with permissive cysteines, arginines, lysines or histidines in proteins resulting in advanced glycation endproducts (AGEs) or advanced lipoxidation endproducts and facilitating protein cross-linking [Citation91]. AGEs can further form under conditions of hyperglycemia through direct adduction of glucose or other sugars to proteins and ROS-dependent conversion to carboxymethyllysine or pentosidine [Citation92,Citation93]. Strong evidence indicates that carbonylated proteins are implicated in the initiation and progression of human pathologies. Accumulation of AGEs is, for example, associated with increased vascular stiffness in diabetic individuals, both in the macro- and microcirculation [Citation94]. Increases in AGEs have also been described in Alzheimer’s disease, where they might contribute to amyloid β crosslinking [Citation95].
Histones are common AGEs/advanced lipoxidation endproducts and H2A modified by 4-HNE has been identified as an autoantigen in SLE [Citation96]. In vitro, histones H1, H2A and H3 that have been modified with 3-deoxyglucosone become less thermostable due to partial unfolding. In vivo, this might lead to alterations in chromatin structure and gene expression [Citation97–99]. In contrast, in vitro glycation of H2A by methylglyoxal led to an increase in alpha-helical structures and stabilization of the protein [Citation100]. These discrepancies might be due to different target residues of the different reactive aldehydes or to the formation of different endproducts.
Several studies indicate that carbonylated histones are lost from chromatin, resulting in an overall reduction in nucleosome content. For example, H1 isolated from livers of diabetic rats displayed a reduced α-helical content and diminished binding to DNA [Citation101], suggesting that the structural alterations described for H1, H2A and H3 in vitro can generally affect association of histones with DNA (A). Similarly, increased cellular 4-ONE might trigger global nucleosome loss via inhibition of nucleosome assembly (A). In contrast to 4-HNE, which targets a variety of cellular proteins, 4-ONE preferentially modifies histones. Among others, adducts are formed with H3K23, H3K27 and H4K79, inhibiting nucleosome assembly in vitro. The H3K27 adduct was also detected in LPS-stimulated macrophages, indicating that it might be a novel ROS-mediated, specific histone mark that stimulates transcription, rather than an unspecific, damaging adduct [Citation102].
Another study in human bronchial epithelial cells described a different mechanism for global histone loss. Free histones are more sensitive to adduct formation with the cigarette smoke component acrolein than histones in nucleosomes. Because acrolein reacts with lysine and arginine residues, acetylation of newly synthesized histones was blocked, inhibiting their nuclear import and incorporation into chromatin (A). Consistently, high concentrations of acrolein led to a global reduction of chromatin-associated H3, which correlated with increased transcription at histone-depleted loci [Citation103]. Moreover, glycated histones are irreversibly damaged and need to be removed by the nuclear proteasome [Citation104], which possibly contributes to the global loss of histones from chromatin (A).
Glutathionylation of histones
Glutathionylation is a common modification of proteins in oxidatively stressed cells. Cysteines oxidized to sulfenic acid can be glutathionylated by glutathione-S-transferases (GSTs), utilizing GSH, to protect the residues from further, irreversible oxidation to sulfonic acid (SO3H) [Citation105,Citation106]. However, oxidative stress also increases the oxidation of GSH to GSSG, decreasing the GSH/GSSG ratio. Glutathionylation of histones H3 and H2B has been shown in MCF7 breast cancer cells and is most likely dependent on GSTP1–1. Enhanced glutathionylation correlated with higher GSH levels and drug resistance, suggesting that it might lead to altered expression of drug resistance genes [Citation107].
In agreement with these findings, histone H3 is glutathionylated in proliferating cells, which is stimulated when the GSH/GSSG ratio is high, but decreases when GSH is depleted (A). Glutathionylation of H3 decreases nucleosome stability, facilitates gene expression and DNA replication [Citation108]. It is conceivable that an oxidative stress-induced reduction in glutathionylation may protect cells from oxidative stress by leading to chromatin compaction and inhibition of replication of potentially damaged DNA. Further, reduced histone glutathionylation might contribute to global gene regulation.
Oxidative damage to DNA
In addition to histones, DNA can also be directly affected by oxidative stress. The most susceptible nucleoside toward oxidation is deoxyguanosine (dG), which becomes easily oxidized to 8-oxo-dG. It is normally bound and excised by OGG1 and then repaired by the base excision repair pathway. Recent studies indicate that this oxidatively ‘damaged’ base can contribute to gene regulation upon oxidative stress.
G-quadruplex (G4) structures, which occur in certain G-rich DNA sequences and consist of guanine tetrads stabilized by hydrogen bonds and monovalent cations, are preferred targets for oxidation. Many oncogenes, like MYC, KRAS, VEGF, BCL2, RB1 or HIF1 are regulated by these G4 structures. Some transcription factors do preferentially bind to these sequences and alter the G4 conformational state to regulate gene expression [Citation109]. Hypoxic stress, which leads to enhanced ROS formation, triggers oxidative damage preferentially in G4-containing regions and about 25% of genes regulated by hypoxia contain G4 structures [Citation110]. This might drive the preferential expression of the aforementioned oncogenes. In another study, a mechanism for 8-oxo-dG-induced transcription has been described. Treatment of airway epithelial cells with TNFα leads to enhanced ROS production and an increase in global 8-oxo-dG. OGG1 is also oxidized under these conditions, rendering it enzymatically inactive. Inactive OGG1 does, however, still bind 8-oxo-dG, recruiting transcription factors and enhancing gene expression (B) [Citation111].
Oxidative DNA damage can also inhibit binding of chromatin proteins. Methyl-CpG-binding proteins normally recognize methylated CpG dinucleotides and mediate transcriptional repression. However, binding of methyl-CpG-binding protein 2 to methylated CpG is inhibited when the guanosine in such a dinucleotide is oxidized or when methylcytosine is oxidatively converted to hydroxymethylcytosine (B) [Citation112].
Effects of oxidative stress on DNA methylation
In addition to directly modifying DNA, ROS can also affect DNA methylation, which is generally linked to repression of transcription. In mammals, the primary methylation targets are cytosines in CpG dinucleotides. While CpGs are usually methylated genome-wide, regions with a high density of CpG dinucleotides (CpG islands) are mostly hypomethylated [Citation113]. Two classes of enzymes control DNA methylation: DNMTs and the demethylases of the TET family. DNMTs utilize S-adenosylmethionine (SAM) to transfer a methyl group to cytosine, while TETs oxidize 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) in an Fe(II)-, α-ketoglutarate- (α-KG), ascorbate- and O2-dependent reaction [Citation114,Citation115].
In many pathologies, like Alzheimer’s disease, diabetes or cancer global DNA hypomethylation has been described [Citation116–119]. However, some specific CpG islands are hypermethylated in diabetes or cancer [Citation117,Citation120] and it has been suggested that both global hypomethylation and specific hypermethylation can be linked to oxidative stress.
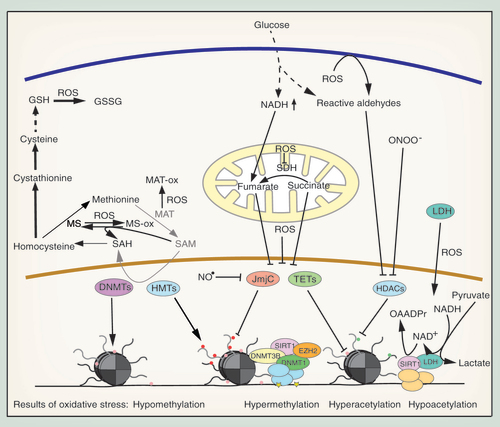
DNA methylation is directly blocked when SAM decreases. SAM is synthesized from methionine by methionine adenosyltransferase (MAT). It is then used for methylation reactions, producing S-adenosylhomocysteine, which is hydrolyzed to homocysteine. Methionine is regenerated from homocysteine by methionine synthase (MS) (). Depletion of SAM can be triggered by oxidative stress via several mechanisms. Firstly, oxidation of MAT leads to its inactivation and to reduced SAM synthesis from methionine [Citation121,Citation122]. Secondly, the cobalamine in MS is easily oxidized, which leads to inactivation of the enzyme. MS can be reactivated by MS reductase utilizing SAM, forming a futile cycle without any net SAM production [Citation123]. And thirdly, the transsulfuration pathway can regenerate glutathione under oxidative stress conditions from SAM via homocysteine, cystathionine and cysteine, depleting SAM in the process () [Citation124–126]. In agreement with this, SAM and DNA methylation were decreased in oxidatively stressed bladder cancer and kidney cell lines. There, supplying SAM, methionine or N-acetylcysteine restored DNA methylation [Citation127]. Similarly, 5-mC levels in liver tissue of hamsters treated with the GSH-depleting agent bromobenzene were reduced. This hypomethylation was rescued, when the animals were supplied with methionine [Citation128]. Both experiments indicate that a reduction in SAM, and not oxidation or modification of DNMTs, was the prime reason for reduced DNA methylation. However, direct oxidation of 5-mC to 5-hmC can also lead to DNA demethylation. 5-hmC inhibits maintenance methyltransferase DNMT1, thus blocking the proper inheritance of methylation patterns and leading to indirect demethylation of CpG sites [Citation129].
A mechanism for DNA hypermethylation upon oxidative stress is the inhibition of TET proteins. As Fe(II)- and α-KG-dependent dioxygenases, they can be inactivated by iron oxidation. During the catalytic cycle, Fe(II) is oxidized to Fe(III) and Fe(IV) [Citation115] and it was hypothesized that oxidized iron is regenerated by ascorbate [Citation114]. Under oxidative stress conditions, this regeneration is perturbed, leading to inactivation of the enzyme and an increase in global 5-mC levels [Citation130]. Consistent with this, 5-hmC is significantly reduced in oxidatively stressed SY5Y neuroblastoma cells [Citation131]. Furthermore, TET1 knockdown enhances cell death in murine cerebellar granule cells, indicating that TET1 inhibition by oxidative stress could be involved in the pathogenesis of neurodegenerative diseases [Citation132]. Additionally, the Krebs cycle intermediates succinate and fumarate inhibit TETs competitively due to their similarity with α-KG [Citation133]. This mechanism might play a role in hyperglycemia, which leads to an increase in fumarate due to impairments in the mitochondrial electron transport chain [Citation134]. An increase in succinate can result from the inhibition of succinate dehydrogenase by oxidative stress () [Citation135].
Both hypermethylation of CpG islands and global hypomethylation can also be mediated by relocalization of DNMTs. Base excision and mismatch repair proteins recognize 8-oxo-dG and recruit maintenance DNA methyltransferase DNMT1, which forms a complex with the SIRT1 deacetylase, the de novo DNA methyltransferase DNMT3B and the histone H3K27 methyltransferase EZH2 [Citation136,Citation137]. The complex is mainly recruited to active CpG island promoter regions, where increased 8-oxo-dG is present, and it enhances DNA methylation and H3K27me3, while reducing H3K4me3 and H4K16ac levels (). Simultaneously, DNMT1, DNMT3B and SIRT1 are lost from other genomic regions. As a consequence, CpG islands of lowly expressed genes were hypermethylated in an in vivo mouse colitis model after exposure to oxidative stress, while housekeeping genes remained unaffected. These same genes have also been shown to be hypermethylated in intestinal cancer, providing a mechanistic explanation for how oxidative stress can drive cancer-specific gene expression [Citation136].
Effects of oxidative stress on histone modifications
Histone PTMs provide an additional layer of complexity for the regulation of chromatin structure and chromatin-regulated processes beyond the simple structural properties of histones. Covalent histone PTMs can directly or through the recognition by chromatin reader proteins influence transcription, chromatin structure, DNA repair or mitosis. Chromatin reader proteins are, for example, adaptor proteins or different enzymes, like chromatin modifiers or chromatin remodeling complexes [Citation13]. Oxidative stress influences histone PTMs in various ways and thus affects a number of chromatin-regulated processes.
Histone lysine methylation
Lysines can be mono-, di- or trimethylated and histone lysine methylation is controlled by HMTs and HDMs. The latter can be divided into LSD and JmjC family enzymes. LSD enzymes catalyze demethylation of mono- and dimethylated lysines, JmjC proteins target all methylation states [Citation138]. Especially JmjC family proteins can be inhibited by oxidative stress, leading to histone hypermethylation. The best-studied methylation marks are H3K4me1/2/3, H3K9me2/3 and H3K27me3. H3K4 methylation is generally associated with transcriptional activation, H3K9me2/3 are mainly found in constitutive heterochromatin and are associated with heterochromatin formation and maintenance, and H3K27me3 is associated with transcriptional silencing and facultative heterochromatin [Citation13]. Alterations in these methylation marks can therefore result in aberrant transcription or changes in chromatin packaging.
Like DNA demethylases of the TET family, HDMs of the JmjC family are Fe(II)- and α-KG-dependent dioxygenases. As such, they are also inhibited by high levels of fumarate, which led to a global increase in H3K4me1, H3K4me3, H3K9me2/3 H3K27me2/3 and H3K79me2 in fumarate hydratase deficient cells [Citation133,Citation139]. Further, iron oxidation blocks the catalytic activity of JmjC HDMs () and especially H3K4me3 was significantly increased globally after exposure of human bronchial epithelial cells to hydrogen peroxide [Citation130].
JmjC demethylases can also be inhibited by nitrosative stress. The NO• radical inhibits the JmjC domain-containing demethylase KDM3A by directly binding to the catalytic iron (). Consequently, treatment of cells with NO• results in an increase in H3K9me2, which is normally targeted by KDM3A. Although NO• caused a simultaneous upregulation of KDM3A, this did not compensate for the NO•-dependent inhibition of the enzyme [Citation140].
Lastly, HMTs could also be affected by oxidative stress. They are dependent on SAM and in oxidatively stressed cells reduced SAM levels are expected to block histone methylation and thus globally alter chromatin structure () [Citation125,Citation141].
It is important to note that oxidative stress does not influence all histone lysine marks in the same way. While some residues are hypermethylated in response to an oxidative insult, others are unaffected or even become hypomethylated [Citation130]. This is intriguing because all known HMTs depend on SAM and all trimethylated residues are exclusively targeted by JmjC family HDMs. The observed specific effects might be a consequence of different enzymes having distinct sensitivities toward oxidation, nitration, competitive inhibition or SAM depletion, depending on structural features of the enzyme or on the microenvironment of an enzyme complex. In this context, it has recently been shown that MATII can be recruited to target genes by the H3K9 methyltransferase SETDB1 [Citation142]. This leads to a local increase in SAM and could facilitate histone methylation even in the absence of high global SAM levels. Hence, specific local rather than global regulation of HMT and HDM activities might predominate in vivo.
Histone acetylation
In contrast to methylation, acetylation of lysine residues neutralizes the positive charge of the histone tail and is therefore generally associated with chromatin relaxation and transcriptional activation. Global changes in histone acetylation patterns do thus have a broad and immediate impact on cell physiology and are intricately linked to several diseases. Histone acetylation is controlled by histone acetyltransferases and histone deacetylases (HDACs). HDACs are grouped into class I, II, III and IV enzymes, depending on their structure, homology to yeast HDACs, subcellular localization and catalytic mechanism [Citation143–145]. Class I, II and VI enzymes possess a catalytic metal ion and produce free acetate [Citation146], while class III HDACs or sirtuins require a structural zinc ion and NAD+, to which they transfer the acetyl group producing O-acetyl-ADP-ribose [Citation147–149].
Oxidative and nitrosative stress are potent modulators of HDAC function at multiple levels. It has been shown that most class I HDACs (HDAC1, HDAC2 and HDAC3) are alkylated and inhibited by several reactive aldehydes, which results in changes in gene expression [Citation150,Citation151]. Moreover, HDAC2 can be nitrosylated [Citation152], the effect of which is being discussed controversially. In one study conducted in neurons, nitrosylation did not influence its activity, but led to displacement from chromatin to activate gene expression [Citation153]. In another study in C2C12 myoblasts, cysteine nitrosylation decreased HDAC2 activity [Citation154]. In this context, it is interesting to note that knockdown or knockout of HDAC1, HDAC2 or HDAC3 can promote the development of hematological malignancies and hepatocellular carcinoma [Citation155–158], indicating that ROS-mediated HDAC inhibition is potentially oncogenic. Furthermore, inhibition of HDACs by oxidative stress might be a physiological response to confer oxidative stress resistance to cells. Treatment of mice with the HDAC inhibitor β-hydroxybutyrate led to upregulation of FOXO3A and metallothioneine 2, two proteins that protect cells from oxidative stress [Citation159].
Several class III HDACs are also inhibited by oxidative stress. SIRT1 can be inactivated as a result of cysteine carbonylation [Citation160], nitrosylation [Citation152] or glutathionylation [Citation161]. Inactivation of SIRT1 during oxidative stress leads to increased acetylation and inhibition of FOXO3. Activation or overexpression of SIRT1, in contrast, protects cells from oxidative stress-induced senescence [Citation162,Citation163], indicating that SIRT1 inhibition negatively affects oxidatively stressed cells. Inactivation of SIRT1 by cigarette smoke results in enhanced acetylation of NFκ-B and the increased expression of proinflammatory genes in macrophages [Citation164]. Similarly, SIRT3 has been shown to be carbonylated and inactivated by 4-HNE [Citation165]. Nuclear SIRT3 is rapidly degraded when cells are exposed to oxidative stress, leading to the upregulation of stress-induced genes [Citation166]. Moreover, the tumor suppressor SIRT6 can be inactivated through peroxynitrite-mediated nitrosylation [Citation167,Citation168], which could be oncogenic.
A different mechanism of oxidative stress regulation has been described for class II HDACs. In cardiac myocytes HDAC4 and HDAC5 translocate from the nucleus to the cytoplasm in an ROS-dependent manner [Citation169]. This process stimulates transcription of MEF2-dependent genes, which has been shown to trigger hypertrophic growth of cardiac myocytes [Citation170].
Although ROS are potent inhibitors of HDACs they can also increase histone deacetylation by directly stimulating HDAC expression [Citation171,Citation172] or by indirectly enhancing HDAC activity. Treatment of HepG2 cells with H2O2 led to an increase in nuclear lactate dehydrogenase, which catalyzes the reduction of pyruvate to lactate using NADH and producing NAD+. Lactate dehydrogenase directly interacts with SIRT1 and provides the enzyme with NAD+ to stimulate deacetylation [Citation173].
Histone phosphorylation
Phosphorylation of different histone serine, threonine and tyrosine residues plays a role in the regulation of gene expression, DNA repair and mitosis [Citation174]. Several studies show that oxidative stress can indirectly influence global histone phosphorylation levels. Oxidative stress leads to the formation of DNA double-strand breaks, which induce the phosphorylation of H2AX to trigger DNA repair [Citation175,Citation176]. Additionally, one study showed that H2O2 treatment increases H2AX phosphorylation in an ATR-dependent way independent of the presence of double-strand breaks, suggesting that it fulfills a different signaling function in oxidatively stressed cells [Citation177]. The effect of ROS on H3S10 phosphorylation is controversial. While oxidative stress increased H3S10p in monocytes and alveolar macrophages in a p38-, MSK1- and IKK2-dependent way [Citation178], it inhibited H3S10 phosphorylation by VRK1 in SY5Y neuroblastoma cells through upregulation of MKP2 [Citation179]. The histone-targeting protein phosphatases PP1 and PP2A can be inhibited by oxidation of their catalytic metal ion [Citation180]. Whether this is a mechanism by which oxidative stress can directly modulate histone phosphorylation levels needs, however, to be seen.
Regulation of chromatin-associated proteins
Regulation of TRIM28
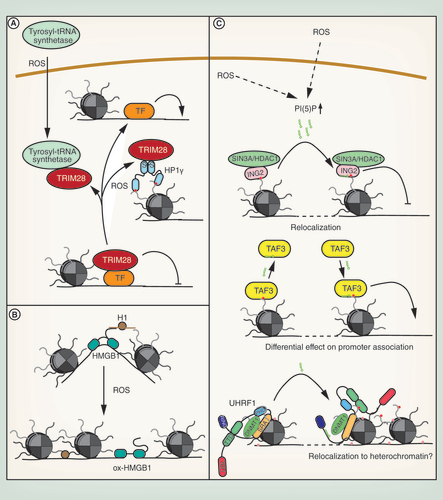
TRIM28 or KAP1 is a global transcriptional co-repressor protein. It can be recruited to a number of transcription factors and interacts with HDACs and HMTs [Citation181]. Two mechanisms of negative regulation of TRIM28 by oxidative stress have been described. The first one involves nuclear import of tyrosyl-tRNA synthetase in oxidatively stressed cells, where it sequesters TRIM28 and HDAC1 to facilitate transcriptional activation of DNA damage repair genes. This protects cells from oxidative stress-induced DNA damage (A) [Citation182].
The second mechanism involves oxidation HP1γ. HP1s bind H3K9me3 and have conventionally been described as essential proteins for heterochromatin formation and maintenance. They are homodimeric proteins, which interact with a number of other chromatin factors. HP1γ is associated with both eu- and heterochromatin. In euchromatin, it has been shown to regulate gene expression, both positively and negatively. It was recently demonstrated that HP1γ is oxidized upon induction of oxidative stress in cultured cells. Oxidation leads to reversible disulfide bond and covalent homodimer formation. This significantly enhances the interaction of HP1γ with TRIM28, sequestering it and inhibiting its repressive function toward a reporter gene (A). Consistent with this, HP1γ is required to retain cell viability after an oxidative stress insult, suggesting that this mechanism also contributes to gene regulation upon oxidative stress [Citation183]. Interestingly, TRIM28 heterozygous mice are obese and show signs of metabolic syndrome, like decreased glucose tolerance [Citation184], indicating that a reduction in TRIM28 levels or availability, for example, by sequestration in oxidatively stressed cells, could be causative for this disease.
Regulation of HMGB1
HMGBs are structural DNA-binding proteins that can bend, loop or unwind DNA. They can mediate nucleosome remodeling, DNA accessibility and histone H1 displacement to facilitate transcriptional activation. HMGB1 interacts with H1 through its highly acidic C terminus [Citation185]. The functions of HMGB1 can be regulated by reversible oxidation of the protein, which leads to disulfide bond formation and conformational changes in the DNA-binding and -bending domain. Consistent with this, oxidized HMGB1 shows a two-fold lower DNA-binding affinity and loses its ability to bend DNA. It does, however, gain a DNA end-joining function, potentially increasing DNA repair after an oxidative insult. It also shows reduced affinity toward histone H1 and its ability to displace H1 from DNA is significantly inhibited (B). In response to oxidative stress, stabilization of H1 on chromatin might repress transcription and enhance chromatin stability [Citation186]. Further, reduced bending of DNA by HMGB1 might result in transcriptional repression through impaired interactions of regulatory elements.
Regulation of chromatin-binding proteins by the second messenger PI(5)P during oxidative stress
Phosphatidylinositol phosphates are important second messengers in a number of signal transduction pathways. They are present in all cellular membrane compartments and, surprisingly, also in the nuclear matrix. Enzymes that produce mono-, di- and triphosphorylated PI have been detected in the nucleus, suggesting that the nuclear pool can be regulated independently of the membrane pool [Citation187,Citation188]. Indeed, recent publications highlight the specific functions of nuclear phosphatidylinositol phosphates as signaling molecules. PI(5)P increases in response to H2O2 and etoposide treatment [Citation189], which mediates its effects by inducing oxidative stress and oxidative stress-dependent DNA damage [Citation190]. PI(5)P might be a general mediator of the oxidative stress response, targeting a number of different chromatin proteins.
PI(5)P binds to ING2 [Citation191], which specifically recognizes H3K4me3 and can associate with the p300 histone acetyltransferases [Citation192] or the SIN3A–HDAC1 complex [Citation193] to stimulate or repress transcription. PI(5)P binding to the ING2 plant homeodomain (PHD) triggers its relocalization to novel chromatin sites and is essential for repression of proproliferative genes [Citation194] and the induction of apoptosis upon etoposide treatment (C) [Citation191]. Alterations in the transcriptional program can also be mediated by PI(5)P-dependent regulation of the general transcription factor TAF3 by either increasing or decreasing binding to promoter elements (C) [Citation195].
Another chromatin protein that interacts with PI(5)P is UHRF1. UHRF1 interacts with DNMT1, it is implicated in maintenance DNA methylation and stimulates proliferation in breast, prostate and lung cancer [Citation196]. UHRF1 is composed of five domains, an N-terminal ubiquitin-like domain, an H3K9me3-binding tandem tudor domain (TTD), a PHD, which binds the unmodified N-terminal histone H3 tail, a hemi-methyl-CpG-binding SRA domain and a C-terminal RING domain. These domains are connected by flexible linkers. PI(5)P binding to the UHRF1 polybasic region in the linker between the SRA and RING domains alters the overall conformation of the protein. While the TTD is blocked in the non-PI(5)P bound protein, enabling binding of the unmodified H3 N-terminus to the PHD only, PI(5)P unblocks the TTD domain, allowing for binding of the TTD to K9me3-modified histone tails (C) [Citation197]. This mechanism might protect the genome from oxidative insults by facilitating heterochromatin and DNA methylation maintenance under conditions of oxidative stress. Furthermore, relocalization of UHRF1 to H3K9me3-marked chromatin might alter gene expression programs in stressed cells.
Conclusion & future perspective
As we have discussed, oxidative stress globally influences chromatin on multiple levels, from DNA and histones to histone modifiers and structural DNA-binding proteins. Also, it is well known that oxidative stress is involved in initiation and progression of neurodegenerative diseases, diabetes, hypertension, cancer and several other pathologies. Nevertheless, for most mechanisms of chromatin regulation by oxidative stress highlighted in this review, a direct link to diseases has not been demonstrated. By and large, general effects of ROS have been described in different tissue culture model systems. Since the chromatin changes induced by oxidative or nitrosative stress observed there are also seen in human diseases, it can be projected that ROS-mediated chromatin alterations play an important role in initiation and progression of the aforementioned pathologies. Future research needs to focus on directly demonstrating how oxidative stress-induced chromatin changes impact on human diseases. Important questions to address are: how do ROS and RNS affect chromatin in specific cells in a disease setting? What are the molecular mechanisms for the observed oxidative stress-induced chromatin changes? What are the cellular effects of these chromatin changes? And what is the pathogenic potential of these chromatin-mediated effects?
Once these issues are resolved we are confident that it will be possible to exploit oxidative stress-mediated chromatin changes in diseases to develop drugs targeting specific ROS-induced chromatin signaling pathways. Antioxidants have already been widely tested as a treatment for neurodegenerative diseases, diabetes and cancer, but results were contradictory, ranging from improvement of the disease to accelerated progression [Citation198–202]. Consistent with this, ROS have both protective and damaging effects on cells. To be effective, it will indeed be important to only target distinct parts of the ROS-dependent response. In this context, chromatin is a very interesting target for drug development because it integrates and mediates most cellular responses to extra- and intracellular stimuli. Furthermore, a number of small molecule inhibitors of chromatin modifying enzymes are already in clinical trials or are even being used as successful therapeutics, making them readily available for therapy. We are looking forward to the many basic connections between ROS, chromatin and human pathologies to be made and to their translation into medical applications.
Oxidative stress
Oxidative stress is an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage. It is linked to the initiation and progression of several common human diseases.
Oxidative stress & global chromatin structure
Depending on the cell type, oxidative stress triggers global heterochromatin loss and cell death or protective stabilization of heterochromatin.
Direct oxidative modifications of histones alter the global chromatin structure, induce changes in transcription, DNA replication and might regulate cell death.
Oxidative stress & DNA methylation
Oxidative stress-induced inhibition of DNA methyltransferases and relocalization of DNA methyltransferases to CpG islands induces global DNA hypomethylation and local promoter hypermethylation.
Inhibition of Fe(II)- and α-KG-dependent TET DNA demethylases by oxidation, fumarate or succinate leads to altered DNA methylation.
Oxidative stress & histone modifications
Oxidative stress inhibits Fe(II)- and α-KG-dependent histone demethylases of the JmjC family and potentially triggers histone hypermethylation.
Oxidative stress directly inhibits histone deacetylases, but also stimulates their expression and locally increases NAD+ to activate SIRT deacetylases.
Oxidative stress regulates DNA-associated proteins
The corepressor TRIM28 is sequestered from the chromatin under conditions of oxidative stress by binding to tyrosyl-tRNA synthetase or oxidized HP1γ, leading to activation of gene expression.
Oxidized HMGB1 loses its DNA-bending activity and its ability to displace histone H1, possibly inhibiting gene expression and stabilizing chromatin. At the same time, it gains a DNA end-joining function, likely contributing to DNA repair after an oxidative insult.
The second messenger PI(5)P is upregulated by oxidative stress and modulates the functions of the corepressor ING2, the general transcription factor TAF3 and the structural chromatin protein UHRF1, thus controlling transcription at multiple levels.
Future perspective
Oxidative stress widely affects chromatin and is implicated in several human diseases. Further studies are required to comprehend the exact molecular connections of these phenomena for exploiting the management of oxidative stress-induced alterations for therapeutic purposes.
Financial & competing interests disclosure
The research reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Chen Z , ZhongC . Oxidative stress in Alzheimer’s disease . Neurosci. Bull.30 ( 2 ), 271 – 281 ( 2014 ).
- Yan MH , WangX , ZhuX . Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease . Free Radic. Biol. Med.62 , 90 – 101 ( 2013 ).
- Poon HF , HensleyK , ThongboonkerdVet al. Redox proteomics analysis of oxidatively modified proteins in G93A–SOD1 transgenic mice – a model of familial amyotrophic lateral sclerosis . Free Radic. Biol. Med.39 ( 4 ), 453 – 462 ( 2005 ).
- Yan L-J . Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress . J. Diabetes Res.2014 , 137919 ( 2014 ).
- Giacco F , BrownleeM . Oxidative stress and diabetic complications . Circ. Res.107 ( 9 ), 1058 – 1070 ( 2010 ).
- Madamanchi NR , VendrovA , RungeMS . Oxidative stress and vascular disease . Arterioscler. Thromb. Vasc. Biol.25 ( 1 ), 29 – 38 ( 2005 ).
- Hernanz R , BrionesAM , SalaicesM , AlonsoMJ . New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension . Clin. Sci.126 ( 2 ), 111 – 121 ( 2014 ).
- Perl A . Oxidative stress in the pathology and treatment of systemic lupus erythematosus . Nat. Rev. Rheumatol.9 ( 11 ), 674 – 686 ( 2013 ).
- Block K , GorinY . Aiding and abetting roles of NOX oxidases in cellular transformation . Nat. Rev. Cancer12 ( 9 ), 627 – 637 ( 2012 ).
- Sabharwal SS , SchumackerPT . Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel?Nat. Rev. Cancer14 ( 11 ), 709 – 721 ( 2014 ).
- Badeaux AI , ShiY . Emerging roles for chromatin as a signal integration and storage platform . Nat. Rev. Mol. Cell Biol.14 ( 4 ), 211 – 224 ( 2013 ).
- Schübeler D . Function and information content of DNA methylation . Nature517 ( 7534 ), 321 – 326 ( 2015 ).
- Bannister AJ , KouzaridesT . Regulation of chromatin by histone modifications . Cell Res.21 ( 3 ), 381 – 395 ( 2011 ).
- Sies H , JonesD . Oxidative Stress . In : Encyclopedia of Stress (Second Edition) . FinkG ( Ed. ). Academic Press , New York, USA , 45 – 48 ( 2007 ).
- Kojer K , RiemerJ . Balancing oxidative protein folding: the influences of reducing pathways on disulfide bond formation . Biochim. Biophys. Acta1844 ( 8 ), 1383 – 1390 ( 2014 ).
- López-Mirabal HR , WintherJR . Redox characteristics of the eukaryotic cytosol . Biochim. Biophys. Acta1783 ( 4 ), 629 – 640 ( 2008 ).
- Espinosa-Diez C , MiguelV , MennerichDet al. Antioxidant responses and cellular adjustments to oxidative stress . Redox Biol.6 , 183 – 197 ( 2015 ).
- Pisoschi AM , PopA . The role of antioxidants in the chemistry of oxidative stress: a review . Eur. J. Med. Chem.97 , 55 – 74 ( 2015 ).
- Go Y-M , JonesDP . Redox control systems in the nucleus: mechanisms and functions . Antioxid. Redox Signal.13 ( 4 ), 489 – 509 ( 2010 ).
- Kowaltowski AJ , de Souza-PintoNC , CastilhoRF , VercesiAE . Mitochondria and reactive oxygen species . Free Radic. Biol. Med.47 ( 4 ), 333 – 343 ( 2009 ).
- Tretter L , Adam-ViziV . Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress . Philos. Trans. R. Soc. B Biol. Sci.360 ( 1464 ), 2335 – 2345 ( 2005 ).
- Murphy MP . How mitochondria produce reactive oxygen species . Biochem. J.417 ( Pt 1 ), 1 – 13 ( 2009 ).
- St-Pierre J , BuckinghamJA , RoebuckSJ , BrandMD . Topology of superoxide production from different sites in the mitochondrial electron transport chain . J. Biol. Chem.277 ( 47 ), 44784 – 44790 ( 2002 ).
- Lambeth JD . NOX enzymes and the biology of reactive oxygen . Nat. Rev. Immunol.4 ( 3 ), 181 – 189 ( 2004 ).
- Radi R . Peroxynitrite, a stealthy biological oxidant . J. Biol. Chem.288 ( 37 ), 26464 – 26472 ( 2013 ).
- Petry A , WeitnauerM , GörlachA . Receptor activation of NADPH oxidases . Antioxid. Redox Signal.13 ( 4 ), 467 – 487 ( 2009 ).
- Matsushima S , KurodaJ , AgoTet al. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy . Circ. Res.112 ( 4 ), 651 – 663 ( 2013 ).
- Schieber M , ChandelNS . ROS function in redox signaling and oxidative stress . Curr. Biol.24 ( 10 ), R453 – R462 ( 2014 ).
- Reczek CR , ChandelNS . ROS-dependent signal transduction . Curr. Opin. Cell Biol.33 , 8 – 13 ( 2015 ).
- Meng T-C , FukadaT , TonksNK . Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo . Mol. Cell9 ( 2 ), 387 – 399 ( 2002 ).
- Lee S-R , YangK-S , KwonJ , LeeC , JeongW , RheeSG . Reversible inactivation of the tumor suppressor PTEN by H2O2 . J. Biol. Chem.277 ( 23 ), 20336 – 20342 ( 2002 ).
- Kamata H , HondaS , MaedaS , ChangL , HirataH , KarinM . Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases . Cell120 ( 5 ), 649 – 661 ( 2005 ).
- Nadeau PJ , CharetteSJ , ToledanoMB , LandryJ . Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis . Mol. Biol. Cell18 ( 10 ), 3903 – 3913 ( 2007 ).
- Marinho HS , RealC , CyrneL , SoaresH , AntunesF . Hydrogen peroxide sensing, signaling and regulation of transcription factors . Redox Biol.2 , 535 – 562 ( 2014 ).
- Nelson KK , MelendezJA . Mitochondrial redox control of matrix metalloproteinases . Free Radic. Biol. Med.37 ( 6 ), 768 – 784 ( 2004 ).
- Provost C , ChoufaniF , AvedanianL , BkailyG , GobeilF , JacquesD . Nitric oxide and reactive oxygen species in the nucleus revisited . Can. J. Physiol. Pharmacol.88 ( 3 ), 296 – 304 ( 2010 ).
- Saluja R , JyotiA , ChatterjeeMet al. Molecular and biochemical characterization of nitric oxide synthase isoforms and their intracellular distribution in human peripheral blood mononuclear cells . Biochim. Biophys. Acta1813 ( 10 ), 1700 – 1707 ( 2011 ).
- Stuehr DJ , SantoliniJ , WangZ-Q , WeiC-C , AdakS . Update on mechanism and catalytic regulation in the NO synthases . J. Biol. Chem.279 ( 35 ), 36167 – 36170 ( 2004 ).
- Hill BG , DrankaBP , BaileySM , LancasterJR , Darley-UsmarVM . What part of NO don’t you understand? Some answers to the cardinal questions in nitric oxide biology . J. Biol. Chem.285 ( 26 ), 19699 – 19704 ( 2010 ).
- Stamati K , MuderaV , CheemaU . Evolution of oxygen utilization in multicellular organisms and implications for cell signalling in tissue engineering . J. Tissue Eng.2 ( 1 ), 2041731411432365 ( 2011 ).
- Crapo JD , OuryT , RabouilleC , SlotJW , ChangLY . Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells . Proc. Natl Acad. Sci. USA89 ( 21 ), 10405 – 10409 ( 1992 ).
- Malhotra JD , KaufmanRJ . Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword?Antioxid. Redox Signal.9 ( 12 ), 2277 – 2293 ( 2007 ).
- Lodhi IJ , SemenkovichCF . Peroxisomes: a nexus for lipid metabolism and cellular signaling . Cell Metab.19 ( 3 ), 380 – 392 ( 2014 ).
- Nordgren M , FransenM . Peroxisomal metabolism and oxidative stress . Biochimie98 , 56 – 62 ( 2014 ).
- Bienert GP , ChaumontF . Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide . Biochim. Biophys. Acta1840 ( 5 ), 1596 – 1604 ( 2014 ).
- Lu J , HolmgrenA . The thioredoxin antioxidant system . Free Radic. Biol. Med.66 , 75 – 87 ( 2014 ).
- Ye Z-W , ZhangJ , TownsendDM , TewKD . Oxidative stress, redox regulation and diseases of cellular differentiation . Biochim. Biophys. Acta1850 ( 8 ), 1607 – 1621 ( 2015 ).
- Wu Y , AntonyS , MeitzlerJL , DoroshowJH . Molecular mechanisms underlying chronic inflammation-associated cancers . Cancer Lett.345 ( 2 ), 164 – 173 ( 2014 ).
- Hou Z , FalconeDJ , SubbaramaiahK , DannenbergAJ . Macrophages induce COX-2 expression in breast cancer cells: role of IL-1β autoamplification . Carcinogenesis32 ( 5 ), 695 – 702 ( 2011 ).
- Berry C , HamiltonCA , BrosnanMJet al. Investigation into the sources of superoxide in human blood vessels: angiotensin II increases superoxide production in human internal mammary arteries . Circulation101 ( 18 ), 2206 – 2212 ( 2000 ).
- Wever RMF , van DamT , van RijnHJM , de GrootF , RabelinkTJ . Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase . Biochem. Biophys. Res. Commun.237 ( 2 ), 340 – 344 ( 1997 ).
- Vásquez-Vivar J , KalyanaramanB , MartásekPet al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors . Proc. Natl Acad. Sci. USA95 ( 16 ), 9220 – 9225 ( 1998 ).
- Lin MI , FultonD , BabbittRet al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production . J. Biol. Chem.278 ( 45 ), 44719 – 44726 ( 2003 ).
- Ou J , OuZ , AckermanAW , OldhamKT , PritchardKAJr . Inhibition of heat shock protein 90 (hsp90) in proliferating endothelial cells uncouples endothelial nitric oxide synthase activity . Free Radic. Biol. Med.34 ( 2 ), 269 – 276 ( 2003 ).
- Yim MB , KangJH , YimHS , KwakHS , ChockPB , StadtmanER . A gain-of-function of an amyotrophic lateral sclerosis-associated Cu, Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide . Proc. Natl Acad. Sci. USA93 ( 12 ), 5709 – 5714 ( 1996 ).
- Crow JP , SampsonJB , ZhuangY , ThompsonJA , BeckmanJS . Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite . J. Neurochem.69 ( 5 ), 1936 – 1944 ( 1997 ).
- Góth L , VitaiM , RassP , SükeiE , PáyA . Detection of a novel familial catalase mutation (Hungarian type D) and the possible risk of inherited catalase deficiency for diabetes mellitus . Electrophoresis26 ( 9 ), 1646 – 1649 ( 2005 ).
- Bostwick DG , AlexanderEE , SinghRet al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer . Cancer89 ( 1 ), 123 – 134 ( 2000 ).
- Min JY , LimS-O , JungG . Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter . FEBS Lett.584 ( 11 ), 2427 – 2432 ( 2010 ).
- Farina M , AvilaDS , da RochaJBT , AschnerM . Metals, oxidative stress and neurodegeneration: a focus on iron, manganese and mercury . Neurochem. Int.62 ( 5 ), 575 – 594 ( 2013 ).
- Doughan AK , HarrisonDG , DikalovSI . Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction . Circ. Res.102 ( 4 ), 488 – 496 ( 2008 ).
- Rathore R , ZhengY-M , NiuC-Fet al. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells . Free Radic. Biol. Med.45 ( 9 ), 1223 – 1231 ( 2008 ).
- Hwang NR , YimS-H , KimYMet al. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions . Biochem. J.423 ( 2 ), 253 – 264 ( 2009 ).
- Martínez-Revelles S , AvendañoMS , García-RedondoABet al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension . Antioxid. Redox Signal.18 ( 1 ), 51 – 65 ( 2012 ).
- Dixon SJ , LembergKM , LamprechtMRet al. Ferroptosis: an iron-dependent form of nonapoptotic cell death . Cell149 ( 5 ), 1060 – 1072 ( 2012 ).
- Circu ML , AwTY . Reactive oxygen species, cellular redox systems and apoptosis . Free Radic. Biol. Med.48 ( 6 ), 749 – 762 ( 2010 ).
- Gehrmann W , ElsnerM , LenzenS . Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β-cells . Diabetes Obes. Metab.12 ( Suppl. 2 ), 149 – 158 ( 2010 ).
- Angelova PR , HorrocksMH , KlenermanD , GandhiS , AbramovAY , ShchepinovMS . Lipid peroxidation is essential for α-synuclein-induced cell death . J. Neurochem.133 ( 4 ), 582 – 589 ( 2015 ).
- Meira LB , BugniJM , GreenSLet al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice . J. Clin. Invest.118 ( 7 ), 2516 – 2525 ( 2008 ).
- Remmen HV , IkenoY , HamiltonMet al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging . Physiol. Genomics16 ( 1 ), 29 – 37 ( 2003 ).
- Mahalingaiah PKS , SinghKP . Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells . PLoS ONE9 ( 1 ), e87371 ( 2014 ).
- Al-Shobaili HA , Al RobaeeAA , AlzolibaniAA , RasheedZ . Antibodies against 4-hydroxy-2-nonenal modified epitopes recognized chromatin and its oxidized forms: role of chromatin, oxidized forms of chromatin and 4-hydroxy-2-nonenal modified epitopes in the etiopathogenesis of SLE . Dis. Markers33 ( 1 ), 19 – 34 ( 2012 ).
- Lunec J , HerbertK , BlountS , GriffithsHR , EmeryP . 8-Hydroxydeoxyguanosine: a marker of oxidative DNA damage in systemic lupus erythematosus . FEBS Lett.348 ( 2 ), 131 – 138 ( 1994 ).
- Görlach A , BrandesRP , NguyenK , AmidiM , DehghaniF , BusseR . A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall . Circ. Res.87 ( 1 ), 26 – 32 ( 2000 ).
- Julienne H , ZoufirA , AuditB , ArneodoA . Human genome replication proceeds through four chromatin states . PLoS Comput. Biol.9 ( 10 ), e1003233 ( 2013 ).
- Yan S-J , LimSJ , ShiS , DuttaP , LiWX . Unphosphorylated STAT and heterochromatin protect genome stability . FASEB J.25 ( 1 ), 232 – 241 ( 2011 ).
- Wang D , ZhouJ , LiuXet al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability . Proc. Natl Acad. Sci. USA110 ( 14 ), 5516 – 5521 ( 2013 ).
- Larson K , YanS-J , TsurumiAet al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis . PLoS Genet.8 ( 1 ), e1002473 ( 2012 ).
- Zhu Q , PaoGM , HuynhAMet al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing . Nature477 ( 7363 ), 179 – 184 ( 2011 ).
- Bernard P , MaureJF , PartridgeJF , GenierS , JaverzatJP , AllshireRC . Requirement of heterochromatin for cohesion at centromeres . Science294 ( 5551 ), 2539 – 2542 ( 2001 ).
- Peters AHFM , O’CarrollD , ScherthanHet al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability . Cell107 ( 3 ), 323 – 337 ( 2001 ).
- Halicka HD , ZhaoH , PodhoreckaM , TraganosF , DarzynkiewiczZ . Cytometric detection of chromatin relaxation, an early reporter of DNA damage response . Cell Cycle Georget. Tex.8 ( 14 ), 2233 – 2237 ( 2009 ).
- Frost B , HembergM , LewisJ , FeanyMB . Tau promotes neurodegeneration through global chromatin relaxation . Nat. Neurosci.17 ( 3 ), 357 – 366 ( 2014 ).
- Khurana V , LuY , SteinhilbML , OldhamS , ShulmanJM , FeanyMB . TOR-mediated cell-cycle activation causes neurodegeneration in a drosophila tauopathy model . Curr. Biol.16 ( 3 ), 230 – 241 ( 2006 ).
- Bosch-Presegué L , Raurell-VilaH , Marazuela-DuqueAet al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection . Mol. Cell42 ( 2 ), 210 – 223 ( 2011 ).
- Dixit K , KhanMA , SharmaYD , Moinuddin , AlamK . Physicochemical studies on peroxynitrite-modified H3 histone . Int. J. Biol. Macromol.46 ( 1 ), 20 – 26 ( 2010 ).
- Khan MA , DixitK , Moinuddin , ArifZ , AlamK . Studies on peroxynitrite-modified H1 histone: implications in systemic lupus erythematosus . Biochimie97 , 104 – 113 ( 2014 ).
- Ayala A , MunozMF , ArguellesS . Lipid Peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal, lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal . Oxidative Med. Cell. Longev. Oxidative Med. Cell. Longev.2014 , e360438 ( 2014 ).
- Beisswenger PJ , HowellSK , SmithK , SzwergoldBS . Glyceraldehyde-3-phosphate dehydrogenase activity as an independent modifier of methylglyoxal levels in diabetes . Biochim. Biophys. Acta1637 ( 1 ), 98 – 106 ( 2003 ).
- Niwa T . 3-Deoxyglucosone: metabolism, analysis, biological activity, and clinical implication . J. Chromatogr. B. Biomed. Sci. App.731 ( 1 ), 23 – 36 ( 1999 ).
- Singh R , BardenA , MoriT , BeilinL . Advanced glycation end-products: a review . Diabetologia44 ( 2 ), 129 – 146 ( 2001 ).
- Nagai R , IkedaK , HigashiTet al. Hydroxyl radical mediates N -(carboxymethyl)lysine formation from amadori product . Biochem. Biophys. Res. Commun.234 ( 1 ), 167 – 172 ( 1997 ).
- Fu MX , Wells-KnechtKJ , BlackledgeJA , LyonsTJ , ThorpeSR , BaynesJW . Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction . Diabetes43 ( 5 ), 676 – 683 ( 1994 ).
- Baumann M . Role of advanced glycation end products in hypertension and cardiovascular risk: human studies . J. Am. Soc. Hypertens. JASH6 ( 6 ), 427 – 435 ( 2012 ).
- Srikanth V , MaczurekA , PhanTet al. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease . Neurobiol. Aging32 ( 5 ), 763 – 777 ( 2011 ).
- Alzolibani AA , Al RobaeeAA , Al-ShobailiHA , RasheedZ . 4-Hydroxy-2-nonenal modified histone-H2A: a possible antigenic stimulus for systemic lupus erythematosus autoantibodies . Cell. Immunol.284 ( 1 ), 154 – 162 ( 2013 ).
- Ashraf JM , AhmadS , RabbaniGet al. Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone . IUBMB Life66 ( 10 ), 686 – 693 ( 2014 ).
- Ashraf JM , RabbaniG , AhmadSet al. Glycation of H1 histone by 3-deoxyglucosone: effects on protein structure and generation of different advanced glycation end products . PLoS ONE10 ( 6 ), e0130630 ( 2015 ).
- Ashraf JM , AhmadS , RabbaniGet al. 3-Deoxyglucosone: a potential glycating agent accountable for structural alteration in H3 histone protein through generation of different AGEs . PLoS ONE10 ( 2 ), e0116804 ( 2015 ).
- Mir AR , UddinM , AlamK , AliA . Methylglyoxal mediated conformational changes in histone H2A-generation of carboxyethylated advanced glycation end products . Int. J. Biol. Macromol.69 , 260 – 266 ( 2014 ).
- Rahmanpour R , BathaieSZ . Histone H1 structural changes and its interaction with DNA in the presence of high glucose concentration in vivo and in vitro . J. Biomol. Struct. Dyn.28 ( 4 ), 575 – 586 ( 2011 ).
- Galligan JJ , RoseKL , BeaversWNet al. Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics . J. Am. Chem. Soc.136 ( 34 ), 11864 – 11866 ( 2014 ).
- Chen D , FangL , LiH , TangM , JinC . Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation . J. Biol. Chem.288 ( 30 ), 21678 – 21687 ( 2013 ).
- Cervantes-Laurean D , RobertsMJ , JacobsonEL , JacobsonMK . Nuclear proteasome activation and degradation of carboxymethylated histones in human keratinocytes following glyoxal treatment . Free Radic. Biol. Med.38 ( 6 ), 786 – 795 ( 2005 ).
- Manevich Y , FeinsteinSI , FisherAB . Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with πGST . Proc. Natl Acad. Sci. USA101 ( 11 ), 3780 – 3785 ( 2004 ).
- Grek CL , ZhangJ , ManevichY , TownsendDM , TewKD . Causes and consequences of cysteine s-glutathionylation . J. Biol. Chem.288 ( 37 ), 26497 – 26504 ( 2013 ).
- de Luca A , MoroniN , SerafinoAet al. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance . Biochem. J.440 ( 2 ), 175 – 183 ( 2011 ).
- García-Giménez JL , OlasoG , HakeSBet al. Histone H3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure . Antioxid. Redox Signal.19 ( 12 ), 1305 – 1320 ( 2013 ).
- Balasubramanian S , HurleyLH , NeidleS . Targeting G-quadruplexes in gene promoters: a novel anticancer strategy?Nat. Rev. Drug Discov.10 ( 4 ), 261 – 275 ( 2011 ).
- Clark DW , PhangT , EdwardsMG , GeraciMW , GillespieMN . Promoter G-quadruplex sequences are targets for base oxidation and strand cleavage during hypoxia-induced transcription . Free Radic. Biol. Med.53 ( 1 ), 51 – 59 ( 2012 ).
- Ba X , BacsiA , LuoJet al. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors . J. Immunol.192 ( 5 ), 2384 – 2394 ( 2014 ).
- Valinluck V , TsaiH-H , RogstadDK , BurdzyA , BirdA , SowersLC . Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) . Nucleic Acids Res.32 ( 14 ), 4100 – 4108 ( 2004 ).
- Deaton AM , BirdA . CpG islands and the regulation of transcription . Genes Dev.25 ( 10 ), 1010 – 1022 ( 2011 ).
- Dickson KM , GustafsonCB , YoungJI , ZüchnerS , WangG . Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate . Biochem. Biophys. Res. Commun.439 ( 4 ), 522 – 527 ( 2013 ).
- Ponnaluri VKC , MaciejewskiJP , MukherjiM . A mechanistic overview of TET-mediated 5-methylcytosine oxidation . Biochem. Biophys. Res. Commun.436 ( 2 ), 115 – 120 ( 2013 ).
- Mastroeni D , GroverA , DelvauxE , WhitesideC , ColemanPD , RogersJ . Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation . Neurobiol. Aging31 ( 12 ), 2025 – 2037 ( 2010 ).
- Volkmar M , DedeurwaerderS , CunhaDAet al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients . EMBO J.31 ( 6 ), 1405 – 1426 ( 2012 ).
- Cheishvili D , StefanskaB , YiCet al. A common promoter hypomethylation signature in invasive breast, liver and prostate cancer cell lines reveals novel targets involved in cancer invasiveness . Oncotarget6 ( 32 ), 33253 – 33268 ( 2015 ).
- Leodolter A , AlonsoS , GonzálezBet al. Somatic DNA hypomethylation in H. pylori-associated high-risk gastritis and gastric cancer: enhanced somatic hypomethylation associates with advanced stage cancer . Clin. Transl. Gastroenterol.6 , e85 ( 2015 ).
- Gravina GL , RanieriG , MuziPet al. Increased levels of DNA methyltransferases are associated with the tumorigenic capacity of prostate cancer cells . Oncol. Rep.29 ( 3 ), 1189 – 1195 ( 2013 ).
- Avila MA , CorralesFJ , RuizFet al. Specific interaction of methionine adenosyltransferase with free radicals . BioFactors8 ( 1 ), 27 – 32 ( 1998 ).
- Pajares MA , DuránC , CorralesF , PliegoMM , MatoJM . Modulation of rat liver S-adenosylmethionine synthetase activity by glutathione . J. Biol. Chem.267 ( 25 ), 17598 – 17605 ( 1992 ).
- Jarrett JT , HooverDM , LudwigML , MatthewsRG . The mechanism of adenosylmethionine-dependent activation of methionine synthase: a rapid kinetic analysis of Intermediates in reductive methylation of Cob(II)alamin enzyme . Biochemistry (Mosc.).37 ( 36 ), 12649 – 12658 ( 1998 ).
- Lu SC . Regulation of glutathione synthesis . Mol. Aspects Med.30 ( 1 ), 42 – 59 ( 2009 ).
- Cyr AR , DomannFE . The redox basis of epigenetic modifications: from mechanisms to functional consequences . Antioxid. Redox Signal.15 ( 2 ), 551 – 589 ( 2011 ).
- Mosharov E , CranfordMR , BanerjeeR . The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes . Biochemistry (Mosc.).39 ( 42 ), 13005 – 13011 ( 2000 ).
- Kloypan C , Srisa-ArtM , MutiranguraA , BoonlaC . LINE-1 hypomethylation induced by reactive oxygen species is mediated via depletion of S-adenosylmethionine . Cell Biochem. Funct.33 ( 6 ), 375 – 384 ( 2015 ).
- Lertratanangkoon K , WuCJ , SavarajN , ThomasML . Alterations of DNA methylation by glutathione depletion . Cancer Lett.120 ( 2 ), 149 – 156 ( 1997 ).
- Valinluck V , SowersLC . Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1 . Cancer Res.67 ( 3 ), 946 – 950 ( 2007 ).
- Niu Y , DesMaraisTL , TongZ , YaoY , CostaM . Oxidative stress alters global histone modification and DNA methylation . Free Radic. Biol. Med.82 , 22 – 28 ( 2015 ).
- Delatte B , JeschkeJ , DefranceMet al. Genome-wide hydroxymethylcytosine pattern changes in response to oxidative stress . Sci. Rep.5 , 12714 ( 2015 ).
- Xin Y-J , YuanB , YuBet al. Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress . Sci. Rep.5 , 7645 ( 2015 ).
- Xiao M , YangH , XuWet al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors . Genes Dev.26 ( 12 ), 1326 – 1338 ( 2012 ).
- Frizzell N , ThomasSA , CarsonJA , BaynesJW . Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity . Biochem. J.445 ( 2 ), 247 – 254 ( 2012 ).
- Tretter L , Adam-ViziV . Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress . J. Neurosci.20 ( 24 ), 8972 – 8979 ( 2000 ).
- O’Hagan HM , WangW , SenSet al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and Polycomb members to promoter CpG islands . Cancer Cell20 ( 5 ), 606 – 619 ( 2011 ).
- Ding N , BonhamEM , HannonBE , AmickTR , BaylinSB , O’HaganHM . Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage . J. Mol. Cell Biol. doi:10.1093/jmcb/mjv050 ( 2015 ) ( Epub ahead of print ).
- Kooistra SM , HelinK . Molecular mechanisms and potential functions of histone demethylases . Nat. Rev. Mol. Cell Biol.13 ( 5 ), 297 – 311 ( 2012 ).
- Sullivan LB , Martinez-GarciaE , NguyenHet al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling . Mol. Cell51 ( 2 ), 236 – 248 ( 2013 ).
- Hickok JR , VasudevanD , AntholineWE , ThomasDD . Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases . J. Biol. Chem.288 ( 22 ), 16004 – 16015 ( 2013 ).
- Hitchler MJ , DomannFE . Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome . Free Radic. Biol. Med.53 ( 11 ), 2178 – 2187 ( 2012 ).
- Kera Y , KatohY , OhtaM , MatsumotoM , Takano-YamamotoT , IgarashiK . Methionine adenosyltransferase II-dependent histone H3K9 methylation at the COX-2 gene locus . J. Biol. Chem.288 ( 19 ), 13592 – 13601 ( 2013 ).
- de Ruijter AJM , Gennip vanAH , CaronHN , KempS , Kuilenburg vanABP . Histone deacetylases (HDACs): characterization of the classical HDAC family . Biochem. J.370 ( 3 ), 737 – 749 ( 2003 ).
- Marks PA , RifkindRA , RichonVM , BreslowR , MillerT , KellyWK . Histone deacetylases and cancer: causes and therapies . Nat. Rev. Cancer1 ( 3 ), 194 – 202 ( 2001 ).
- Delcuve GP , KhanDH , DavieJR . Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors . Clin. Epigenetics4 ( 1 ), 5 ( 2012 ).
- Lombardi PM , ColeKE , DowlingDP , ChristiansonDW . Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes . Curr. Opin. Struct. Biol.21 ( 6 ), 735 – 743 ( 2011 ).
- Khan O , La ThangueNB . HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications . Immunol. Cell Biol.90 ( 1 ), 85 – 94 ( 2012 ).
- Tanny JC , MoazedD . Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown product . Proc. Natl Acad. Sci. USA98 ( 2 ), 415 – 420 ( 2001 ).
- Tanner KG , LandryJ , SternglanzR , DenuJM . Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose . Proc. Natl Acad. Sci. USA97 ( 26 ), 14178 – 14182 ( 2000 ).
- Moodie FM , MarwickJA , AndersonCSet al. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells . FASEB J.18 ( 15 ), 1897 – 1899 ( 2004 ).
- Doyle K , FitzpatrickFA . Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function . J. Biol. Chem.285 ( 23 ), 17417 – 17424 ( 2010 ).
- Kornberg MD , SenN , HaraMRet al. GAPDH mediates nitrosylation of nuclear proteins . Nat. Cell Biol.12 ( 11 ), 1094 – 1100 ( 2010 ).
- Nott A , WatsonPM , RobinsonJD , CrepaldiL , RiccioA . S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons . Nature455 ( 7211 ), 411 – 415 ( 2008 ).
- Colussi C , MozzettaC , GurtnerAet al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment . Proc. Natl Acad. Sci. USA105 ( 49 ), 19183 – 19187 ( 2008 ).
- Bhaskara S , KnutsonSK , JiangGet al. Hdac3 is essential for the maintenance of chromatin structure and genome stability . Cancer Cell18 ( 5 ), 436 – 447 ( 2010 ).
- Dovey OM , FosterCT , ConteNet al. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice . Blood121 ( 8 ), 1335 – 1344 ( 2013 ).
- Heideman MR , WiltingRH , YanoverEet al. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function . Blood121 ( 11 ), 2038 – 2050 ( 2013 ).
- Santoro F , BotrugnoOA , ZuffoRDet al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance . Blood121 ( 17 ), 3459 – 3468 ( 2013 ).
- Shimazu T , HirscheyMD , NewmanJet al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor . Science339 ( 6116 ), 211 – 214 ( 2013 ).
- Caito S , RajendrasozhanS , CookSet al. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress . FASEB J.24 ( 9 ), 3145 – 3159 ( 2010 ).
- Shao D , FryJL , HanJet al. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress . J. Biol. Chem.289 ( 11 ), 7293 – 7306 ( 2014 ).
- Yao H , ChungS , HwangJ-Wet al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice . J. Clin. Invest.122 ( 6 ), 2032 – 2045 ( 2012 ).
- Ota H , EtoM , KanoMRet al. Cilostazol Inhibits oxidative stress–induced premature senescence via upregulation of Sirt1 in human endothelial cells . Arterioscler. Thromb. Vasc. Biol.28 ( 9 ), 1634 – 1639 ( 2008 ).
- Yang S-R , WrightJ , BauterM , SeweryniakK , KodeA , RahmanI . Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging . Am. J. Physiol. Lung Cell Mol. Physiol.292 ( 2 ), L567 – L576 ( 2007 ).
- Fritz KS , GalliganJJ , SmathersRLet al. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification . Chem. Res. Toxicol.24 ( 5 ), 651 – 662 ( 2011 ).
- Iwahara T , BonasioR , NarendraV , ReinbergD . SIRT3 functions in the nucleus in the control of stress-related gene expression . Mol. Cell Biol.32 ( 24 ), 5022 – 5034 ( 2012 ).
- Hu S , LiuH , HaYet al. Posttranslational modification of Sirt6 activity by peroxynitrite . Free Radic. Biol. Med.79 , 176 – 185 ( 2015 ).
- Sebastián C , ZwaansBMM , SilbermanDMet al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism . Cell151 ( 6 ), 1185 – 1199 ( 2012 ).
- Liu Y , Hernández-OchoaEO , RandallWR , SchneiderMF . NOX2-dependent ROS is required for HDAC5 nuclear efflux and contributes to HDAC4 nuclear efflux during intense repetitive activity of fast skeletal muscle fibers . Am. J. Physiol. Cell Physiol.303 ( 3 ), C334 – C347 ( 2012 ).
- Lu J , McKinseyTA , NicolRL , OlsonEN . Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases . Proc. Natl Acad. Sci. USA97 ( 8 ), 4070 – 4075 ( 2000 ).
- Hwang J , YaoH , CaitoS , SundarIK , RahmanI . Redox regulation of SIRT1 in inflammation and cellular senescence . Free Radic. Biol. Med.61 , 95 – 110 ( 2013 ).
- Agudelo M , GandhiN , SaiyedZet al. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA) . Alcohol. Clin. Exp. Res.35 ( 8 ), 1550 – 1556 ( 2011 ).
- Castonguay Z , AugerC , ThomasSC , ChahmaM’hamed , AppannaVD . Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes . Biochem. Biophys. Res. Commun.454 ( 1 ), 172 – 177 ( 2014 ).
- Banerjee T , ChakravartiD . A peek into the complex realm of histone phosphorylation . Mol. Cell Biol.31 ( 24 ), 4858 – 4873 ( 2011 ).
- Ye B , HouN , XiaoL , XuY , XuH , LiF . Dynamic monitoring of oxidative DNA double-strand break and repair in cardiomyocytes . Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol.25 ( 2 ), 93 – 100 ( 2016 ).
- Mah L-J , El-OstaA , KaragiannisTC . gammaH2AX: a sensitive molecular marker of DNA damage and repair . Leukemia24 ( 4 ), 679 – 686 ( 2010 ).
- Katsube T , MoriM , TsujiHet al. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks . J. Biochem.156 ( 2 ), 85 – 95 ( 2014 ).
- Marwick JA , TudorC , KhorasaniN , MichaeloudesC , BhavsarPK , ChungKF . Oxidants induce a corticosteroid-insensitive phosphorylation of histone 3 at serine 10 in monocytes . PLoS ONE10 ( 4 ), e0124961 ( 2015 ).
- Jeong M-W , KangT-H , KimW , ChoiYH , KimK-T . Mitogen-activated protein kinase phosphatase 2 regulates histone H3 phosphorylation via interaction with vaccinia-related kinase 1 . Mol. Biol. Cell24 ( 3 ), 373 – 384 ( 2013 ).
- Rusnak F , ReiterT . Sensing electrons: protein phosphatase redox regulation . Trends Biochem. Sci.25 ( 11 ), 527 – 529 ( 2000 ).
- Iyengar S , FarnhamPJ . KAP1 protein: an enigmatic master regulator of the genome . J. Biol. Chem.286 ( 30 ), 26267 – 26276 ( 2011 ).
- Wei N , ShiY , TruongLNet al. Oxidative stress diverts tRNA synthetase to nucleus for protection against DNA damage . Mol. Cell56 ( 2 ), 323 – 332 ( 2014 ).
- Higo S , AsanoY , KatoHet al. Isoform-specific intermolecular disulfide bond formation of heterochromatin protein 1 (HP1) . J. Biol. Chem.285 ( 41 ), 31337 – 31347 ( 2010 ).
- Whitelaw NC , ChongS , MorganDKet al. Reduced levels of two modifiers of epigenetic gene silencing, Dnmt3a and Trim28, cause increased phenotypic noise . Genome Biol.11 ( 11 ), R111 ( 2010 ).
- Štros M . HMGB proteins: interactions with DNA and chromatin . Biochim. Biophys. Acta BBA1799 ( 1 ), 101 – 113 ( 2010 ).
- Polanská E , Pospíšilovአ, ŠtrosM . Binding of histone H1 to DNA is differentially modulated by redox state of HMGB1 . PLoS ONE9 ( 2 ), e89070 ( 2014 ).
- Fiume R , Stijf-BultsmaY , ShahZHet al. PIP4K and the role of nuclear phosphoinositides in tumour suppression . Biochim. Biophys. Acta1851 ( 6 ), 898 – 910 ( 2015 ).
- Keune jan W , BultsmaY , SommerL , JonesD , DivechaN . Phosphoinositide signalling in the nucleus . Adv. Enzyme Regul.51 ( 1 ), 91 – 99 ( 2011 ).
- Jones DR , BultsmaY , KeuneW-Jet al. Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta . Mol. Cell23 ( 5 ), 685 – 695 ( 2006 ).
- Attia SM , AhmadSF , HarisaGI , MansourAM , El SayedESM , BakheetSA . Wogonin attenuates etoposide-induced oxidative DNA damage and apoptosis via suppression of oxidative DNA stress and modulation of OGG1 expression . Food Chem. Toxicol.59 , 724 – 730 ( 2013 ).
- Gozani O , KarumanP , JonesDRet al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor . Cell114 ( 1 ), 99 – 111 ( 2003 ).
- Pedeux R , SenguptaS , ShenJCet al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation . Mol. Cell Biol.25 ( 15 ), 6639 – 6648 ( 2005 ).
- Doyon Y , CayrouC , UllahMet al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation . Mol. Cell21 ( 1 ), 51 – 64 ( 2006 ).
- Bua DJ , MartinGM , BindaO , GozaniO . Nuclear phosphatidylinositol-5-phosphate regulates ING2 stability at discrete chromatin targets in response to DNA damage . Sci. Rep.3 , 2137 ( 2013 ).
- Stijf-Bultsma Y , SommerL , TauberMet al. The basal transcription complex component TAF3 transduces changes in nuclear phosphoinositides into transcriptional output . Mol. Cell58 ( 3 ), 453 – 467 ( 2015 ).
- Bronner C . Control of DNMT1 abundance in epigenetic inheritance by acetylation, ubiquitylation, and the histone code . Sci. Signal.4 ( 157 ), pe3 ( 2011 ).
- Gelato KA , TauberM , OngMSet al. Accessibility of different histone H3-binding domains of UHRF1 is allosterically regulated by phosphatidylinositol 5-phosphate . Mol. Cell54 ( 6 ), 905 – 919 ( 2014 ).
- Montero D , WaltherG , StehouwerCDA , HoubenAJHM , BeckmanJA , VinetA . Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: a systematic review and meta-analysis of randomized controlled trials . Obes. Rev.15 ( 2 ), 107 – 116 ( 2014 ).
- Rafieian-Kopaie M , NasriH . On the Occasion of World Cancer Day 2015; the possibility of cancer prevention or treatment with antioxidants: the ongoing cancer prevention researches . Int. J. Prev. Med.6 , 108 ( 2015 ).
- Galasko DR , PeskindE , ClarkCMet al. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures . Arch. Neurol.69 ( 7 ), 836 – 841 ( 2012 ).
- Harrison FE , AllardJ , BixlerRet al. Antioxidants and cognitive training interact to affect oxidative stress and memory in APP/PSEN1 mice . Nutr. Neurosci.12 ( 5 ), 203 – 218 ( 2009 ).
- Jin H , KanthasamyA , GhoshA , AnantharamV , KalyanaramanB , KanthasamyAG . Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes . Biochim. Biophys. Acta1842 ( 8 ), 1282 – 1294 ( 2014 ).