
Abstract
Aim: The role of TMEM176A methylation in lung cancer and its therapeutic application remains unclear. Materials and methods: Nine lung cancer cell lines and 123 cases of cancer tissue samples were employed. Results:TMEM176A was methylated in 53.66% of primary lung cancer. Restoration of TMEM176A expression induced cell apoptosis and G2/M phase arrest, and inhibited colony formation, cell proliferation, migration and invasion. TMEM176A suppressed H1299 cell xenograft growth in mice. Methylation of TMEM176A activated ERK signaling and sensitized H1299 and H23 cells to AZD0156, an ATM inhibitor. Conclusion: The expression of TMEM176A is regulated by promoter region methylation. Methylation of TMEM176A is a potential lung cancer diagnostic marker and a novel synthetic lethal therapeutic marker for AZD0156.
Lay abstract
The TMEM176A gene is often methylated in human lung cancer by addition of a methyl group to the gene promotor region. This regulates the expression of TMEM176A. We found that TMEM176A suppressed lung cancer growth both invitro and invivo by inhibiting ERK signaling. Methylation of TMEM176A sensitized H1299 and H23 cells to AZD0156, an ATM kinase inhibitor used to induce tumor cell death. Re-expression of TMEM176A reduced the sensitivity of these cells to AZD0156. Methylation of TMEM176A is a novel synthetic lethality therapeutic marker of AZD0156 in human lung cancer.
Lung cancer is one of the most common cancers and the primary cause of cancer-related death worldwide [Citation1]. Tobacco consumption is considered the most important risk factor for the development of lung cancer [Citation2], although it is currently well established that an important percentage of people who have never smoked are diagnosed with lung cancer [Citation3]. Many other risk factors have been described, including radon, asbestos exposure, domestic fuel smoke and human papillomavirus infection [Citation2,Citation3]. The mechanism of lung cancer development remains unclear. Aberrant genetic and epigenetic changes are involved in tumorigenesis and progression [Citation4,Citation5].
The identification of actionable oncogenic mutations has greatly improved the treatment of different cancers [Citation6–8]. In nonsmall-cell lung cancers (NSCLCs), the discovery of activating mutations in the EGFR gene have ushered in a new era of genomics-guided precision targeted therapy in lung cancer [Citation9]. Advances in the knowledge of pathway, newly developed drugs to block the activities of signaling pathways in recent years have allowed the physicians to tailor the treatment options [Citation8,Citation10]. Nevertheless, the unprecedented benefit from current standard therapies is still only observed in a minority of patients.
Aberrant epigenetic changes have been reported in various cancers, including lung cancer [Citation4,Citation5,Citation11,Citation12]. It is now well established that epigenetic alterations are driver events in the pathogenesis of human cancers. In contrast to ‘mut-driver genes’, a greater number of ‘epi-driver genes’ are abnormally expressed by aberrant epigenetic changes in human cancers [Citation13]. Cancer-related signaling pathways may be disrupted by a key component aberrant methylation. Currently, epigenetic therapies are successfully used in the clinic to treat hematological malignancies. However, little success has been achieved in treating solid tumors. Novel epigenome-based therapeutic strategies are developing, including ‘synthetic lethality’ [Citation10,Citation14–16]. The concept of synthetic lethality originates from studies in Drosophila model systems in which a combination of mutations in two or more separate genes leads to cell death [Citation17]. In 2005, two groups described the synthetic lethality interaction between PARP inhibition and BRCA1 and BRCA2 mutation, and developed a novel treatment strategy for BRCA-mutant tumors [Citation18,Citation19]. The synthetic lethality approach has been applied in various dysfunctional BRCA1 and BRCA2 cancers [Citation20]. ‘BRCAness’ means a homologous recombination repair (HR) defect phenotype beyond the narrow scope of BRCA1 or BRCA2 mutation. It is similar to gene mutations that epigenetic silencing of tumor suppressor genes may cause inactivation or ‘loss-of-function’ in these genes. Thus, epigenetics joins Knudson’s two-hit theory. DNA damage repair (DDR) and cell cycle regulator genes were found to be frequently methylated in human cancers. It is reasonable to apply the aberrant epigenetic changes to synthetic lethal therapy in human cancers. However, epigenetic-based synthetic lethality has not been extensively studied. Thus, it is important to identify the predictive response biomarker and/or DDR resistance inhibitors.
TMEM176A was first identified by screening tumor related antigens in hepatocellular carcinoma (HCC) [Citation21,Citation22]. Most of studies were mainly focused on its function in development and the immune system [Citation23–25]. Our previous study found that the expression of TMEM176A was reduced in human colorectal cancer tissue compared with normal colorectal mucosa according to RNA-seq [Citation26]. TMEM176A was found to be frequently methylated in human colorectal, hepatic and esophageal cancers and was thought to be a tumor suppressor [Citation26–28]. The role of TMEM176A in lung cancer remains to be elucidated.
In this study, we found that TMEM176A is frequently methylated in human lung cancer and the expression of TMEM176A is regulated by promoter region methylation. TMEM176A suppressed lung cancer growth both in vitro and in vivo by inhibiting ERK signaling. Further study found that methylation of TMEM176A sensitized H1299 and H23 cells to AZD0156, an ATM inhibitor, and the re-expression of TMEM176A reduced the sensitivity of these cells to AZD0156. Methylation of TMEM176A is a novel synthetic lethality therapeutic marker of ATM inhibitors in human lung cancer.
Materials & methods
Human tissue samples & cell lines
One hundred and twenty-three primary lung cancer samples were collected from the Chinese PLA General Hospital. The median age of the cancer patients was 60 years old (range: 29–79 years). Fifteen cases of normal lung tissue were collected from the Chinese PLA General Hospital. Among 123 cancer samples, only 40 cases were available for paraffin samples with matched cancer and adjacent tissue. Nine lung cancer cell lines (H157 H1563, H727, H358, H446, H460, H23, H1299 and A549) were previously established from primary lung cancer and grown in RPMI-1640 (Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (Hyclone, UT, USA) and 1% penicillin/streptomycin solution (Sigma-Aldrich, MO, USA) [Citation29–31]. All these cell lines were ATCC origin and the STR reports are available (identified by the BioWings company).
5-Aza-2-deoxycytidine & SCH772984 treatment
For methylation regulation analysis, lung cancer cell lines were split to low density (30% confluence) 12 h before treatment. Cells were treated with 5-Aza-2-deoxycytidine (DAC, Sigma-Aldrich), a DNA demethylation reagent, at a concentration of 2 μM in the growth medium, which was exchanged every 24 h for a total of 96 h and cultured at 37°C in a 5% CO2 incubator. At the end of the treatment period, cells were prepared for extraction of total RNA. To verify the role of TMEM176A in ERK signaling, SCH772984, an ERK inhibitor, was added to TMEM176A knocking down H727 cell at 2 μM/l for 24 h (MedChemExpress, NJ, USA) [Citation32].
RNA isolation & semiquantitative reverse transcription PCR
Total RNA was extracted using Trizol Reagent (Life Technologies, CA, USA). Agarose gel electrophoresis and spectrophotometric analysis were used to detect RNA quality and quantity. First strand cDNA was synthesized according to manufacturer’s instructions (Invitrogen). Five micrograms of RNA were used to synthesize first strand cDNA. The reaction mixture was diluted to 100 μl with water, and then 2 μl of diluted cDNA was used for 25 μl PCR reaction. The PCR primer sequences for TMEM176A were as follows: 5′-GGG AAC AGC CGA CA G TGA T-3′ (F) and 5′-GCC AGC GTT AGC AGA GTC CT-3′(R). PCR cycle conditions were as follows: 95°C 5 min, one cycle; (95, 60 and 72°C, all for 30 s) 32 cycles; 72°C 5 min, one cycle. PCR product size is 369 bp. GAPDH was amplified for 25 cycles as an internal control. The GAPDH primer sequences were as follows: 5′-GAC CAC AGT CCA TGC CAT CAC-3′ (F), and 5′-GTC CAC CAC CCT GTT GCT GTA-3′ (R). PCR cycle conditions were as follows: 95°C 5 min, one cycle; (95, 60 and 72°C, all for 30 s) 25 cycles; 72°C 5 min, one cycle. PCR product size is 448 bp. The amplified PCR products were examined by 2% agarose gels.
DNA extraction, bisulfite modification, methylation-specific PCR
Genomic DNA from lung cancer cell lines and lung cancer tissue samples were prepared using the proteinase-K method. Normal lymphocyte DNA was prepared from healthy donor blood lymphocytes by proteinase-K method [Citation33]. Normal lymphocyte DNA (NL) was used as a control for unmethylation and in vitro methylated DNA (IVD) was used as a methylation control. IVD was prepared using SssI methylase (New England Biolabs, MA, USA) following the manufacturer’s instructions. Methylation-specific PCR (MSP) primers were designed according to genomic sequences inside the CpG islands in the TMEM176A gene promoter region.
MSP primers for TMEM176A were designed -362 to -203 bp upstream of the transcription start site (TSS) and synthesized to detect methylated (M) and unmethylated (U) alleles. The detected region has been previously reported to be hypermethylated and associated with low expression [Citation27]. MSP primers for TMEM176A were as follows: 5′-GTT TCG TTT AGG TTG CGC GGT TTT TC-3′ (MF), 5′-CCA AAA CCG ACG TAC AAA TAT ACG CG-3′ (MR); 5′-TGG TTT TGT TTA GGT TGT GTG GTT TTT T-3′ (UF), 5′-CAA CCA AAA CCA ACA TAC AAA TAT ACA CA-3′ (UR).
PCR cycle conditions were as follows: 95°C 5 min, one cycle; (95, 60 and 72°C, all for 30 s) 35 cycles; 72°C 5 min, one cycle.
Bisulfite sequencing (BSSQ) primers encompassed a 231-bp region upstream of the TMEM176A transcription start site (-388 bp to -157 bp) and included the region analyzed by MSP. BSSQ primers were designed as follows:
5′-GAG ACG GTA GAT GTA CGG GT-3′ (F);
5′-AAC RAA CRA CCC TAA AAA AAC CC-3′ (R). PCR cycle conditions were as follows: 95°C 5 min, one cycle; (95°C 30 s, 55°C 30 s and 72°C 30 s) 35 cycles; 72°C 5 min, one cycle.
Immunohistochemistry
Immunohistochemistry (IHC) was performed in primary lung cancer samples and matched adjacent tissue samples. TMEM176A antibody was diluted to 1:50 (cat. no.: HPA008770; Sigma-Aldrich). The expression of MMP-2, MMP-9 and p-ERK1/2 was detected in H1299 cell xenografts. MMP-2, MMP-9 and p-ERK1/2 antibody was diluted to 1:100, 1:100 (Protein Tech Group, IL, USA) and 1:400 (Cell Signaling Technology, MA, USA). The procedure was performed as described previously [Citation34]. The staining intensity and extent of the staining area were scored using the German semiquantitative scoring systems as previously described [Citation34–36]. Staining intensity of the membrane and/or cytoplasm was characterized as follows: no staining = 0, weak staining = 1, moderate staining = 2 and strong staining = 3; the extent of staining was defined as 0% = 0, 1–24% = 1, 25–49% = 2, 50–74% = 3 and 75–100% = 4. The final immunereactive score (0–12) was determined by multiplying the intensity score by the extent of staining score.
Construction of lentiviral TMEM176A expression vectors & selection of stable expression cells
The human full length TMEM176A cDNA (NM-018487.2) was cloned into the pLenti6 vector. Primers were as follows: 5′-CTT AGG ATC CGC CAC CAT GGG AAC AGC CGAC-3′ (F) and 5′-ACT TAG TCG ACC TAG ATT CCA CTC ACT TCC-3′(R). The HEK-293T cell line was maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum. TMEM176A expressing Lentiviral vector was transfected into HEK-293T cells (5.5 × 106 per 100-mm dish) using lipofectamine 3000 Reagent (Invitrogen) at a ratio of 1:3 (DNA mass: Lipo mass). Viral supernatant was collected and filtered after 48 h. H23 and H1299 cells were then infected with viral supernatant. H23 and H1299 cells stably expressing TMEM176A were screened by Blasticidin (Life Technologies) at concentrations of 6.0 μg/ml and 5.0 μg/ml for 2 weeks, respectively. Then TMEM176A expressed H23 and H1299 monoclonal cells were screened by 96-well and thereafter by 6-well plates.
RNA interference assay
Two sets of targeting siRNA for TMEM176A and one set of RNAi scrambled control duplex sequence are as follows:
siRNA#1 duplex for TMEM176A (sense: 5′-GGC UAC UCU UAU UAC AAC ATT-3′; antisense: UGU UGU AAU AAG AGU AGC CTT-3′),
siRNA#2 duplex for TMEM176A (sense: 5′-CUG UAC UGC UGG AGA AUG UTT-3′; antisense: 5′-ACA UUC UCC AGC AGU ACA GTT-3′),
TMEM176A siRNA scrambled duplex (scrambled siTMEM176A, sense: 5′-ACA UUC UCC AGC AGU ACA GTT-3′; antisense: 5′-ACG UGA CAC GUU CGG AGA ATT-3′). SiRNA#2 was found more effective than siRNA#1, and siRNA#2 was applied to further study (GenePharma Co. Shanghai, China).
Cell viability detection
H23 and H1299 cells were seeded into 96-well plates before and after re-expression of TMEM176A at 1 × 103 cells/well. H727 cell were plated into 96-well plates before and after knockdown of TMEM176A at a density of 5 × 103 cells/well. The cell viability was measured by MTT (3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di-phenytetrazoliumromide) assay at 0, 24, 48, 72 and 96 h (KeyGen Biotech, Nanjing, China). Absorbance was measured on a microplate reader (Thermo Multiskan MK3, MA, USA) at a wavelength of 490 nm. Each experiment was repeated three times.
H23 and H1299 cells were seeded into 96-well plates at 4 × 103 cells/well and 3 × 103 cells/well, respectively. The IC50 value was detected by the MTT assay before and after re-expression of TMEM176A. H727 cell were plated into 96-well plates at a density of 8 × 103 cells/well before and after knockdown of TMEM176A. Cells were treated with AZD0156 for 24 h at 0, 0.001, 0.01, 0.1, 1, 10, 100 and 1000 μM/l after beingseeded for 24 h [Citation37]. Each experiment was repeated for three times.
Colony formation assay
TMEM176A unexpressed and re-expressed H23 and H1299 cells were seeded in 6-well plates at a density of 200 cells per well. H727 cells were seeded into 6-well plates at a density of 300 cells per well, before and after TMEM176A knockdown. After 2 weeks, cells were fixed with 75% ethanol for 30 min. Colonies were then stained with 0.5% crystal violet solution and counted. The experiment was performed in triplicate.
Flow cytometry
TMEM176A unexpressed and stably expressed H23 and H1299 cells were starved for 12 h to synchronize, and cells were restimulated with 10% FBS for 24 h. Cells were fixed with 70% ethanol and treated using the Cell Cycle Detection Kit (KeyGen Biotech). Cells were then detected using a FACS Caliber flow cytometer (BD Biosciences). The cell cycle was analyzed also for H727 cells with or without knocking down TMEM176A. Cell phase distribution was analyzed using the Modfit software (Verity Software House, ME, USA).
To increase the sensitivity of apoptosis detection, TMEM176A unexpressed and stably re-expressed H23 and H1299 cells were treated with doxorubicin at 0.8 μg/ml and 0.6 μg/ml for 24 h respectively [Citation38]. Apoptosis was also analyzed in H727 cell with or without knockdown of TMEM176A. The cells were prepared using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, NJ, USA) following the manufacturer’s instructions and then sorted by FACS Calibur (BD Biosciences). Each experiment was repeated three times.
Transwell assay
For migration study, cells were suspended in 200 μl serum-free RPMI 1640 media, and 5 × 104 H23 and 2 × 104 H1299 cells were added to the upper chamber of an 8.0-μm pore size transwell apparatus (COSTAR Transwell Corning Incorporated, MA, USA). Cells were stained with crystal violet and counted in three independent high-power fields (×100) after incubation for 16 h (H23 cells) or 16 h (H1299). Each experiment was repeated three times.
For invasion assay, H23 cells (1 × 105) and H1299 cells (5 × 104) were seeded to the upper chamber of a transwell apparatus coated with Matrigel (BD Biosciences) and incubated for 36 h (H23) and 36 h (H1299). Each experiment was repeated three times.
Western blot
Cells were collected 48 h after transfection and cell lysates were prepared using ice-cold Tris buffer (20 mmol/l Tris; pH 7.5) containing 137 mmol/l NaCl, 2 mmol/l EDTA, 1% Triton X, 10% glycerol, 50 mmol/l NaF, 1 mmol/l DTT, PMSF, and a protein phosphatases inhibitor (Applygen Tech. Beijing, China). For extracellular signal-regulated kinase (ERK) signaling analysis, cells were starved with serum-free medium for 24 h after transfection. These cells were then stimulated with medium containing 10% serum for 60 min before collection. To analyze the sensitivity of AZD0156, cells were exposed to UV 20 mJ/cm2 for 2 h [Citation39] before treatment with 0.5 μM/l AZD0156 or ethanol (control), and cells were collected after 24 h treatment. western blot was performed as described previously [Citation34]. Quantification of blots was performed with Image J.
Primary antibodies were as follows: TMEM176A (Sigma-Aldrich), cleaved CASP3 (Protein Tech Group, IL, USA), Bcl2 (Protein Tech Group), CASP3 (Protein Tech Group), Bax (Protein Tech Group), MMP-2 (Protein Tech Group), MMP-9 (Protein Tech Group), cyclin B1 (Protein Tech Group), CDC2 (Protein Tech Group), ATM (HuaXingBoChuang, China),γ-H2AX (HuaXingBoChuang), p-CHK2 (Zhengneng, China), ERK1/2 (Protein Tech Group), p-ERK1/2 (Cell Signaling Technology, MA, USA), SAR1A (Protein Tech Group) and β-actin (Beyotime Biotech, Nanjing, China).
Lung cancer cell xenograft mouse model
H1299 cell lines stably transfected with plenti6 vector or plenti6-TMEM176A vector (1 × 107 cells diluted in phosphate-buffered saline) were injected subcutaneously into the dorsal left side of 4-week-old female Balb/c nude mice. Each group included six mice. Tumor volume was measured every 4 days. Tumor volume was calculated according to the formula: V = L × W2/2, in which V represents volume (mm3), L represents biggest diameter (mm), and W represents the smallest diameter (mm). Mice were sacrificed on the 24th day after inoculation and tumors were weighed. All procedures were approved by the Animal Ethics Committee of the Chinese PLA General Hospital.
Data Analysis
RNASeq data for TMEM176A gene expression in the dataset of lung cancer and normal tissues were downloaded from the Cancer Genome Atlas (TCGA; http://xena.ucsc.edu/; 16 September 2019). Statistical analysis was performed using SPSS 17.0 software (SPSS, IL, USA) and Graphpad Prism (CA, USA). Chi-square test was used to evaluate the relationship between methylation status and clinicopathological characteristics. Two-tailed independent samples t-test was applied to determine the statistical significance of the differences between the two experimental groups. Two-sided tests were used to determine the statistical difference, and p < 0.05 was considered statistically significance.
Results
TMEM176A is frequently methylated in human NSCL cancer & the expression of TMEM176A is regulated by promoter region hypermethylation
TCGA database was employed to explore whether expression of TMEM176A is regulated by promoter region methylation. TMEM176A mRNA expression and promoter region methylation data were extracted from the database (http://xena.ucsc.edu/). Methylation of TMEM176A was analyzed by Illumina Infinium Human Methylation 450 (HM450). In the 457 cases of lung adenocarcinoma samples and 372 cases of lung squamous carcinoma samples, reduced expression of TMEM176A was associated with promoter region hypermethylation at multiple probe () and most strongly at cg19336959 in the promoter region (). The results indicate that the expression of TMEM176A is downregulated by promoter region methylation.
(A) The association of TMEM176A expression and methylation at each CpG site of promoter region. (B) Scatter plots show reduced TMEM176A expression is associated with methylation status of cg19336959 in 457 cases of lung adenocarcinoma and 372 cases of lung squamous cell carcinoma. (C) Semiquantitative reverse transciptase PCR and western blot show the levels of TMEM176A expression in lung cancer cells (H157, H358, H1299, H23, H1563, A549, H446, H460 and H727) both in RNA and protein levels. (D) Methylation status of TMEM176A in lung cancer cells. (E) BSSQ results in H1299, H23, H727 cells and normal lung. Filled circles: methylation; open circles: unmethylation.
***p < 0.001.
BSSQ: Sodium bisulfite sequence; (-): without DAC; (+): with DAC; AD: Lung adenocarcinoma; GAPDH: Internal control; H2O: Double-distilled water; IVD: Invitral methylated DNA; M: Methylation; MSP: Methylation-specific PCR; NL: Normal lymph cell DNA; SCC: Lung squamous cell carcinoma; TSS: Transcription start site; U: Unmethylation.
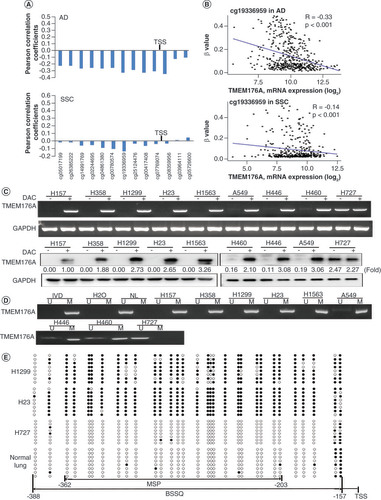
Epigenetic regulation of TMEM176A expression was further examined in experiments in lung cancer cell lines and tissue samples. The expression of TMEM176A was examined in human lung cancer cells by semiquantitative RT-PCR and western blot. TMEM176A was highly expressed in H727 cell, reduced expression was observed in A549, H446 and H460 cells, and no expression was found in H157, H1563, H358, H1299 and H23 cells both in RNA and protein levels. Re-expression of TMEM176A was found in H157, H1563, H358, H1299 and H23 cells and increased expression of TMEM176A was observed in A549, H446 and H460 cells after treatment with DAC, a DNA methyltransferase inhibitor. No expression changes were found in H727 cell before and after DAC treatment both in RNA and protein levels (). The promoter region methylation was examined by MSP. Unmethylation was detected in H727, partial methylation was observed in A549, H446 and H460 cells, and complete methylation was found in H157, H1563, H358, H1299 and H23 cells (). These results demonstrate that loss of/reduced expression of TMEM176A was correlated with promoter region methylation.
To further validate the efficiency of MSP primers and explore the methylation density in lung cancer, sodium bisulfite sequence (BSSQ) was performed in H1299, H23, H727 cells and normal lung tissue samples. Dense methylation was observed in the promoter region of TMEM176A in H1299 and H23 cells, unmethylation was detected in H727 cells and normal lung tissue samples ().
The methylation status of TMEM176A was also detected by MSP in 123 cases of primary human lung cancer and 15 cases of noncancerous lung tissue samples. TMEM176A was methylated in 53.66% (67/123) of human primary lung cancer, and no methylation was found in noncancerous lung tissue samples (). No association was found between TMEM176A methylation and age, gender, alcohol abuse, smoking, tumor size, lymph node metastasis, differentiation and tumor-node-metastasis (TNM) stage (, all p > 0.05).
(A) Representative methylation-specific PCR results. (B) TMEM176A staining in lung cancer and adjacent tissue samples (top: 200×; bottom: 400×). (C) Box plots for TMEM176A expression, horizontal lines represent the median score; the bottom and top of the boxes represent the 25th and 75th percentiles, respectively; vertical bars represent expression levels. (D) Bar diagram: the levels of TMEM176A expression and methylation status.
*p < 0.05; ***p < 0.001.
IVD: In vitro methylated DNA; LC: Primary lung cancer; M: Methylation; N: Normal lung tissue samples; NL: Normal lymph cell DNA; U: Unmethylation.
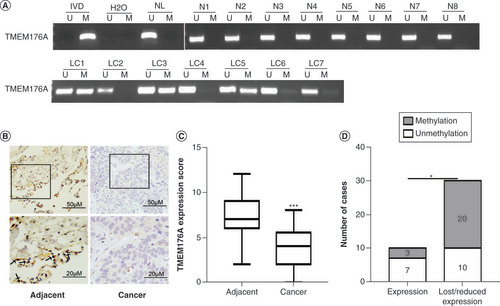
Table 1. The association of TMEM176A methylation and clinical factors in non-small-cell lung cancer.
The expression of TMEM176A was evaluated by immunohistochemistry in 40 cases of available matched lung cancer and adjacent tissue samples. TMEM176A staining was found mainly in cytoplasm (). The expression levels of TMEM176A were reduced in cancer compared with adjacent tissue samples (, p < 0.05). Lower level expression of TMEM176A was found in 30 cases of cancer tissue samples. Among the 30 cases that had reduced expression of TMEM176A, 20 cases were methylated. The reduced expression of TMEM176A was significantly associated with promoter region methylation (, p < 0.05). These data indicate that the expression of TMEM176A is regulated by promoter region methylation in human primary lung cancer.
TMEM176A inhibits lung cancer cells proliferation
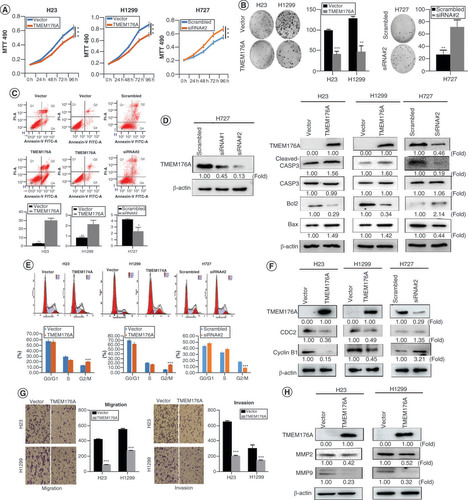
MTT and colony formation assays were used to evaluate the effects of TMEM176A on cell proliferation. TMEM176A stably expressed cells were established by transfection assay and TMEM176A highly expressed cells were knocked down by siRNA. The OD values were 0.648 ± 0.006 versus 0.527 ± 0.005 in H23 cells and 0.878 ± 0.010 versus 0.700 ± 0.008 in H1299 cells before and after restoration of TMEM176A expression. The OD values were reduced significantly after restoration of TMEM176A expression in H23 and H1299 cells (both p < 0.001, ). The OD values were 0.550 ± 0.040 versus 0.673 ± 0.025 in H727 before and after knockdown of TMEM176A. The OD values increased significantly after knockdown of TMEM176A expression in H727 cell (p < 0.001, ). These results demonstrated that TMEM176A inhibits cell proliferation in lung cancer cells. The clone numbers were 99.6 ± 3.5 versus 42.0 ± 7.5 (p < 0.001) in H23 cells and 128.7 ± 5.9 versus 47.7 ± 5.7 in H1299 cells (p < 0.01) before and after restoration of TMEM176A expression (). The clone numbers were 26 ± 7.9 versus 71.3 ± 11.3 in H727 cells (p < 0.01) before and after knockdown of TMEM176A (). These data suggest that TMEM176A suppresses cell growth in lung cancer.
(A) Growth curves represent cell viability. (B) Colony formation results. Bar diagram: average number of tumor clones. (C) Flow cytometry shows apoptosis results. (D) The effect of TMEM176A on caspase-3, cleaved caspase-3, Bcl2 and Bax expression in H23, H1299 and H727 cells. siRNA#1 and siRNA#2: siRNA for TMEM176A. (E) Cell phase distribution in TMEM176A unexpressed and expressed H23, H1299 and H727 cells. (F) The effect of TMEM176A on CDC2 and cyclin B1 expression in H23, H1299 and H727 cells. (G) The migration and invasion assays for TMEM176A expressed and unexpressed H23 and H1299 cells. (H) The effect of TMEM176A on MMP-2 and MMP-9 expression in H23 and H1299 cells.
*p < 0.05; **p < 0.01; ***p < 0.001.
β-actin: Internal control; Scrambled: Scrambled siRNA serve as negative control; siRNA#2: siRNA for TMEM176A; TMEM176A: TMEM176A expressing vector; Vector: Control vector.
TMEM176A induces lung cancer cells apoptosis
The effect of TMEM176A on apoptosis was analyzed by flow cytometry. Under doxorubicin treatment, the ratios of apoptotic cells in TMEM176A unexpressed and re-expressed cells were 3.20 ± 0.01% versus 29.80 ± 0.03% in H23 cells, and 0.80 ± 0.00% versus 2.10 ± 0.00% in H1299 cells. The ratio of apoptotic cells increased significantly after re-expression of TMEM176A (all p < 0.05, ). In H727 cell, the ratios of apoptotic cells were 4.17 ± 0.00% versus 2.30 ± 0.01% before and after knockdown of TMEM176A. The ratio of apoptotic cells decreased significantly after knockdown of TMEM176A (p < 0.05, ).
By detecting the efficiency of siRNA for TMEM176A, siRNA#2 was found more effective than siRNA#1, and siRNA#2 was applied to further study. To further validate the effect of TMEM176A on apoptosis, caspase-3, cleaved caspase-3, Bcl2 and Bax expression were analyzed with or without TMEM176A expression in lung cancer cells. The levels of caspase-3 were not apparent different with or without TMEM176A expression (). The levels of cleaved caspase-3 and Bax increased after re-expression of TMEM176A in H23 and H1299 cells, decreased after knockdown of TMEM176A in H727 cell (). The levels of Bcl2 decreased after re-expression of TMEM176A in H23 and H1299 cells, increased after knockdown of TMEM176A in H727 cell (). These results demonstrate that TMEM176A induces apoptosis in lung cancer cells.
TMEM176A induces G2/M phase arrest in lung cancer cells
To evaluate the effect of TMEM176A on the cell cycle, flow cytometry was employed. The percentage of cell phases in TMEM176A unexpressed and re-expressed H23 cell lines was 56.95 ± 0.07% versus 56.16 ± 0.06% in G0/G1 phase, 29.35 ± 0.16% versus 23.46 ± 0.30% in S phase, and 13.70 ± 0.19% versus 20.38 ± 0.26% in G2/M phase. The G2/M phase was increased significantly after re-expression of TMEM176A (p < 0.001; ). In H1299 cells, the percentage of the cell phase was 70.66 ± 1.10% versus 63.28% ± 0.54% in G0/G1 phase, 21.97 ± 0.51% versus 18.72 ± 0.45% in S phase, and 7.37 ± 0.80% versus 18.00 ± 0.24% in G2/M phase before and after restoration of TMEM176A expression. The G2/M phase was increased significantly after re-expression of TMEM176A in H1299 cells (p < 0.001; ). The effect of TMEM176A on cell cycle was further validated by knocking down TMEM176A in TMEM176A highly expressed H727 cells. The distribution of cell phases was 45.86 ± 0.50% versus 50.46 ± 0.66% in G0/G1 phase, 34.95 ± 0.78% versus 40.75 ± 0.32% in S phase, and 19.19 ± 0.61% versus 9.02 ± 0.13% in G2/M phase. The G2/M phase was significantly reduced by knocking down TMEM176A in H727 cells (p < 0.001; ). The preceding results suggest that TMEM176A induced G2/M phase arrest. The levels of cyclinB1 and CDC2 were also detected by western blot. As shown in , the levels of cyclinB1 and CDC2 were reduced after re-expression of TMEM176A in H23 and H1299 cells. The levels of cyclinB1 and CDC2 were increased after knockdown of TMEM176A in H727 cells. These results further suggest that TMEM176A induces G2/M phase arrest in human lung cancer cells.
TMEM176A inhibits lung cancer cell migration & invasion
To evaluate the effects of TMEM176A on cell migration and invasion, transwell assays were used. The numbers of migration cells were 431.5 ± 8.22 versus 98.0 ± 4.83 in H23 cells and 562.0 ± 15.03 versus 285.0 ± 3.46 in H1299 cells before and after restoration of TMEM176A expression. The number of migration cells decreased significantly after re-expression of TMEM176A in H23 and H1299 cells (both p < 0.001; ). The numbers of invasion cells were 652.0 ± 16.15 versus 209.3 ± 10.24 in H23 cells and 306.7 ± 48.21 versus 152.0 ± 7.30 in H1299 cells before and after restoration of TMEM176A expression. The cell number decreased significantly after re-expression of TMEM176A in H23 and H1299 cells (both p < 0.001; ). These results suggest that TMEM176A suppresses lung cancer cell migration and invasion. To further explore the mechanism of TMEM176A on cell migration and invasion, MMP-2 and MMP-9 expression were measured by western blot. The expression levels of MMP-2 and MMP-9 were reduced after re-expression of TMEM176A in H23 and H1299 cells. These results suggest that TMEM176A inhibits cell invasion in lung cancer cells (). As H727 is carcinoid cell and the invasive and migration ability are very poor. Therefore, the transwell assay was not performed.
TMEM176A inhibits ERK signaling pathway in lung cancer cells
TMEM176A was previously shown to be involved in ERK signaling in hepatic cancer [Citation27], prompting examination in human lung cancer. As the effect on ERK phosphylation is really only seen when the pathway is active, with serum stimulation. Total ERK1/2 and phosphorylated ERK1/2 (p-ERK1/2) were determined by western blot in serum stimulated lung cancer cells with or without TMEM176A expression. As shown in , although the levels of ERK1/2 did not differ before and after re-expression of TMEM176A in H23 and H1299 cells, p-ERK1/2 was reduced after re-expression of TMEM176A in serum stimulated H23 and H1299 cells. The levels of ERK1/2 in TMEM176A expressing H727 cells were also not altered by TMEM176A siRNA knock down, but p-ERK1/2 was increased after knockdown of TMEM176A in H727 cells. These results suggest that TMEM176A inhibits ERK signaling in lung cancer cells.
(A) western blots results. β-actin: internal control. (-): no serum stimulation; (+): serum stimulation. (B) Growth curves represent cell viability. SiRNA#2: siRNA for TMEM176A. (C) western blot results. SCH772984: p-ERK1/2 inhibitor; (-): absence of SCH772984; (+): presence of SCH772984. (D) MTT assays show OD value for IC50. ATMi: ATM inhibitor. (E) Epigenetic silencing TMEM176A sensitized lung cancer cells to UV and AZD0156. western blot shows the levels of TMEM176A, ATM, p-CHK2, γ-H2AX, RAD51 and β-actin.
***p < 0.001.
AZD0156: ATM inhibitor; (-): absence of AZD0156 or UV; (+): presence of AZD0156 or UV.
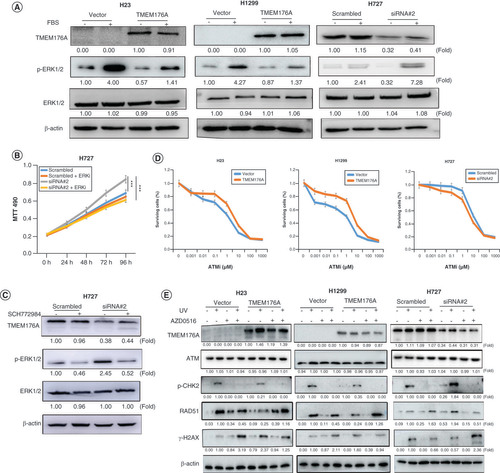
To confirm the role of TMEM176A in ERK signaling, we used an ERK signaling inhibitor (SCH772984) and assessed cell growth by MTT. There was no significant difference was found between control cells, control plus SCH772984 and siTMEM176A plus SCH772984 treatment group (p > 0.05) in H727 cells. However, growth is reduced significantly in siTMEM176A plus SCH772984 treatment group compared with siTMEM176A group in H727 cells (p < 0.001; ), suggesting that ERK inhibition is only effective with reduction in TMEM176A and resulting ERK activation, as seen in . This increase in p-ERK1/2 is significantly reduced in plus SCH772984 treatment (). These results further demonstrate that TMEM176A inhibits ERK signaling in lung cancer.
Epigenetic silencing of TMEM176A sensitized lung cancer cells to AZD0156
ERK signaling was reported as a positive regulator of ATM-dependent DNA homologous recombination repair (HR) [Citation40]. Since we found TMEM176A can inhibit ERK signaling in human lung cancer, we tested the sensitivity of TMEM176A unexpressed and re-expressed H1299 cells and H23 cells to the ATM inhibitor AZD0156. The IC50 of AZD0156 was 1.35 ± 0.05 versus 4.00 ± 0.15 μM/l in TMEM176A unexpressed and re-expressed H23 cells, and 1.88 ± 0.09 versus 4.76 ± 0.08 μM/l in TMEM176A unexpressed and re-expressed H1299 cells, respectively. The IC50 was reduced significantly in human lung cancer cells with silencing of TMEM176A by promoter region hypermethylation (all p < 0.05; ). This effect was confirmed by siRNA knocking down in TMEM176A expressing lung cancer cells. The IC50 was 7.61 ± 0.07 versus 4.39 ± 0.13 μM/l in H727 cells before and after knockdown of TMEM176A, with the IC50 was significantly reduced after knockdown of TMEM176A (p < 0.05; ). The preceding results demonstrated that loss of TMEM176A expression sensitized cells to the ATM inhibitor AZD0156.
To further validate the effect of TMEM176A on DDR, the levels of ATM, p-CHK2, γ-H2AX and RAD51 were detected by western blot in TMEM176A unexpressed and re-expressed lung cancer cells. The protein of p-CHK2 is the key factor downstream of ATM pathway. As shown in , p-CHK2, the active form key downstream factor of ATM pathway was detected before and after re-expression of TMEM176A in H23 and H1299 cells under UV treatment. The levels of p-CHK2 were reduced after re-expression of TMEM176A. After exposed to UV for 2 h, lung cancer cells were treated with AZD0156 for 24 h. The levels of γ-H2AX were higher in TMEM176A unexpressed H23 and H1299 cells compared with re-expressed H23 and H1299 cells. The levels of RAD51 were lower in TMEM176A unexpressed H23 and H1299 cells compared to re-expressed H23 and H1299 cells (). The levels of ATM, p-CHK2, γ-H2AX and RAD51 were detected in TMEM176A highly expressed H727 cells before and after siRNA knocking down. The levels of p-CHK2 were increased after knockdown of TMEM176A in H727 cells under UV treatment (). The levels of γ-H2AX were reduced and the levels of RAD51 were increased after knocking down of TMEM176A in H727 cells under UV treatment (). After exposed to UV for 2 h, H727 cells were treated with AZD0156 for 24 h. The levels of γ-H2AX were increased and the levels of RAD51 were reduced after knocking down of TMEM176A in H727 cells (). The preceding results suggest that methylation of TMEM176A may serve as a lethal therapeutic marker for AZD0156 and UV treatment.
As the significant overlap between the ATR (AT and Rad3-related protein) and ATM pathway, we analyzed the effect of ATR inhibitor (AZD6738) on TMEM176A expressed and unexpressed lung cancer cells. The IC50 of AZD6738 was 11.15 ± 1.4 versus 10.69 ± 1.63 μM/l in TMEM176A unexpressed and re-expressed H23 cells, and 12.83 ± 2.1 versus 12.03 ± 1.66 μM/l in TMEM176A unexpressed and re-expressed H1299 cells, respectively (all p > 0.05). There is no significant difference between TMEM176A unexpressed and re-expressed cancer cells. The results suggest that TMEM176A is not involved in ATR pathway through ERK signaling.
TMEM176A suppresses human lung cancer cell xenograft growth by inhibiting ERK signaling
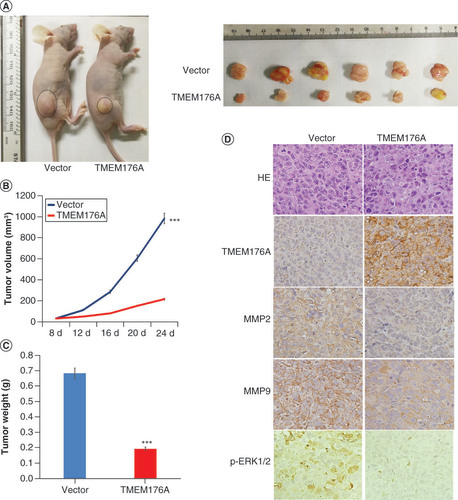
To further evaluate the effect of TMEM176A in human lung cancer, TMEM176A unexpressed and re-expressed H1299 cells were used in xenograft mouse studies (). The tumor volume was 983.32 ± 101.76 versus 216.06 ± 86.96 mm3 in TMEM176A unexpressed and re-expressed H1299 cell xenografts (), with a significantly reduced tumor volume in TMEM176A re-expressed H1299 cell xenograft mice (p < 0.001). The tumor weight was 0.68 ± 0.11 versus 0.20 ± 0.07 g in TMEM176A unexpressed and re-expressed H1299 cell xenograft mice (). The tumor weight was also reduced significantly in TMEM176A re-expressed H1299 cells xenograft mice (p < 0.001). These results indicate that TMEM176A suppresses lung cancer cell growth in vivo. To further validate the effect of TMEM176A on tumor metastasis, the expression of MMP-2 and MMP-9 were examined by IHC in xenograft tumors. The expression levels of MMP-2 and MMP-9 were decreased in TMEM176A re-expressed H1299 cell xenografts compared to TMEM176A unexpressed H1299 cells (). To further validate that TMEM176A inhibits ERK signaling in vivo, p-ERK1/2 was detected by IHC staining in TMEM176A unexpressed and re-expressed H1299 cell xenografts, with decreased p-ERK1/2 in TMEM176A re-expressed H1299 cell xenografts ().
(A) TMEM176A unexpressed and re-expressed H1299 cell xenografts in mice. (B) Growth curves for TMEM176A unexpressed and re-expressed H1299 cell xenografts. (C) Tumor weights: TMEM176A unexpressed and re-expressed H1299 cell xenografts. (D) TMEM176A affects the levels of MMP-2, MMP-9 and p-ERK1/2 in H1299 cell xenografts.
***p < 0.001.
Discussion
TMEM176A is located in human chromosome 7q36.1, a region where there is a frequent loss of heterozygosity in human cancers [Citation41,Citation42]. By RNA-seq analysis, we found that the expression of TMEM176A was reduced in human colorectal cancer [Citation26]. TMEM176A was found frequently methylated in human colorectal, esophageal and hepatic cancers and may be serves as a potential tumor suppressor in these cancers [Citation26–28]. TMEM176A is also found to be involved in ERK signaling in hepatic cancer in our previous study [Citation27]. Further, a recently report demonstrated that the expression of TMEM176A was increased in primary human glioblastoma and TMEM176A promoted glioblastoma cell proliferation by activating ERK signaling. They also found that both TMEM176A and TMEM176B were increased expression in glioblastoma [Citation43]. The expression pattern of TMEM176A and TMEM176B are possibly discrepant in different tissues. TMEM176A and TMEM176B may play different roles in different microenvironment. Drujont et al. found that the expression of TMEM176A was increased in TMEM176b-/- cells compared with wild-type cells of Th17 in mice [Citation44]. Epigenetic changes can display cancer specific [Citation10,Citation45]. Thus, we explored the epigenetic regulation and function of TMEM176A in human lung cancer.
Although there are approaches targeting ‘epigenetic machinery’ inhibitors in ongoing clinical trials [Citation45], there are many challenges to be resolved for efficient use of epidrugs in the treatment of human cancer, including the lack of specificity, disappointing success in solid tumors and the acquisition of drug chemo-resistance leading to higher risk of tumor relapse. Because no specific clinical detective marker of histone modifications in tumor detection, the efficiency of targeting histone modifier therapy remains very limited [Citation10]. With the great understanding of ‘synthetic lethality’ based on DNA damage repair signaling defects, synthetic lethality becomes a hot topic in cancer therapeutic study, but few mutations in DDR genes have been found in human lung cancer. Notably, synthetic lethality strategy allows the therapeutic exploitation of nondruggable mutated tumor suppressor genes and directly difficult to target (hard-druggable) oncogenes, via targeting their synthetic lethality partners [Citation46]. ‘BRCAnesss’ based ‘synthetic lethality’ has been put forwarded [Citation47]. Wnt, PI3K-ERK and other cancer-related signaling pathways were reported to be involved in cell cycle and DDR [Citation14,Citation48,Citation49]. Aberrant epigenetic changes of the key components in the cell fate determining signaling pathways may offer more opportunities for synthetic lethal therapy. In this study, we found that TMEM176A is frequently methylated in human lung cancer and the expression of TMEM176A is regulated by promoter region methylation. Methylation of TMEM176A may serve as a potential lung cancer diagnostic marker. TMEM176A inhibits cell proliferation, invasion, migration, colony formation, induces G2/M phase arrest and apoptosis in lung cancer. TMEM176A suppresses H1299 lung cancer cell xenografts growth in mice. Similar to our previous study in hepatic cancer, TMEM176A suppressed lung cancer growth by inhibiting ERK signaling both in vitro and in vivo.
Aberrant epigenetic changes occur more frequently compared to gene mutations in human cancers. Beyond ‘BRCAness’, understanding the causative epigenetic changes of ‘loss of function’ may develop more therapeutic strategies in cancer, and epigenetic-based synthetic lethal therapy may be more specific and more efficient [Citation14,Citation15].
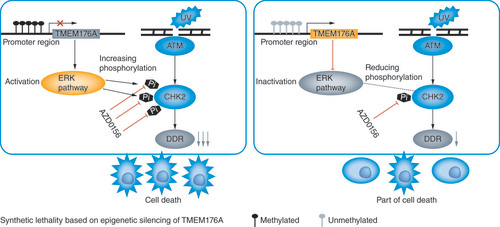
The role of ERK was clearly defined as a positive regulator of HR and HR is the most important DDR in late S and G2/M phases of the cell cycle [Citation40]. ATM and ATR are two major kinases to take part in DDR, and ATM specifically regulates HR [Citation50]. TMEM176A induced G2/M arrest in lung cancer cells. Thus, we analyzed the role of TMEM176A in ATM pathway. Our study demonstrated that methylation of TMEM176A sensitized H1299 and H23 cells to AZD0156, and restoration of TMEM176A expression decreased their sensitivities to AZD0156. The results suggest that methylation of TMEM176A is a sensitive marker of ATM inhibitors for lung cancer cells. There is considerable overlap of the phosphorylation substrates of both ATR and ATM. Then, we detected the sensitivity of ATR inhibitor, AZD6738, TMEM176A expressed and unexpressed lung cancer cells. No difference was found between TMEM176A expressed and unexpressed lung cancer cells for AZD6738. No synthetic effect was found between ATR inhibitors and defect of TMEM176A. Our results suggest that methylation of TMEM176A is a synthetic lethal marker for ATM inhibitors in lung cancer. illustrates the schematic synthetic lethality between TMEM176A methylation and ATM inhibitors in lung cancer.
Conclusion
TMEM176A suppresses lung cancer growth both in vitro and in vivo by inhibiting ERK signaling pathway. TMEM176A is a potential tumor suppressor in human lung cancer. Methylation of TMEM176A is a potential diagnostic marker and a novel synthetic lethal therapeutic marker for ATM inhibitor in human lung cancer.
Future perspective
Synthetic lethality is a new strategy of cancer therapy. Methylation of TMEM176A activated ERK signaling, and sensitized H1299 and H23 cells to AZD0156, an ATM inhibitor. Methylation of TMEM176A is a potential lung cancer diagnostic marker and a novel synthetic lethal therapeutic marker for AZD0156 treatment. Epigenetic-based synthetic lethality may provide more opportunities for cancer prevention and treatment.
The expression of TMEM176A was found to be regulated by promoter region methylation in lung cancer cells.
TMEM176A was methylated in 53.66% (66/123) of primary lung cancer.
Restoration of TMEM176A expression induced cell apoptosis and G2/M phase arrest and inhibited colony formation, cell proliferation, migration and invasion in H1299 and H23 cells.
TMEM176A suppressed H1299 cell xenograft growth in mice.
Methylation of TMEM176A activated ERK signaling, and sensitized H1299 and H23 cells to AZD0156, an ATM inhibitor.
Author contributions
H Li performed the research and analyzed the data. H Li and MZ Guo wrote the manuscript. MZ Guo made substantial contributions to the conception and design of the study, W Yang, M Zhang, T He, F Zhou and JG Herman provided manuscript and experimental advice. L Hu supervised the study. All authors read and approved the final manuscript.
Ethical conduct of research
The authors state that all samples were collected following the guidelines approved by the Institutional Review Board of the Chinese PLA General Hospital. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved. The ethics approval statements for animal work were provided by the Institutional Animal Care and Use Committee of Chinese PLA General Hospital.
Financial & competing interests disclosure
This work was supported by grants from National Key Research and Development Program of China (2018YFA0208902, 2020YFC2002705); National Science Foundation of China (NSFC no. U1604281, 81672138); Beijing Science Foundation of China (BJSFC no. 7171008); National Key Scientific Instrument Special Program of China (grant no. 2011YQ03013405). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Bray F , FerlayJ , SoerjomataramIet al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.68(6), 394–424 (2018).
- Reck M , RabeKF. Precision diagnosis and treatment for advanced non-small-cell lung cancer. New Engl. J. Med.377(9), 849–861 (2017).
- Corrales L , RosellR , CardonaAFet al. Lung cancer in never smokers: the role of different risk factors other than tobacco smoking. Crit. Rev. Oncol. Hematol.148, 102895 (2020).
- Brock MV , HookerCM , Ota-MachidaEet al. DNA methylation markers and early recurrence in stage I lung cancer. New Engl. J. Med.358(11), 1118–1128 (2008).
- Guo M , RenJ , HouseMGet al. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin. Cancer Res.12(15), 4515–4522 (2006).
- Yuan M , HuangLL , ChenJH , WuJ , XuQ. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal transduction and targeted therapy4, 61–75 (2019).
- Guo M , LiuS , LuF. Gefitinib-sensitizing mutations in esophageal carcinoma. New Engl. J. Med.354(20), 2193–2194 (2006).
- Herbst RS , MorgenszternD , BoshoffC. The biology and management of non-small cell lung cancer. Nature553(7689), 446–454 (2018).
- Lim ZF , MaPC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol.12(1), 134 (2019).
- Guo M , PengY , GaoAet al. Epigenetic heterogeneity in cancer. Biomark Res.7, 23 (2019).
- Guo M , AlumkalJ , DrachovaTet al. CHFR methylation strongly correlates with methylation of DNA damage repair and apoptotic pathway genes in non-small cell lung cancer. Discov. Med.19(104), 151–158 (2015).
- Yu Y , YanW , LiuXet al. DACT2 is frequently methylated in human gastric cancer and methylation of DACT2 activated Wnt signaling. Am. J. Cancer Res.4(6), 710–724 (2014).
- Dolsten M , SøgaardM. Precision medicine: an approach to R&D for delivering superior medicines to patients. Clin. Transl. Med.1(1), 7 (2012).
- Gao A , GuoM. Epigenetic based synthetic lethal strategies in human cancers. Biomark Res.8, 44 (2020).
- Hu Y , GuoM. Synthetic lethality strategies: beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci.111(9), 3111–3121 (2020).
- Du W , GaoA , HermanJGet al. Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in esophageal cancer. Cancer Sci.112(7), 2870–2883 (2021).
- Mclornan DP , ListA , MuftiGJ. Applying synthetic lethality for the selective targeting of cancer. New Engl. J. Med.371(18), 1725–1735 (2014).
- Bryant HE , SchultzN , ThomasHDet al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature434(7035), 913–917 (2005).
- Farmer H , MccabeN , LordCJet al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature434(7035), 917–921 (2005).
- Hu Y , WenZ , LiuSet al. Ibrutinib suppresses intracellular mycobacterium tuberculosis growth by inducing macrophage autophagy. J. Infection80(6), e19–e26 (2020).
- Gehrau R , MalufD , ArcherKet al. Molecular pathways differentiate hepatitis C virus (HCV) recurrence from acute cellular rejection in HCV liver recipients. Mole. Med. (Cambridge, Mass.)17(7–8), 824–833 (2011).
- Nakajima H , TakenakaM , KaimoriJYet al. Gene expression profile of renal proximal tubules regulated by proteinuria. Kidney Int.61(5), 1577–1587 (2002).
- Zuccolo J , DengL , UnruhTLet al. Expression of MS4A and TMEM176 genes in human B lymphocytes. Front. Immunol.4, 195 (2013).
- Condamine T , LeTexier L , HowieDet al. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J. Leukoc. Biol.88(3), 507–515 (2010).
- Grunin M , Hagbi-LeviS , RinskyBet al. Transcriptome analysis on monocytes from patients with neovascular age-related macular degeneration. Sci. Rep.6, 29046 (2016).
- Gao D , HanY , YangYet al. Methylation of TMEM176A is an independent prognostic marker and is involved in human colorectal cancer development. Epigenetics12(7), 575–583 (2017).
- Li H , ZhangM , LinghuEet al. Epigenetic silencing of TMEM176A activates ERK signaling in human hepatocellular carcinoma. Clin. Epigenetics10(1), 137 (2018).
- Wang Y , ZhangY , HermanJG , LinghuE , GuoM. Epigenetic silencing of TMEM176A promotes esophageal squamous cell cancer development. Oncotarget.8(41), 70035–70048 (2017).
- Guo M , AkiyamaY , HouseMGet al. Hypermethylation of the GATA genes in lung cancer. Clin. Cancer Res.10(23), 7917–7924 (2004).
- Guo M , HouseMG , SuzukiHet al. Epigenetic silencing of CDX2 is a feature of squamous esophageal cancer. Int. J. Cancer121(6), 1219–1226 (2007).
- Liu X , ZhangX , ZhanQet al. CDX2 serves as a Wnt signaling inhibitor and is frequently methylated in lung cancer. Cancer Biol. Ther.13(12), 1152–1157 (2012).
- Morris EJ , JhaS , RestainoCRet al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov.3(7), 742–750 (2013).
- Zheng R , GaoD , HeTet al. Methylation of DIRAS1 promotes colorectal cancer progression and may serve as a marker for poor prognosis. Clin. Epigenetics9, 50 (2017).
- Yan W , WuK , HermanJGet al. Epigenetic regulation of DACH1, a novel Wnt signaling component in colorectal cancer. Epigenetics8(12), 1373–1383 (2013).
- Jia Y , YangY , LiuSet al. SOX17 antagonizes WNT/β-catenin signaling pathway in hepatocellular carcinoma. Epigenetics5(8), 743–749 (2010).
- Cui YGD , LinghuE , ZhanQet al. Epigenetic changes and functional study of HOXA11 in human gastric cancer. Epigenomics7(2), 201–213 (2015).
- Brown JS , O’CarriganB , JacksonSP , YapTA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov.7(1), 20–37 (2017).
- Yang MC , LinRW , HuangSBet al. Bim directly antagonizes Bcl-xl in doxorubicin-induced prostate cancer cell apoptosis independently of p53. Cell Cycle15(3), 394–402 (2016).
- Hegedűs C , BorosG , FidrusEet al. PARP1 inhibition augments UVB-mediated mitochondrial changes–impications for UV-induced DNA repair and photocarcinogenesis. Cancers12(1), (2019).
- Golding SE , RosenbergE , NeillSet al. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res.67(3), 1046–1053 (2007).
- Riegman PH , BurgartLJ , WangKKet al. Allelic imbalance of 7q32.3-q36.1 during tumorigenesis in Barrett’s esophagus. Cancer Res.62(5), 1531–1533 (2002).
- Kimmel RR , ZhaoLP , NguyenDet al. Microarray comparative genomic hybridization reveals genome-wide patterns of DNA gains and losses in post-Chernobyl thyroid cancer. Radiat. Res.166(3), 519–531 (2006).
- Liu Z , AnH , SongPet al. Potential targets of TMEM176A in the growth of glioblastoma cells. OncoTarget. Ther.11, 7763–7775 (2018).
- Drujont L , LemoineA , MoreauAet al. RORgammat+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci. Rep.6, 23682 (2016).
- Yan W , HermanJG , GuoM. Epigenome-based personalized medicine in human cancer. Epigenomics8(1), 119–133 (2016).
- Pfister SX , AshworthA. Marked for death: targeting epigenetic changes in cancer. Nat. Rev. Drug Discov.16(4), 241–263 (2017).
- Lord CJ , AshworthA. BRCAness revisited. Nat. Rev. Cancer16(2), 110–120 (2016).
- Zhong Z , VirshupDM. Wnt signaling and drug resistance in cancer. Mol. Pharmacol.97(2), 72–89 (2020).
- Huang TT , LampertEJ , CootsC , LeeJM. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treatment Rev.86, 102021 (2020).
- Morrison C , SonodaE , TakaoN et al. The controlling role of ATM in homologous recombinational repair of DNA damage. The EMBO J.19(3), 463–471 (2000).