
Abstract
Aim: To explore the N6-methyladenosine (m6A) methylation of mRNAs and its roles in a mouse model of scleroderma. Materials & methods: To evaluate whether the mouse model of scleroderma could meet the experimental requirements, we examined skin tissue specimens by pathological staining and identified the related indicators by quantitative PCR (qPCR). m6A-tagged mRNAs were identified via m6A epitranscriptomic microarray, and m6A-RNA-immunoprecipitation qPCR and qPCR were performed to confirm microarray data. Results: There were differences in m6A methylation among 843 mRNAs. Further, there were significant differences among Hras, Saa1, Ccl3, Ccl9 and Il1b in terms of methylation and expression. Conclusion: The m6A methylation spectrum in a mouse model of scleroderma may explain the occurrence of scleroderma.
Lay abstract
Scleroderma is an autoimmune disease with an unknown etiology. At present there is no effective treatment, and the prognosis is poor. Although m6A methylation is the most common RNA modification and affects cell biological function by influencing mRNA expression levels, the role of m6A methylation in scleroderma is unknown. Based on a mouse model of scleroderma, we screened and validated mRNAs related to m6A regulation, including Hras, Lama3, Stat3, Tnc, Ccl3, Ccl9, Saa1 and Il1b. While the results are encouraging, they are only preliminary. We plan to explore the altered pattern of m6A methylation in skin specimens from patients with scleroderma. This study is important for developing biomarkers for diagnosis and treatment of scleroderma in the future.
Scleroderma is an autoimmune connective tissue disease characterized by organ fibrosis, immune abnormalities and vascular injury [Citation1]. Depending on the extent of the lesion involved, the condition can present as localized scleroderma or systemic scleroderma [Citation2]. Systemic scleroderma involves other tissues in addition to the skin; the lung, liver and kidney can also be involved, and destruction of the standard structure of the tissue may be observed. Fibrosis is the most typical characteristic of scleroderma; however, immune disorder and vascular injury precede and aggravate fibrosis [Citation3]. Although scleroderma is a rare disease with low prevalence and morbidity, its mortality remains high [Citation4]. Interstitial lung disease and pulmonary hypertension are the most common causes of death [Citation5]. In addition, treatment for the condition has not changed significantly in the past 40 years and is still dominated by traditional symptomatic treatment [Citation6]. As the pathogenesis of scleroderma remains unknown, it cannot be targeted to reverse the progression of fibrosis, highlighting unmet medical needs [Citation7,Citation8].
Epigenetics is a branch of genetics that deals with heritable changes in gene expression which do not involve any changes in the DNA sequence [Citation9]. The genome contains two types of genetic information, namely DNA genetic information and epigenetic information, explaining why the limited genetic information has so many biological functions. For example, individuals can be genetically susceptible to scleroderma, but environmental factors such as viral infection and drug use are associated with the development of the condition. These factors may affect gene expression profiles through epigenetic mechanisms, thus altering various cells [Citation10–12].
RNAs can be chemically modified to more than 100 distinct types that can enrich genetic diversity, such as N1-methyladenosine (m1A), N6-methyladenosine (m6A), N8-methyladenosine (m8A), N1-methylguanosine (m1G) and N2-methlyguanosine (m2G) [Citation13–16]. RNA methylation affects every aspect of RNA regulation, including RNA stability, localization, transport, splicing and translation [Citation17]. RNA methylation is a reversible process, and its dynamic equilibrium is dependent on the regulation of methyltransferases (METTL3, METTL14 and WTAP) and demethylases (FTO and ALKBH5), which are called ‘writers’ and ‘erasers’, respectively [Citation18]. These enzymes regulate binding proteins (‘readers’), such as YTHDF1, YTHDF2, YTHDF3, YTHDC1 and YTHDC2, which can further identify RNA methylation modification information [Citation19]. When the N6 position of adenine is methylated, it is referred to as m6A. m6A methylation is the most common RNA modification [Citation20], accounting for about 50% of methylated ribonucleotides [Citation21]. It is located near stop codons and in 3′ UTRs [Citation22].
Although m6A methylation was discovered in the 1960s [Citation23,Citation24], research on m6A methylation has lagged behind compared with that of other modified bases, the main reason behind this being the lack of feasible methods to determine m6A sites [Citation25]. Similarly, although high-throughput genomic technologies have revealed changes in noncoding RNAs (ncRNAs; miRNA, circRNA and long ncRNA [lncRNA]) and mRNA expression among scleroderma [Citation26–29], few high-throughput epigenomic studies have reported on RNA m6A methylation in scleroderma [Citation30]. Excitingly, scientists have found a way to combine m6A antibodies with m6A-containing RNA and perform high-throughput sequencing to identify the areas of RNA with high levels of m6A; this technique is called m6A-RNA-immunoprecipitation sequencing (MeRIP-seq) [Citation20,Citation25].
MeRIP-seq has helped scholars to reveal the role of abnormal m6A modification in diseases and particularly in oncology, including hepatocellular cancer [Citation31], breast cancer [Citation32] and pancreatic cancer [Citation33]. In the same way, it is of great value to explore the role of m6A methylation in diseases outside of malignancy, especially fibrosis. Although the way in which m6A modification regulates fibrosis is not well understood, multiple studies have suggested that abnormal m6A modifications are involved in multiple organ fibrosis. In idiopathic pulmonary fibrosis, m6A methylation is upregulated in patients, a bleomycin (BLM)-induced pulmonary fibrosis mouse model and fibroblast-to-myofibroblast transition-derived myofibroblasts, and silencing METTL3 can suppress fibroblast-to-myofibroblast transition in vitro and in vivo [Citation34]. In a mouse model of renal interstitial fibrosis, the m6A level is time-dependent and decreased within 1 week [Citation18]. Similar alterations are observed in METTL3, METTL14 and WTAP [Citation18]. In a mouse model of liver fibrosis, differential m6A-modified peaks are involved in oxidative stress and cytoplasmic metabolism during fibrogenesis, while differential m6A-modified peaks are enriched in immune response and apoptosis during fibrosis regression [Citation9]. In a mouse model of myocardial infarction, Fto overexpression alleviates myocardial fibrosis [Citation35]. However, its effects on skin fibrosis remain unknown. m6A methylation is closely associated with autoimmune diseases such as psoriasis [Citation36], systemic lupus erythematosus [Citation37,Citation38] and rheumatoid arthritis [Citation39], but little is known about how it affects immune abnormalities in scleroderma. Based on these two perspectives, we are interested in m6A methylation in scleroderma.
Although human scleroderma samples can be obtained from pathological examination, there may be significant differences between samples based on the degree of the disease and the site of skin lesions; therefore a reliable animal model to limit confounders is necessary. We selected the BLM-induced scleroderma model [Citation40] and performed m6A epitranscriptomic microarray for analyzing the m6A methylation profile in a mouse model of scleroderma. Compared with MeRIP-seq, m6A epitranscriptomic microarray can estimate the levels of different types of RNAs, such as mRNA, lncRNA and circRNA. Also, MeRIP-seq requires 120 μg of total RNA, which may make it unsuitable for valuable clinical samples. Our study will provide a basis for further research on how m6A methylation regulates the occurrence and development of scleroderma, and provide potential therapeutic directions.
Materials & methods
Establishment of a mouse model of scleroderma
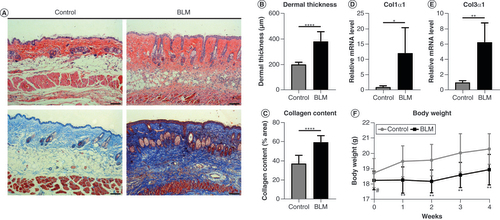
Female Balb/c mice aged 6–8 weeks were acquired from Charles River Laboratories (Shanghai, China). Sixteen mice were randomly divided into two groups: the control and a BLM-induced (BLM) group. In the BLM group, 100 μl BLM (1 mg/ml; Thermo Fisher Scientific, MA, USA) was injected subcutaneously into the back of the mice daily for 28 consecutive days. Similarly, mice in the control group were injected with 100 μl phosphate-buffered saline daily for 28 days. In the BLM group, a circular depilation area with a diameter of 0.1 cm was observed at the injection site, along with scar formation. Compared with the control group mice, the BLM group mice gained less weight (F).
(A) Skin specimens were stained with hematoxylin and eosin (top) and Masson’s trichrome (bottom). Original magnification ×100. Scale bar = 100 μm. (B) Dermal thickness and (C) collagen content were quantified from the hematoxylin and eosin and Masson’s trichrome staining data, respectively (n = 8 per group). (D)Col1α1 and (E)Col3α1 mRNA levels were examined by quantitative PCR (n = 4 per group). (F) Changes in body weight of mice in the control group and BLM-treated group within 4 weeks. Mean ± standard deviation.
*p < 0.05; **p < 0.01; ****p < 0.001 compared with the control group.
#No significance compared with the control group.
BLM: Bleomycin.
Histological analysis
All mice were sacrificed by cervical dislocation to allow for histological analysis. The skin samples were obtained from the lower right of the back, where BLM was injected. Skin samples (n = 8, each group) were fixed with 4% paraformaldehyde for 24 h at 4°C and then paraffin-embedded. The skin samples were stained with hematoxylin and eosin and Masson’s trichrome stain to assess the dermal thickness and collagen content, respectively. ‘Dermal thickness’ refers to the distance between the epidermal–dermal junction and the dermal–subcutaneous fat junction. ‘Collagen content’ refers to the ratio of collagen deposition area to the tissue area. Sections were observed using a light microscope (Olympus, Tokyo, Japan) and photographed using a digital camera. The evaluation was conducted by two independent examiners using ImageJ software (NIH, MD, USA), and three areas in each section were randomly selected.
Quantitative real-time PCR
Total RNA extraction was performed using TRIzol™ reagent (Sigma Aldrich, MO, USA), following the manufacturer’s instructions. cDNA was reverse transcribed using SuperScript™ III Reverse Transcriptase kit (Invitrogen, CA, USA), and quantitative PCR (qPCR) was performed with 2X PCR master mix (Arraystar, MD, USA) on the ViiA 7 Real-time PCR System (Thermo Fisher Scientific). The mRNA levels of target genes were normalized to the mRNA levels of ACTB or GAPDH via the 2-ΔΔCt method. The primers used are presented in Supplementary Table 1. We conducted qPCR experiments twice; the first time, Col1α1 and Col3α1 were normalized to ACTB (n = 4, each group), and the second time, the other indicators were normalized to GAPDH (n = 4, each group).
RNA extraction & quality control
Skin tissue was cut into small pieces and homogenized in TRIzol reagent to isolate total RNA. The quantification and quality of RNA were examined using NanoDrop ND-1000 (Thermo Fisher Scientific, Shanghai, China). The extracted total RNA was stored at -80°C for the subsequent experiments. The quantification and quality of RNA are shown in Supplementary Figure 1 & Supplementary Table 2.
m6A immunoprecipitation
The extracted RNA was immunoprecipitated with an anti-m6A antibody according to the manufacturer’s protocol. For this, 1–3 μg RNA and m6A spike-in control mixture was added to 300 μl 1× immunoprecipitation (IP) buffer containing 2 μg anti-m6A rabbit polyclonal antibody (Synaptic Systems, Göttingen, Germany). The reaction was incubated with head-over-tail rotation at 4°C for 2 h. Following this, 20 μl of Dynabeads™ M-280 sheep anti-rabbit IgG suspension (Thermo Fisher Scientific) per sample was blocked with freshly prepared 0.5% bovine serum ALB at 4°C for 2 h, washed thrice with 300 μl 1× IP buffer and subsequently resuspended in the prepared RNA–antibody mixture. RNA binding to the m6A antibody beads was carried out with head-over-tail rotation at 4°C for 2 h. The beads were then washed thrice with 500 μl 1× IP buffer and twice with 500 μl wash buffer. Finally, the enriched RNA was eluted with 200 μl elution buffer at 50°C for 1 h. The RNA was extracted by the acid phenol–chloroform and ethanol precipitation method.
Labeling & hybridization
An equal amount of calibration spike-in control RNA was added to the immunoprecipitated RNA (IP) and supernatant RNA (Sup), following which they were amplified and labeled with Cy3 (for Sup) and Cy5 (for IP) using Arraystar Super RNA Labeling Kit (Arraystar). The synthesized cRNAs were purified using RNeasy Mini Kit (Sigma Aldrich). The cRNA concentration and specific activity were measured using NanoDrop™ ND-1000 (Thermo Fisher Scientific; Supplementary Table 3).
Next, 2.5 μg of the Cy3- and Cy5-labeled cRNAs were mixed. The cRNA mixture was fragmented by adding 5 μl 10× blocking agent and 1 μl of 25× fragmentation buffer, heating at 60°C for 30 min and combining with 25 μl 2× hybridization buffer. Next, 50 μl hybridization solution was dispensed into the gasket slide and assembled to the m6A epitranscriptomic microarray slide. The slides were incubated at 65°C for 17 h in an Agilent hybridization oven (Agilent, CA, USA). Finally, the hybridized arrays were washed, fixed and scanned using an Agilent G2505C scanner (Agilent).
Epitranscriptomic microarray data analysis
The array images were analyzed using Agilent Feature Extraction software (Agilent). The raw intensities of IP and Sup were normalized with an average of log2-scaled spike-in RNA intensities. Following normalization, probe signals with Present or Marginal QC flags in at least four out of eight samples were retained for further m6A methylation level, m6A quantity and expression level analyses. For determining the m6A methylation level, the percentage modification was calculated based on the normalized intensities of IP and Sup. For determining the m6A quantity, the m6A methylation amount was calculated based on normalized intensities of IP. The expression level was calculated based on the total normalized intensities of IP and Sup. An additional quantile normalization method in the limma package was used to normalize the RNA expression level between arrays before probe flag screening.
Filtering with fold change ≥1.5 and p < 0.05 (unadjusted p-values) was used to identify differentially m6A-methylated RNAs and differentially expressed RNAs between the two comparison groups.
MeRIP-qPCR
IP (n = 4, each group) was used to validate the m6A epitranscriptomic microarray data. Then MeRIP-qPCR was used to quantify the RNA enrichment via 2-ΔΔct analysis. The primers used are described in Supplementary Table 1.
Statistical analysis
All data were presented as mean ± standard deviation. A two-sided t-test was used to compare the two unpaired groups using GraphPad Prism 8.0 (GraphPad, CA, USA). A p-value of <0.05 was considered statistically significant.
Results
Progression of Balb/c mouse skin fibrosis induced by BLM
After the subcutaneous injection of BLM for 4 weeks, a mouse model of scleroderma was successfully established. Compared with the control group, hematoxylin and eosin staining (A) showed that the distinction between epidermal, dermal and subcutaneous tissues was not clear in the BLM group. Further, dermal thickness had increased in the BLM group (B). In the BLM group, Masson’s trichrome staining (A) showed that excessive collagen was deposited in the dermis, which further extended to the subcutaneous fat and even substituted fat tissues. Collagen content analysis also revealed the same result (C). To evaluate the changes in extracellular matrix (ECM) production, we selected Col1α1 and Col3α1 as indicators, because they are mainly distributed in the skin. Compared with the control group, the mRNA expression of Col1α1 and Col3α1 was significantly increased in the BLM group (D & E). In summary, a mouse model of scleroderma was successfully established, making it possible for the subsequent experiments to succeed.
Alteration of m6A-methylated mRNAs in the BLM-induced scleroderma
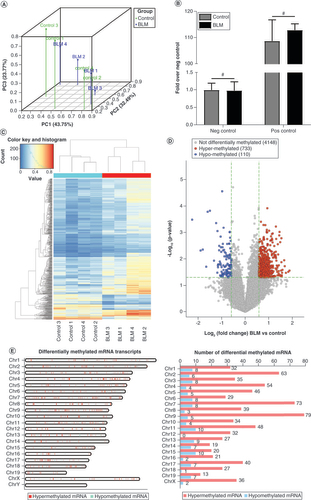
The results of three-dimensional principal components analysis revealed distinct clustering and segregation between the control and BLM groups. The percentages of variance associated with principal component (PC) 1, PC2 and PC3 were 43.75, 32.94 and 23.77%, respectively (A). Moreover, a corresponding MeRIP enrichment %(MeRIP/Input) of positive and negative spike-in controls was present between the control and the BLM groups, indicating a rigorous analysis of the m6A epitranscriptomic microarray (B). Hierarchical clustering analysis revealed the intragroup similarities and intergroup differences in methylated mRNA patterns (C). A volcano plot revealed that compared with the control group, 843 mRNAs in the BLM group had significantly different m6A modification, including 733 hypermethylated mRNAs and 110 hypomethylated mRNAs, all with fold change ≥1.5 and p < 0.05 (D). The differentially m6A-methylated mRNAs were mapped to all chromosomes except Y (E), because the Balb/c mice were female. The top five chromosomes with the most hypermethylated and hypomethylated m6A mRNAs (F) were: chromosomes 2 (63), 4 (54), 7 (73), 9 (79), 11 (48); and 1 (8), 3 (8), 6 (8), 7 (8), 11 (10), 13 (9) and 15 (10), respectively.
(A) PC analysis was conducted to classify the differentially methylated mRNAs of the control group and the BLM group. (B) Amplification of positive control primer set (Pos) and negative control primer set (Neg) by quantitative PCR assay indicated comparable m6A-RNA-immunoprecipitation enrichment % (m6A-RNA-immunoprecipitation/Input) between the control group and the BLM group. (C) The heat map shows opposite m6A-methylated mRNAs between the control group and the BLM group. (D) The volcano plot shows the differently m6A-methylated mRNAs between the control and BLM groups with fold change ≥1.5 and p < 0.05. (E) Chromosomal distribution of m6A-methylated mRNAs. n = 4, each group. Mean ± standard deviation.
#No significance compared with the control group.
BLM: Bleomycin; Chr: Chromosome; m6A: N6-methyladenosine; Neg: Negative; PC: Principal componant; Pos: Positive.
Association analysis of m6A methylation & mRNA expression in the BLM-induced scleroderma
The microarray data revealed significant differences in the global mRNA expression between the control and BLM groups. As shown in Supplementary Figure 2, the heat map and volcano plot showed 1991 mRNAs with increased expression and 1993 mRNAs with decreased expression (fold change ≥1.5 and p < 0.05).
The differentially expressed mRNAs were analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses to explore the general biological functional pathways in scleroderma of the mRNAs. GO analysis showed that the differentially upregulated mRNAs were significantly associated with immune system process (biological process), cellular anatomical entity (cellular component) and protein binding (molecular function; Supplementary Figure 3A). The differentially downregulated mRNAs were significantly associated with receptor cellular process (biological process), intracellular (cellular component) and binding (molecular function; Supplementary Figure 3B). As shown in Supplementary Figure 3C, the differentially upregulated mRNAs were most significantly associated with malaria, cell adhesion molecules, Kaposi sarcoma-associated herpes virus infection and cytokine–cytokine receptor interaction. The differentially downregulated mRNAs were most significantly associated with tight junction, Hippo signaling pathway, melanogenesis and basal cell carcinoma (Supplementary Figure 3D).
Compared with markers for skin fibrosis, the expression of markers for vascular damage and inflammation was increased in all tested samples, indicating a successful mouse model (). The top 20 differentially expressed mRNAs are listed in .
Table 1. Specific gene expression associated with scleroderma.
Table 2. The top 20 differentially expressed mRNAs in the mouse model of scleroderma.
The association analysis of m6A methylation and mRNA expression with fold change ≥1.5 revealed that the BLM group had 37 m6A-hypermethylated and upregulated mRNAs, 1164 m6A-hypermethylated and downregulated mRNAs, 27 m6A-hypomethylated and downregulated mRNAs and 358 m6A-hypomethylated and upregulated mRNAs (A). A study indicated that m6A methylation could affect the stability of mRNA [Citation41]. Following the steps described in that study, we found more hyper/down and hypo/up mRNAs than hyper/up and hypo/down mRNAs, suggesting that m6A methylation was negatively correlated with mRNA expression in the mouse model of scleroderma.
(A) The four-quadrant and Venn diagram showing the N6-methyladenosine-methylated and expressed mRNAs (fold change ≥1.5). (B) The protein–protein interaction network showed the connection between mRNAs in the combined analysis (fold change ≥2).
BLM: Bleomycin.
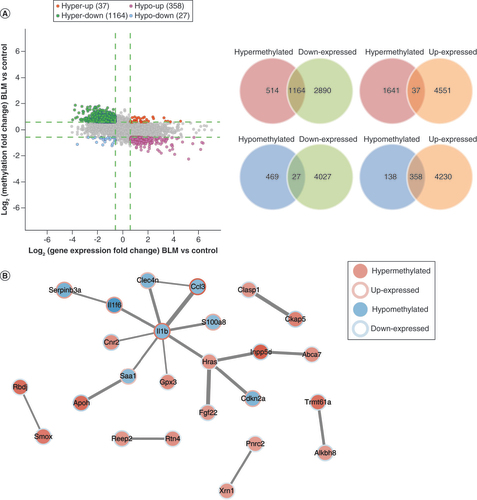
The protein–protein interaction network was established to show the connection between mRNAs (fold change ≥2) in the combined analysis (B).
Differentially m6A-methylated mRNAs were involved in important pathways
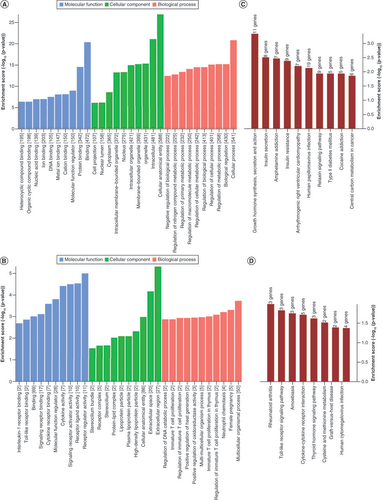
GO analysis showed that the differentially m6A-hypermethylated mRNAs were significantly associated with binding (biological process), cellular anatomical entity (cellular component) and cellular process (molecular function; A). In addition, the differentially m6A-hypomethylated mRNAs were significantly associated with receptor regulator activity (biological process), extracellular region (cellular component) and multicellular organismal process (molecular function; B). As shown in C, the differentially m6A-hypermethylated mRNAs were most significantly associated with growth hormone synthesis, secretion and action, insulin secretion and amphetamine addiction. In addition, the differentially m6A-hypomethylated mRNAs were most significantly associated with rheumatoid arthritis, Toll-like receptor signaling pathway and amoebiasis (D).
(A) The top ten Gene Ontology terms of hypermethylated mRNAs, ranked by enrichment score. (B) The top ten Gene Ontology terms of hypomethylated mRNAs, ranked by enrichment score. (C) The top ten significantly enriched pathways of hypermethylated mRNAs, ranked by enrichment score. (D) The top ten significantly enriched pathways of hypomethylated mRNAs, ranked by enrichment score.
Validation of the m6A epitranscriptomic microarray by MeRIP-qPCR & quantitative real-time PCR
To validate the m6A epitranscriptomic microarray data, we conducted MeRIP-qPCR for four hypermethylated mRNAs (Hras, Lama3, Stat3 and Tnc) and four hypomethylated mRNAs (Ccl3, Ccl9, Saa1 and Il1b). These have been preliminarily studied in fibrosis, but not deeply ().
Table 3. Review for the selected genes in scleroderma.
We selected hypermethylated mRNAs from the perspective of KEGG analyses. Because collagen deposition is the most important pathological process in scleroderma, mRNAs involved in the ECM–receptor interaction pathway were analyzed. We sorted fold change from high to low, and then picked two mRNAs (). Although COL4A1 ranked the second highest, we did not select this mRNA because it has received little attention in skin fibrosis compared with another two collagen proteins, COL1A1 and COL3A1, which have been intensively studied. Instead, we selected novel LAMA3 and TNC, both of which play relatively clear roles in skin fibrosis. As the pathways with the highest enrichment score, growth hormone synthesis, secretion and action were also selected. These mRNAs included Pik3cd, Cacna1s, Pik3r2, Ghrl, Atf6b and Atf2 (). Except for Pik3cd [Citation53] and Atf2 [Citation54,Citation55], the other four mRNAs are not involved in skin fibrosis. A report linking Pik3r2 with scleroderma [Citation56] has been published since the completion of our study. In contrast, there have been many studies on Stat3 in scleroderma, and there are two publications on DNA methylation [Citation57,Citation58]; hence we chose Stat3 for verification.
Table 4. Differentially hypermethylated mRNAs involved in the extracellular matrix–receptor interaction pathway.
Table 5. Differentially hypermethylated mRNAs involved in the growth hormone synthesis, secretion and action pathway.
We selected hypomethylated mRNAs from the perspective of GO/KEGG analyses. Scleroderma is an autoimmune disease, so we selected the duplicated mRNAs in cytokine–cytokine receptor interaction pathways and the inflammatory response (biological process), namely Il1f6, Il1b, Ccl3 and Ccl9. Although Il1f6 ranked the lowest, there are no publications related to its association with fibrosis. Thus we chose Saa1 because its fold change was second only to that of Ccl3 ( &).
Table 6. Differentially hypomethylated mRNAs involved in cytokine–cytokine receptor interaction pathways.
Table 7. Differentially hypomethylated mRNAs involved in inflammatory response (biological process).
As shown in A, the results of MeRIP-qPCR revealed a significant methylation increase in Hras, Lama3 and Tnc (but not Stat3: p = 0.052) and a significant methylation decrease in Ccl3, Ccl9, Saa1 and Il1b in the BLM group compared with the control group. As shown in B, these results showed an identical trend of m6A-modified mRNA changes between microarray and MeRIP-qPCR (except for Lama3 and Stat3). Fold change in Lama3 in the BLM group compared with the control group was 1.32, which was less than 1.5.
(A) The relative m6A level of the selected mRNAs was detected using MeRIP-qPCR. (B) The comparison between microarray data and MeRIP-qPCR of the selected mRNAs. (C) The six mRNAs were analyzed using qPCR. (D) The comparison between microarray data and qPCR of the selected mRNAs. (E) Identification of RNA binding proteins to the validated m6A-methylated mRNAs differentially. n = 4, each group. Mean ± standard deviation.
#No significance.
*p < 0.05; **p < 0.01; ***p < 0.001 compared with the control group.
BLM: Bleomycin; m6A: N6-methyladenosine; MeRIP: m6A-RNA-immunoprecipitation; qPCR: Quantitative PCR.
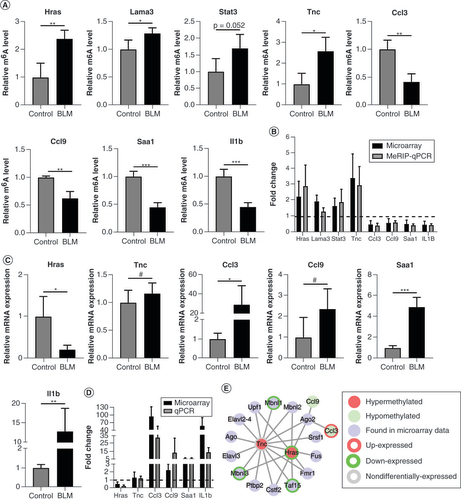
Next, we analyzed the mRNA expression of Hras, Tnc, Ccl3, Ccl9, Saa1 and Ilb1 to determine whether mRNA expression was regulated by m6A methylation. However, qPCR showed no statistically significant differences in the expression levels of Tnc and Ccl9 between the BLM and control groups (C). Compared with the control group, the m6A modification and mRNA expression of Hras, Ccl3, Saa1 and Ilb1 revealed the opposite trend in the BLM group. The results also showed identical trends of mRNA expression between microarray and qPCR results (D).
Finally, we used RMBase 2.0 to explore the potential RNA-binding proteins bound to the validated m6A-methylated mRNAs [Citation59]. As shown in E, Hras, Tnc, Ccl3 and Ccl9 had corresponding RNA-binding proteins. Among them, Mbnl1, Mbnl3 and Taf15 transcripts were also included in our microarray data, and their mRNA expression levels showed statistically significant differences between the BLM and control groups.
Discussion
Scleroderma is an autoimmune disease with a poor prognosis whose pathogenesis remains unclear. Compared with 0.03% incidence in the general population, the incidence of scleroderma in families with a history of scleroderma is 1.5% [Citation60], revealing that heredity is a significant risk factor. Several studies have shown that scleroderma is closely related to epigenetics, and the main research directions involve the investigation of post-translational modifications, DNA methylation, mRNAs and lncRNAs [Citation61]. However, the RNA m6A modification landscape in scleroderma remains largely unknown. This study elucidated the m6A landscape in the BLM-induced mouse model of scleroderma. First, we demonstrated that BLM induced skin collagen deposition and inflammation in mice. Next, we performed m6A epitranscriptomic microarray studies to identify differentially methylated and expressed mRNAs. GO and KEGG pathway analyses of differentially m6A-methylated genes identified their potential roles in physiological and pathological mechanisms. Subsequently, we performed an association analysis of m6A methylation and mRNA expression. Finally, we verified the candidate mRNAs via MeRIP-qPCR and qPCR. The selected methyltransferases and demethylases were verified using qPCR. The above findings better our understanding of the role of RNA m6A methylation in scleroderma.
KEGG analysis revealed that the differentially hypermethylated m6A mRNAs were enriched in pathways involved in diabetes, such as insulin secretion, insulin resistance and Type II diabetes mellitus. A nationwide cohort study in Taiwan found that the incidence of diabetes was 10.02% in 2671 scleroderma patients, compared to 24.81% in 7769 controls [Citation62]. A similar study in Minnesota, USA reported that the incidence of diabetes was 2.56% in 78 scleroderma patients, compared to 12.18% in 125 non-scleroderma patients [Citation63]. These two studies revealed epidemiologically a lower prevalence of diabetes in scleroderma patients. Some studies have identified that an increase of IL-10 [Citation64,Citation65] and IL-13 [Citation66,Citation67] led to immune system abnormalities in scleroderma, which was a protective factor for diabetes. TNF-related apoptosis-inducing ligand (TRAIL) is decreased in diabetic patients, and the inhibition of TRAIL intensifies the apoptosis of pancreatic β cells; TRAIL is elevated in scleroderma and might be beneficial for scleroderma [Citation68]. Metformin, a routine treatment for diabetes, could also reduce skin fibrosis and inhibit inflammatory cytokines such as IL-1β, IL-6, IL-17 and TNF-α [Citation69]. A new technique of metabolomic profiling has found that the excessive deposition of ECM depends on metabolic disturbances [Citation70]. Hesperidin 165 (NCT03377140) [Citation71] and Omacor 166 (NCT03018041) [Citation72] have been identified as candidate antifibrotic drugs given their effects in regulating glucose metabolism. As differential mRNAs were enriched in the pathways of diabetes, we speculate that m6A methylation is related to the above-mentioned relationship between diabetes and scleroderma.
Unlike the hypermethylated mRNAs, the differentially hypomethylated m6A mRNAs were enriched in the pathways of immune regulation, such as rheumatoid arthritis, Toll-like receptor signaling pathway, cytokine–cytokine receptor interaction and graft-versus-host disease, revealing that m6A methylation plays a role in immunoregulatory alteration. For example, Saa1 plays the following key role in inflammation and fibrosis: Saa1 knockdown can suppress the process of macrophage-to-myofibroblast transition, which can inhibit renal interstitial fibrosis [Citation51]. Elevated SAA1 is associated with a fibrotic pulmonary phenotype [Citation73]. Different plasma levels of SAA1 were related to different grades of inflammation and stages of fibrosis in the livers of patients with autoimmune hepatitis [Citation74]. In our study, Saa1 was enriched in inflammatory response (biological process) and was highly expressed in skin fibrosis, similar to the above findings, and was differentially hypermethylated, indicating that methylation may be the cause of its role in fibrosis.
As shown in Supplementary Figure 4, a KEGG network showed some critical pathways in scleroderma, which are mainly involved in immune response and cell proliferation. Further studies are warranted to explain the exact pathogenesis.
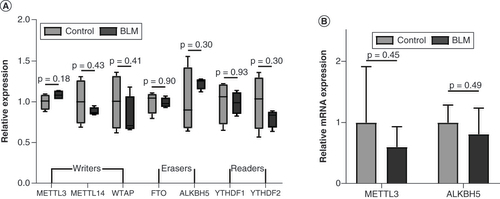
m6A methylation is a reversible process, dynamically regulated by key m6A enzymes. However, in the present study the expression levels of methyltransferases (writers), demethylases (erasers) and binding proteins (readers) were unaltered between the control group and BLM group (A). There were no statistically significant differences in the expression levels of Mettl3 and Alkbh5 between the BLM and control groups (B). Our sample size may be too small, warranting further studies with larger sample sizes to explore the reasons underlying m6A methylation alterations in the mouse model of scleroderma. In addition, no other studies have investigated the involvement of these key enzymes in scleroderma, making comparison of our results difficult. Therefore functional experiments involving the knockdown/overexpression of key enzymes are needed to confirm the regulatory role of m6A methylation in scleroderma.
(A) The relative expression of writers, erasers and readers in microarray data. (B)Mettl3 and Alkbh5 were analyzed using quantitative PCR. n = 4, each group. Mean ± standard deviation.
BLM: Bleomycin.
Conclusion
Our study reports on differentially m6A-methylated mRNAs in a BLM-induced mouse model of scleroderma, demonstrating a potential strong connection between RNA m6A methylation and the regulation of fibrosis and inflammation. This study hence provides novel insights into the pathogenesis of scleroderma.
Future perspective
Our study revealed alterations in Hras, Lama3, Stat3, Tnc, Ccl3, Ccl9, Saa1 and Il1b, which require further investigation. These mRNAs may play a critical role in epigenetic mechanisms, contributing to the identification of diagnostic markers and therapeutic targets for scleroderma.
A total of differentially 733 hypermethylated N6-methyladenosine (m6A) mRNAs and 110 hypomethylated m6A mRNAs were identified in the bleomycin-induced mouse model of scleroderma.
The hypermethylated m6A mRNAs were enriched in pathways of diabetes, such as insulin secretion, insulin resistance and Type II diabetes mellitus and growth hormone synthesis, secretion and action.
The hypomethylated m6A mRNAs were enriched in pathways of immunoregulation, such as rheumatoid arthritis, Toll-like receptor signaling pathway, cytokine–cytokine receptor interaction and graft-versus-host disease.
m6A-RNA-immunoprecipitation quantitative PCR (qPCR) verified that Hras and Tnc were hypermethylated m6A mRNAs, while Ccl3, Ccl9, Saa1 and Il1b were hypomethylated m6A mRNAs. qPCR verified that Hras was upregulated, while Ccl3, Saa1 and Ilb1 were downregulated.
Using RMBase 2.0, MBNL1, MBN3 and TAF15 were predicted to be RNA-binding proteins of Hras and Tnc.
There were no statistically significant differences in the expression levels of methyltransferases, demethylases or binding proteins between the control and bleomycin-treated groups.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all animal experimental investigations, and all experiments were approved and performed following the guidelines of the Tongji University Ethic committee of China (no. TJ-HB-LAC-2020-83).
Supplemental Document
Download Zip (6 MB)Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2021-0369
Financial & competing interests disclosure
This work is supported by the grants from the National Natural Science Foundation of China, which is a subsidiary of the Ministry of Science and Technology, PRC. The General Program number is 81874240. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was provided by Enago and was funded by the National Natural Science Foundation of China (program no. 81874240).
Additional information
Funding
References
- Denton CP , KhannaD. Systemic sclerosis. Lancet390(10103), 1685–1699 (2017).
- Knobler R , MoinzadehP , HunzelmannNet al. European Dermatology Forum S1 guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 1: localized scleroderma, systemic sclerosis and overlap syndromes. J. Eur. Acad. Dermatol. Venereol.31(9), 1401–1424 (2017).
- Gabrielli A , AvvedimentoEV , KriegT. Scleroderma. N. Engl. J. Med.360(19), 1989–2003 (2009).
- Royle JG , LanyonPC , GraingeMJ , AbhishekA , PearceFA. The incidence, prevalence, and survival of systemic sclerosis in the UK Clinical Practice Research Datalink. Clin. Rheumatol.37(8), 2103–2111 (2018).
- Cottin V , BrownKK. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir. Res.20(1), 13 (2019).
- Giuggioli D , ManfrediA , LumettiF , ColaciM , FerriC. Scleroderma skin ulcers definition, classification and treatment strategies our experience and review of the literature. Autoimmun. Rev.17(2), 155–164 (2018).
- Bhattacharyya S , WeiJ , VargaJ. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol.8(1), 42–54 (2011).
- Hinchcliff M , O’ReillyS. Current and potential new targets in systemic sclerosis therapy: a new hope. Curr. Rheumatol. Rep.22(8), 42 (2020).
- Cui Z , HuangN , LiuLet al. Dynamic analysis of m6A methylation spectroscopy during progression and reversal of hepatic fibrosis. Epigenomics12(19), 1707–1723 (2020).
- Tsou PS , SawalhaAH. Unfolding the pathogenesis of scleroderma through genomics and epigenomics. J. Autoimmun.83, 73–94 (2017).
- Altorok N , KahalehB. Epigenetics and systemic sclerosis. Semin. Immunopathol.37(5), 453–462 (2015).
- Tsou PS , VargaJ , O’ReillyS. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat. Rev. Rheumatol.17(10), 596–607 (2021).
- Cantara WA , CrainPF , RozenskiJet al. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res.39(Database issue), D195–D201 (2011).
- Globisch D , PearsonD , HienzschAet al. Systems-based analysis of modified tRNA bases. Angew. Chem. Int. Ed. Engl.50(41), 9739–9742 (2011).
- Boccaletto P , MachnickaMA , PurtaEet al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res.46(D1), D303–d307 (2018).
- Hu J , LinY. Fusarium infection alters the m6A-modified transcript landscape in the cornea. Exp. Eye Res.200, 108216 (2020).
- Zaccara S , RiesRJ , JaffreySR. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol.20(10), 608–624 (2019).
- Li X , FanX , YinX , LiuH , YangY. Alteration of N6-methyladenosine epitranscriptome profile in unilateral ureteral obstructive nephropathy. Epigenomics12(14), 1157–1173 (2020).
- Shi H , WeiJ , HeC. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell74(4), 640–650 (2019).
- Dominissini D , Moshitch-MoshkovitzS , SchwartzSet al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature485(7397), 201–206 (2012).
- Wei CM , GershowitzA , MossB. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell4(4), 379–386 (1975).
- Lan Q , LiuPY , HaaseJ , BellJL , HüttelmaierS , LiuT. The critical role of RNA m6A methylation in cancer. Cancer Res.79(7), 1285–1292 (2019).
- Saneyoshi M , HaradaF , NishimuraS. Isolation and characterization of N6-methyladenosine from Escherichia coli valine transfer RNA. Biochim. Biophys. Acta190(2), 264–273 (1969).
- Iwanami Y , BrownGM. Methylated bases of ribosomal ribonucleic acid from HeLa cells. Arch. Biochem. Biophys.126(1), 8–15 (1968).
- Meyer KD , SaletoreY , ZumboP , ElementoO , MasonCE , JaffreySR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell149(7), 1635–1646 (2012).
- Chen C , WangD , MoshaveriniaAet al. Mesenchymal stem cell transplantation in tight-skin mice identifies miR-151-5p as a therapeutic target for systemic sclerosis. Cell Res.27(4), 559–577 (2017).
- Wermuth PJ , Piera-VelazquezS , RosenbloomJ , JimenezSA. Existing and novel biomarkers for precision medicine in systemic sclerosis. Nat. Rev. Rheumatol.14(7), 421–432 (2018).
- Henry TW , MendozaFA , JimenezSA. Role of microRNA in the pathogenesis of systemic sclerosis tissue fibrosis and vasculopathy. Autoimmun. Rev.18(11), 102396 (2019).
- Atarod S , NordenJ , BibbyLAet al. Differential microRNA expression levels in cutaneous acute graft-versus-host disease. Front. Immunol.9, 1485 (2018).
- Wang Y , MaoJ , WangXet al. Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics11(7), 805–819 (2019).
- Ma JZ , YangF , ZhouCCet al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary microRNA processing. Hepatology65(2), 529–543 (2017).
- Zhang C , ZhiWI , LuHet al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget7(40), 64527–64542 (2016).
- He Y , HuH , WangYet al. ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell. Physiol. Biochem.48(2), 838–846 (2018).
- Zhang JX , HuangPJ , WangDPet al. m6A modification regulates lung fibroblast-to-myofibroblast transition through modulating KCNH6 mRNA translation. Mol. Ther. doi:10.1016/j.ymthe.2021.06.008 (2021) ( Epub ahead of print).
- Mathiyalagan P , AdamiakM , MayourianJet al. FTO-dependent N6-methyladenosine regulates cardiac function during remodeling and repair. Circulation139(4), 518–532 (2019).
- Wang YN , JinHZ. Transcriptome-wide m6A methylation in skin lesions from patients with psoriasis vulgaris. Front. Cell Dev. Biol.8, 591629(2020).
- Luo Q , FuB , ZhangL , GuoY , HuangZ , LiJ. Decreased peripheral blood ALKBH5 correlates with markers of autoimmune response in systemic lupus erythematosus. Dis. Markers2020, 8193895 (2020).
- Luo Q , RaoJ , ZhangLet al. The study of METTL14, ALKBH5, and YTHDF2 in peripheral blood mononuclear cells from systemic lupus erythematosus. Mol. Genet. Genomic Med.8(9), e1298 (2020).
- Jiang H , CaoK , FanC , CuiX , MaY , LiuJ. Transcriptome-wide high-throughput m6A sequencing of differential m6A methylation patterns in the human rheumatoid arthritis fibroblast-like synoviocytes cell line MH7A. J. Inflamm. Res.14, 575–586 (2021).
- Lagares D , HinzB. Animal and human models of tissue repair and fibrosis: an introduction. Methods Mol. Biol.2299, 277–290 (2021).
- Geula S , Moshitch-MoshkovitzS , DominissiniDet al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science347(6225), 1002–1006 (2015).
- Marut W , KavianN , ServettazAet al. Amelioration of systemic fibrosis in mice by angiotensin II receptor blockade. Arthritis Rheum.65(5), 1367–1377 (2013).
- Pesch M , KönigS , AumailleyM. Targeted disruption of the Lama3 gene in adult mice is sufficient to induce skin inflammation and fibrosis. J. Invest. Dermatol.137(2), 332–340 (2017).
- Moon SJ , BaeJM , ParkKS , TagkopoulosI , KimKJ. Compendium of skin molecular signatures identifies key pathological features associated with fibrosis in systemic sclerosis. Ann. Rheum. Dis.78(6), 817–825 (2019).
- Ummarino D . Systemic sclerosis: tenascin C perpetuates tissue fibrosis. Nat. Rev. Rheumatol.12(7), 375 (2016).
- Brissett M , VeraldiKL , PilewskiJM , MedsgerTAJr , Feghali-BostwickCA. Localized expression of tenascin in systemic sclerosis-associated pulmonary fibrosis and its regulation by insulin-like growth factor binding protein 3. Arthritis Rheum.64(1), 272–280 (2012).
- McHugh J . Systemic sclerosis: STAT3 – a key integrator of profibrotic signalling. Nat. Rev. Rheumatol.13(12), 693 (2017).
- O’Reilly S , CiechomskaM , CantR , Van LaarJM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem.289(14), 9952–9960 (2014).
- Lim JY , RyuDB , LeeSE , ParkG , MinCK. Mesenchymal stem cells (MSCs) attenuate cutaneous sclerodermatous graft-versus-host disease (Scl-GVHD) through inhibition of immune cell infiltration in a mouse model. J. Invest. Dermatol.137(9), 1895–1904 (2017).
- Brass DM , McgeeSP , DunkelMKet al. Gender influences the response to experimental silica-induced lung fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol.299(5), L664–671 (2010).
- Feng Y , GuoF , XiaZet al. Inhibition of fatty acid-binding protein 4 attenuated kidney fibrosis by mediating macrophage-to-myofibroblast transition. Front. Immunol.11, 566535 (2020).
- Pendergrass SA , HayesE , FarinaGet al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE5(8), e12106 (2010).
- Paz K , FlynnR , DuJet al. Targeting PI3Kδ function for amelioration of murine chronic graft-versus-host disease. Am. J. Transplant.19(6), 1820–1830 (2019).
- Akiyama Y , OgawaF , IwataYet al. Autoantibody against activating transcription factor-2 in patients with systemic sclerosis. Clin. Exp. Rheumatol.27(5), 751–757 (2009).
- Daian T , OhtsuruA , RogounovitchTet al. Insulin-like growth factor-I enhances transforming growth factor-beta-induced extracellular matrix protein production through the P38/activating transcription factor-2 signaling pathway in keloid fibroblasts. J. Invest. Dermatol.120(6), 956–962 (2003).
- Wang Y , SunJ , KahalehB. Epigenetic down-regulation of microRNA-126 in scleroderma endothelial cells is associated with impaired responses to VEGF and defective angiogenesis. J. Cell. Mol. Med.25(14), 7078–7088 (2021).
- Dees C , PötterS , ZhangYet al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Invest.130(5), 2347–2363 (2020).
- Coit P , SchollaertKL , MirizioEM , TorokKS , SawalhaAH. DNA methylation patterns in juvenile systemic sclerosis and localized scleroderma. Clin. Immunol.228, 108756 (2021).
- Xuan JJ , SunWJ , LinPHet al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res.46(D1), D327–d334 (2018).
- Tsou PS , VargaJ , O’ReillyS. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat. Rev. Rheumatol.17(10), 596–607 (2021).
- Angiolilli C , MarutW , VanDer Kroef M , ChouriE , ReedquistKA , RadstakeT. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol.14(11), 657–673 (2018).
- Tseng CC , ChangSJ , TsaiWCet al. Reduced incidence of Type 1 diabetes and Type 2 diabetes in systemic sclerosis: a nationwide cohort study. Joint Bone Spine83(3), 307–313 (2016).
- Kurmann RD , SandhuAS , CrowsonCSet al. Cardiovascular risk factors and atherosclerotic cardiovascular events among incident cases of systemic sclerosis: results from a population-based cohort (1980–2016). Mayo Clin. Proc.95(7), 1369–1378 (2020).
- Dziankowska-Bartkowiak B , ZalewskaA , Sysa-JedrzejowskaA. Duration of Raynaud’s phenomenon is negatively correlated with serum levels of interleukin 10 (IL-10), soluble receptor of interleukin 2 (sIL2R), and sFas in systemic sclerosis patients. Med. Sci. Monit.10(5), Cr202–208 (2004).
- Kikodze N , PantsulaiaI , RekhviashviliKet al. Cytokines and T regulatory cells in the pathogenesis of Type 1 diabetes. Georgian Med. News (222), 29–35 (2013).
- Vettori S , CuomoG , IudiciMet al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J. Clin. Immunol.34(6), 663–668 (2014).
- Rasche SS , PhillipsM , McInerneyMF , SercarzEE , QuinnA. IL-13Rα1 expression on β-cell-specific T cells in NOD mice. Diabetes60(6), 1716–1725 (2011).
- Kang S , ParkSY , LeeHJ , YooYH. TRAIL upregulates decoy receptor 1 and mediates resistance to apoptosis in insulin-secreting INS-1 cells. Biochem. Biophys. Res. Commun.396(3), 731–735 (2010).
- Moon J , LeeSY , ChoiJWet al. Metformin ameliorates scleroderma via inhibiting Th17 cells and reducing mTOR-STAT3 signaling in skin fibroblasts. J. Transl. Med.19(1), 192 (2021).
- Kang YP , LeeSB , LeeJMet al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J. Proteome Res.15(5), 1717–1724 (2016).
- Jung UJ , LeeMK , JeongKS , ChoiMS. The hypoglycemic effects of hesperidin and naringin are partly mediated by hepatic glucose-regulating enzymes in C57BL/KsJ-db/db mice. J. Nutr.134(10), 2499–2503 (2004).
- Zhang Z , LiuH , LiuJ. Akt activation: a potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract.156, 107092 (2019).
- Beijer E , Roodenburg-BenschopC , SchimmelpenninkMC , GruttersJC , MeekB , VeltkampM. Elevated serum amyloid a levels are not specific for sarcoidosis but associate with a fibrotic pulmonary phenotype. Cells10(3), 585 (2021).
- Wang H , YanW , FengZet al. Plasma proteomic analysis of autoimmune hepatitis in an improved AIH mouse model. J. Transl. Med.18(1), 3 (2020).