
Abstract
Aims & methods: The aim of this study was to characterize the methylation level of a polymorphically imprinted gene, VTRNA2-1/nc886, in human populations and somatic tissues.48 datasets, consisting of more than 30 tissues and >30,000 individuals, were used. Results:nc886 methylation status is associated with twin status and ethnic background, but the variation between populations is limited. Monozygotic twin pairs present concordant methylation, whereas ∼30% of dizygotic twin pairs present discordant methylation in the nc886 locus. The methylation levels of nc886 are uniform across somatic tissues, except in cerebellum and skeletal muscle. Conclusion: The nc886 imprint may be established in the oocyte, and, after implantation, the methylation status is stable, excluding a few specific tissues.
Tweetable abstract
Methylation status of a polymorphically imprinted gene, VTRNA2-1/nc886, is stable in human populations (48 cohorts, n > 30,000) and in somatic tissues, except in cerebellum and skeletal muscle. Twin data suggest it may already be established in the oocyte.
Genomic imprinting can be defined as the expression of a gene from only the maternal or the paternal allele, while the corresponding allele in the other chromosome is silenced via epigenetic mechanisms, including DNA methylation [Citation1]. The epigenetic profiles maintaining the imprinted status are established during gametogenesis when the DNA methylation pattern is erased, and then the parent-of-origin-related DNA methylation pattern is created. For males, the DNA methylation profile of the sperm, including imprints, is completed in the primordial germ cells, before the birth of the male child. On the other hand, in oocytes, de novo DNA methylation will begin only after the birth of the female child, during the follicular growth phase, with gene-specific timepoints for imprinting reported [Citation2–4]. Canonically imprinted genes, approximately 130 of which exist in humans, retain the parent-of-origin-related expression pattern throughout an individual’s life in all of their somatic tissues, although tissue or developmental stage-specific imprinting can be seen, for example, in the placenta [Citation5–7]. The significance of intact genetic imprints is highlighted by the severe disorders caused by imprinting defects [Citation8].
In humans, the locus harboring noncoding 886 (nc886, also known as VTRNA2-1) is polymorphically imprinted, with approximately 75% of individuals having a methylated maternal allele (i.e., imprinted nc886 locus) and the remaining ~25% having both maternal and paternal nc886 allele unmethylated [Citation9–11]. According to current literature, this pattern is not due to genetic variation [Citation9,Citation12,Citation13]. We have also previously identified individuals who escape this bimodal methylation pattern. We found they present either intermediate methylation levels (methylation beta value 0.20–0.40; i.e., methylation level of 20–40%, in approximately 1–5% of the population) or methylation beta values >0.60 (methylation level of >60%), indicating that also the paternal allele has gained methylation in somatic tissues (in ~0.1% of the population) [Citation11]. The nc886 locus, flanked by two CTCF-binding sites, suggested to be important for its imprinting [Citation14], codes for a 102nt long noncoding RNA, which might then be cleaved into miRNA-like short noncoding RNAs, although the nature of these RNAs is still widely debated [Citation15,Citation16].
The establishment of the nc886 imprint has been suggested to be an early event in the zygote, happening between 4 and 6 days after fertilization [Citation13]. Recently, it was suggested that the methylation pattern of nc886 is already established in the preconceptional oocyte [Citation10]. Early establishment of nc886 methylation status is supported by the fact that it has been shown to be uniform across analyzed somatic tissues [Citation10,Citation17,Citation18]. Given that the methylation pattern of nc886 had been reported to be concordant in monozygotic twin pairs (MZ, 97 twin pairs) but not in dizygotic twin pairs (DZ, 162 twin pairs), genetic factors were hypothesized to influence the methylation pattern [Citation9], which was later shown not to be the case [Citation10,Citation11,Citation19]. Once established, the methylation status of nc886 is stable from childhood to adolescence [Citation17] and from adolescence to adulthood [Citation11].
Changes in the proportion of individuals with methylated or unmethylated maternal nc886 allele have been associated with maternal age [Citation9,Citation11], maternal socioeconomic status [Citation11] and maternal alcohol consumption [Citation10], as well as the season of conception in rural Gambia [Citation9,Citation17]. Furthermore, the methylation level or status of nc886 or level of nc886 RNAs transcribed from the locus have been associated with childhood BMI [Citation20], adiposity and cholesterol levels [Citation11], as well as allergies [Citation21], asthma [Citation22], infections [Citation23] and inflammation [Citation24]. Interestingly, the nc886 methylation status and the level of nc886 RNAs have also been associated with indicators of glucose metabolism [Citation11,Citation25]. These findings suggest that the methylation status of nc886 is a potential molecular mediator of the developmental origins of health and disease (DOHaD) hypothesis (also known as the Barker hypothesis) [Citation26].
Detailed understanding of the determinants and functions of the methylation status of nc886 is still lacking. In vitro methods are of limited feasibility, as both carcinogenesis [Citation13] and pluripotency induction [Citation11] have an effect on the methylation pattern at the nc886 locus. Currently, no animal models are available to study nc886 because most species, including mice and rats, do not harbor the gene, and in species harboring the nc886 gene, the locus is not polymorphically imprinted [Citation14]. Thus, we wanted to use available resources – the numerous existing DNA methylation datasets from humans – to gain insight on the methylation of nc886, a unique polymorphically imprinted gene and a potential molecular mediator of DOHaD. Furthermore, patterns of imprinting of nc886 across populations and tissues could reveal factors important to the establishment and maintenance of other maternally imprinted genes in humans.
More precisely, we wanted to investigate the prevalence of nc886 DNA methylation status groups in a large number of human populations (with a total n > 30,000) with divergent historical and geographic origins to identify the factors associating with the existing variation; the nc886 methylation status in MZ and DZ twin pairs to elucidate the contribution of shared gametes versus unique gametes in shared prenatal environment to the establishment of nc886 methylation pattern; and the nc886 methylation patterns in a larger variety of tissues, including brain regions and placenta, which have been shown to express a multitude of imprinted genes, as well as present dynamic imprinting [Citation5,Citation27,Citation28] to have insights on the potential function of nc886 and the stability of this polymorphic imprint in human tissues.
Materials & methods
Datasets
This study used 48 DNA methylation datasets, including DILGOM, FTC, ERMA, KORA, LURIC, NELLI, SATSA and YFS as well as 39 datasets available in the Gene Expression Omnibus (GEO) [Citation29] consisting of >30 tissues and >30,000 individuals. (Supplementary Table 1). These datasets were used to study the methylation of nc886 locus across populations, in twin pairs and across different tissues, with some datasets used in multiple settings. Datasets from GEO were retrieved between January 2021 and August 2022. Of these datasets, for the population analyses, datasets with n > 300 were included, and cancerous tissues were excluded. For tissue analyses, cancerous tissues and cell culture experiments were excluded. For all included datasets, DNA methylation analysis was performed with either Illumina Infinium MethylationEPIC or Methylation450K BeadChip.
For the population analysis, 32 datasets were included, with the number of individuals ranging from 131 to 2711 (total n = 30,347). In these datasets, the DNA methylation data was available from blood [Citation11,Citation30–48], separated peripheral blood cells [Citation49–51], blood spots [Citation20], umbilical cord buffy coats [Citation52], fetal cord tissue [Citation53] or buccal swabs [Citation52]. Association of zygosity (MZ vs DZ pairs) with nc886 methylation was analyzed in five datasets [Citation36,Citation47,Citation48,Citation54,Citation55]. A dataset containing infants with multilocus methylation disturbance (MLID), single locus imprinting disorder (SLID) and controls [Citation56] were included to analyze the effect of MLID on nc886 methylation.
To analyze the methylation of nc886 across different tissues, 17 datasets were used. These included a dataset consisting of 30 tissues from a 112-year-old female [Citation57], as well as datasets consisting of different brain regions [Citation58] (GSE134379), adipose tissue [Citation54,Citation59], muscle [Citation46,Citation55,Citation56,Citation57,Citation60], (GSE142141, GSE171140), liver [Citation59], buccal swabs [Citation52,Citation62], skin [Citation63], sperm [Citation64,Citation65] and placenta [Citation66–69]. All datasets are described in detail in Supplementary Table 1.
DILGOM [Citation44] was collected as an extension of the FINRISK 2007 survey. FINRISK surveys consist of cross-sectional, population-based studies conducted to monitor the risk of chronic diseases in Finland [Citation70]. The data used for the research were obtained from the THL biobank (study no. THLBB2021_22).
The FTC study includes three longitudinal cohorts [Citation71,Citation72]: the Older Twin cohort [Citation72], FinnTwin16 (FT16) [Citation73] and FinnTwin12 (FT12) [Citation74]. In this study, two subsets of FTC samples were included: a smaller subset of individuals from FT12 and FT16 for whom DNA methylation data was available from muscle, adipose tissue and blood [Citation61] and a larger subset including individuals from the Older, FT16 and FT12 cohorts with methylation data only available from blood [Citation54]. For 49 MZ twin pairs, the information on whether they were dichorionic and diamniotic (DCDA, 22 pairs), monochorionic and diamniotic (MCDA, 9 pairs) or monochorionic and monoamniotic (MCMA, 18 pairs) was available. Participants of FTC were born before 1988 in Finland.
ERMA was a prospective cohort study, the aim of which was to reveal how hormonal differences over the menopausal stages in middle-aged females affect their physiological and psychological functioning [Citation75]. The cohort includes 47- to 55-year-old females from city of Jyväskylä or neighboring municipalities in Finland. For this study, a subset of 47 individuals were included from whom whole blood and muscle tissue samples were available [Citation61].
The KORA cohorts were collected as a series of population-based epidemiological surveys and follow-up examinations in the region of Augsburg and two adjacent counties in southern Germany [Citation76]. Here we have included KORA-F4 (2006/2007) and FF4 (2013/2014) cohorts, both of which are follow-up studies of the KORA Survey S4, conducted in 1999–2001.
The LURIC cohort includes patients of German ancestry from a tertiary care centre in southwestern Germany who underwent coronary angiography between 1997 and 2000 [Citation77]. For this study, 2423 individuals with DNA methylation data were included in the present work.
The NELLI cohort consists of pregnant mothers participating in an intervention study aimed at preventing gestational diabetes from 14 municipalities in southern Finland [Citation78]. Included in this study are data from children of these mothers, collected at a 7-year follow-up [Citation62]. The children participating in this study were born in 2007–2008 in Finland
The SATSA cohort is drawn from the Swedish Twin Registry and includes same-sex pairs of twins who were reared together and pairs who were separated before age 11, collected between 1984 and 2014 [Citation79], 478 of whom had DNA methylation data available.
The YFS is a multicenter follow-up study on cardiovascular risk from childhood to adulthood, launched in 1980 [Citation80]. DNA methylation data used here are from the 30-year follow-up in 2011, including 1714 individuals. The participants in this study were born between 1962 and 1977 in Finland.
DNA methylation data processing
The majority of datasets in GEO were available as processed data, and these datasets were used as such. Datasets GSE61454, GSE71678, GSE134379 and GSE157896 were downloaded as raw idat-files, extracted with minfi package function read.metharray.exp and quantile normalized with default settings [Citation81]. For DILGOM [Citation44], FTC [Citation54,Citation61], ERMA [Citation61], KORA [Citation11,Citation45], LURIC [Citation46], SATSA [Citation47] and YFS [Citation11], the processing of DNA methylation data has been described in detail in previous publications referenced here. For all datasets, information on sample material, Illumina array type (MethylationEPIC or Methylation450K BeadChip) and the processing methods used are provided in Supplementary Table 1.
For NELLI, genomic DNA from buccal swabs was extracted with Gentra Buccal Cell Kit (QIAGEN, cat. no. 158845) and stored at -20°C. DNA in 1-μg aliquots were subjected to bisulphite conversion, after which a 4-μl aliquot of this bisulphite-converted DNA was subjected to whole-genome amplification. This was followed by enzymatic fragmentation and hybridization onto an Illumina Infinium MethylationEPIC BeadChip at Helmholtz Zentrum (Munich, Germany). The arrays were scanned with the iScan reader (Illumina). For all the samples, the sum of detection p-values across all the probes was <0.01. Log2-transformed median of methylated and unmethylated intensities of the analyzed samples were inspected visually, and these clustered well, except for one outlier that was removed from further analyses. Samples were checked for discrepancies between the reported and the predicted sex, and none failed the test. Normalization was done with a stratified quantile normalization method with preprocessQuantile function from minfi R/Bioconductor package [Citation81,Citation82]. Probes were filtered out if the detection p-value was >0.01 in 99% of the samples, if they were classified as cross-reactive or were associated with an SNP [Citation83,Citation84]. All aforementioned procedures were performed with the minfi R/Bioconductor package [Citation81].
Clustering of individuals according to nc886 locus methylation level
For all the included datasets, the methylation values for 14 CpGs (cg07158503, cg04515200, cg13581155, cg11978884, cg11608150, cg06478886, cg04481923, cg18678645, cg06536614, cg25340688, cg26896946, cg00124993, cg08745965 and cg18797653) shown to display bimodal DNA methylation pattern in the nc886 locus were retrieved [Citation9,Citation11]. See for the structure of nc886 locus. For some datasets, the methylation data were not available from all 14 CpGs. The number of probes available for each dataset is provided in Supplementary Table 1.
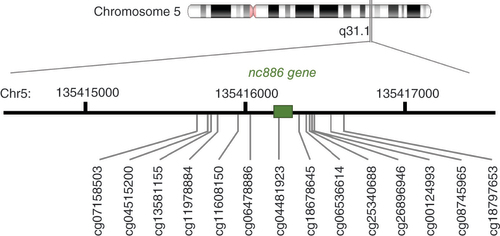
On the basis of the methylation levels of the 14 CpGs, individuals were clustered into three groups by k-means clustering; on the basis of the median methylation level of the cluster, nc886 methylation status groups were defined as imprinted (typical methylation β 0.40–0.60, indicative of monoallelic methylation), nonmethylated (typical methylation β < 0.15, indicative of two unmethylated alleles) and intermediately methylated (typical methylation β = 0.15–0.40). Data were visualized to verify that the clustering had identified the groups as expected. The intermediately methylated individuals could not be detected in all datasets. In the datasets in which they were identified, the proportion ranged from 2 to 6%.
To verify that the clustering is reproducible across different datasets, the clustering results within one dataset processed were compared with different methods. It was established that although the imprinted group could be reliably identified across normalization methods, there were some inconsistencies between intermediately methylated individuals and nonmethylated individuals (Supplementary Data 1). Therefore, for analyses in which proportions of nc886 status groups were investigated across datasets, the intermediately methylated and nonmethylated clusters were combined; that is, nc886 status is described as ‘imprinted’ or as ‘other’, the latter group including both intermediately methylated and nonmethylated individuals. Only when comparing individuals within a dataset – namely, in the case of MZ twin pairs, are all three status groups in the analyses kept together.
Comparison to established imprinted genes
When analyzing different tissues, the methylation level of six known imprinted genes (DIRAS3, KCNQ10T1, MEG3, MEST, NAP1L5, PEG10 and ZNF597) were also examined [Citation85]. This was done in tissues not presenting bimodal nc886 pattern to ensure that imprinted genes, in general, do not display atypical methylation patterns in these tissues.
Statistical analyses
Differences in the proportion of individuals with imprinted nc886 locus between sexes or in a case–control setting (depression [GSE125105], assisted reproductive technologies [GSE157896], rheumatoid arthritis [GSE42861], gestational diabetes mellitus [GSE141065], schizophrenia [GSE80417, GSE84727], inflammatory bowel disease [GSE87648], childhood abuse [GSE132203] and Parkinson disease [GSE111629]) were investigated with the χ2 test, with a threshold for significance set at p < 0.05 (Supplementary Table 2). For DZ twin pairs, the mathematically estimated proportion of miss-matched pairs (i.e., one twin imprinted, one intermediately methylated or nonmethylated) was calculated. The difference between the observed number of miss-matched pairs and the estimated numbers was then investigated with the χ2 test, with a threshold for significance set at p < 0.05.
In tissues not presenting bimodal nc886 methylation pattern, the difference in methylation levels between imprinted, and other groups (as clustered according to a tissue presenting the expected methylation pattern from the same individuals) were analyzed, with Mann–Whitney U-test between the median methylation levels of the 14 CpG sites with the threshold for significance set at p < 0.05. The median methylation levels in the nc886 locus were correlated between different tissues with Spearman correlation, with the threshold for significance set at p < 0.05.
Results
Methylation status of nc886 across population cohorts
The methylation status of the nc886 locus was characterized in 32 cohorts consisting of 30,347 individuals in total. In the majority of the cohorts, the participants were described as being of European descent, White or Caucasian, hereafter referred to as White. In the included datasets, DNA methylation data was from whole blood, blood cells or blood cell subtypes, buccal swabs or fetal cord tissue (Supplementary Table 2). In these tissues, the methylation level of the nc886 locus followed the expected bimodal pattern, and thus the individuals could be clustered into nc886 methylation status groups (Supplementary Figure 1). Across all datasets, the proportion of imprinted individuals (individuals with the methylation level indicative of monoallelic methylation) varied between 65.8 and 83.5%, with an average percentage of 75.3% ( & Supplementary Table 2). When considering only cohorts consisting of White singletons, the proportion of imprinted individuals varied less and was between 72.6 and 77.6% ( & Supplementary Table 2).
Individuals were clustered as imprinted and other (including nonmethylated and intermediately methylated). Ethnicity was not specified for all cohorts used. Details of each cohort can be found from Supplementary Tables 1 & 2.
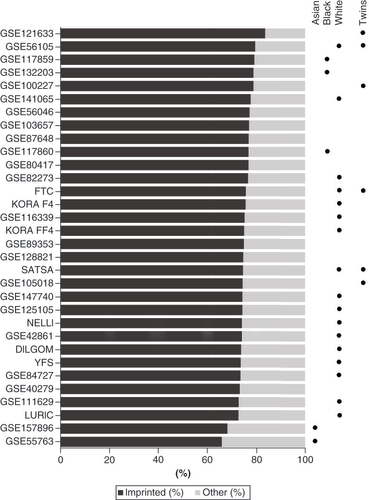
The lowest proportion of imprinted individuals was observed in datasets GSE157896 and GSE55763, with 68.2 and 65.8% of imprinted individuals, respectively ( & Supplementary Table 2). GSE157896 consists of newborns whose mothers were Singaporean citizens or permanent residents, with self-reported homogenous Chinese, Indian or Malay ancestry [Citation53], and GSE55763 consists of individuals with Indian Asian ancestry living in the UK [Citation86].
In contrast, datasets consisting primarily of African–American individuals (GSE117859 and GSE132203) had the third and fourth highest proportions of imprinted individuals – 79.1 and 78.7%, respectively. A third cohort consisting primarily of African–American individuals (GSE117860) did not stand out in this regard, with 76.8% of imprinted individuals. Because these three datasets consist of other ethnicities in addition to African–Americans, whether there is a difference in the proportion of imprinted individuals across ethnic groups was tested. In two of the datasets – GSE117859 and GSE117860 – the proportion of imprinted individuals was significantly higher in African–Americans than in White individuals (χ2-test p < 0.05, Supplementary Figure 2).
The highest proportion of imprinted individuals was observed in datasets GSE121633 and GSE56105, with 83.5% and 79.5% of imprinted individuals, respectively. Both GSE121633 and GSE56105 consist of twins. Dataset GSE100227, also consisting of twins, has the fifth-highest percentage of imprinted individuals with 78.7%. However, some datasets consisting of twins had an average proportion of imprinted individuals (74.3% in GSE105018, 75.5% in FTC and 74.5% in SATSA, & Supplementary Table 2).
No difference was found (χ2-test p > 0.05) in the proportion of imprinted individuals between males and females in 26 datasets (Supplementary Table 2). The only exception was the dataset GSE82273, in which the proportion of imprinted individuals was 73.3% (total N = 505) in males and 80.7% (total N = 384) in females (χ2-test p = 0.009). In addition, no statistically significant differences were found in the proportion of imprinted individuals in any of the case–control settings reported in the datasets included (χ2-test p > 0.05, Supplementary Table 2) and no bias was seen according to the array type used (EPIC vs 450K).
In individuals suffering from MLID the clear binomial methylation pattern of nc886 was lost, and these individuals could not be reliably classified as nonmethylated or imprinted. In this cohort, 35% (6 out of 17) individuals present median methylation β levels between 0.20 and 0.40 in the nc886 locus (Supplementary Figure 3).
The methylation of nc886 in MZ & DZ twin pairs
In five datasets analyzed, almost all MZ pairs were concordant regarding the nc886 locus methylation level, whereas a large proportion of DZ twins were discordant for the nc886 locus methylation level ( & Supplementary Figures 4–7).
Table 1. Number of twin pairs discordant for nc886 methylation status and the absolute difference in the methylation level across twin pairs.
Of the total 1250 MZ twin pairs investigated, 17 pairs (1.3%) that were clustered to different nc886 methylation status groups, when individuals were classified as imprinted and other, were identified (). In datasets in which the intermediately methylated individuals (GSE61496, GSE105018, in total 582 twin pairs) could be identified, 13 (2.2%) were clustered to different nc886 status groups. Of these discordant pairs, one co-twin was always intermediately methylated, whereas the other co-twin was either imprinted or nonmethylated in all cases – that is, no twin pairs were identified in which one co-twin was imprinted and the other was nonmethylated (Supplementary Table 3).
Across all twin pair datasets, the absolute difference in the nc886 methylation level between MZ co-twins was below 0.05 for 88.2% of the pairs. Only 1.0% of pairs had a methylation beta value difference greater than 0.20. Only one MZ pair across all datasets presented a methylation beta value difference over 0.30 (). For this pair, the methylation beta values for nc886 locus were 0.38 and 0.71, suggesting that one twin was imprinted and the other had gained methylation also in the paternal allele of nc886. These results are in line with our finding that there were no imprinted–nonmethylated MZ twin pairs.
The absolute difference in the nc886 methylation level in MZ twin pairs for whom there was information on chorionicity and amnionicity was investigated. In DCDA (separated between days 1 and 3 after fertilization, 22 pairs) and MCDA (separated after day 3, but before implantation, nine pairs) twin pairs, it was observed that in 64.5% of the twin pairs the within-pair difference in their median methylation beta values was below 0.025. For the remaining twin pairs, the within-pair difference was 0.025–0.05 in 19.4% of the pairs and above 0.05 in 16.1% of the pairs. In contrast, in MCMA twin pairs (18 pairs), which are separated only after implantation, the within-pair difference in median methylation value was below 0.025 for all pairs.
Four of the five twin cohorts also contained DZ twin pairs. Of these, 30.3, 35.0, 29.0 and 32.1% were nc886 methylation status (imprinted/other) discordant pairs (). Given the proportions of the nc886 methylation status groups in these four datasets, with random pairings, the expected proportion of discordant pairs would be 39.8, 37.7, 35.7, and 38.8%, respectively (details on proportions and expected proportions in Supplementary Table 4). For all four datasets, the proportion of identified discordant pairs was lower than expected, and for FTC, the largest of the available cohorts, the difference was statistically significant (29.0% vs 35.7%, χ2-test p = 0.02).
The methylation level of nc886 differs in the cerebellum & in skeletal muscle
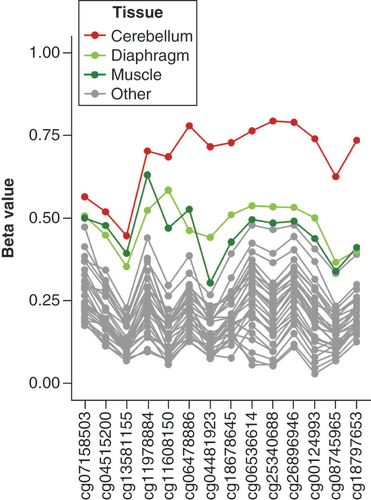
As a starting point for analyzing nc886 methylation in different tissues, dataset GSE64491, consisting of DNA methylation data for 30 tissues from a 112-year-old female, was used. As seen in & Supplementary Figure 8, the methylation level of the nc886 locus was higher in the cerebellum compared with other tissues, with the methylation beta value above 0.70 for most probes in this locus. In addition, muscle and diaphragm showed slightly higher methylation beta values compared with other tissues. For other tissues in dataset GSE64491, variation in the level of methylation at the nc886 locus between tissues was similar in magnitude to what can be observed between the replicates in the data (Supplementary Table 5). Interestingly, unlike skeletal muscle and diaphragm, the nc886 methylation level of heart was not elevated but was comparable to other tissues in the dataset (Supplementary Figure 8). A marked difference in the methylation level between the cerebellum or muscle and other tissues was not observed in the methylation level of six known imprinted genes (Supplementary Figure 9). For these six genes, variation in the methylation level between tissues and between the replicates was smaller as compared to the nc886 locus (Supplementary Table 5).
In this individual, cerebellum has a considerably higher level of methylation compared with other tissues. Muscle and diaphragm also show slightly elevated levels of methylation compared with other tissues. For a figure with all tissues presented in color, see Supplementary Figure 8.
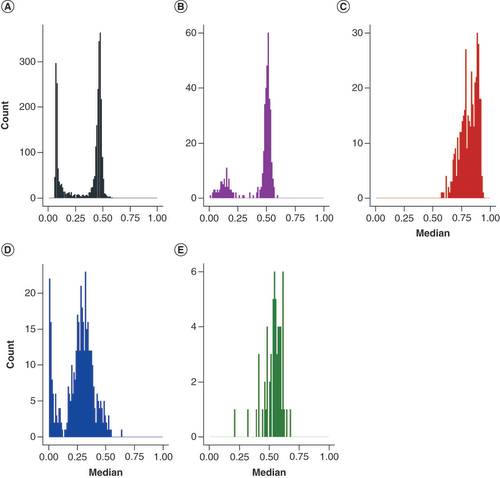
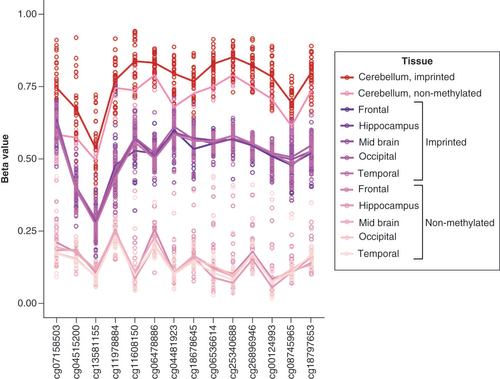
To verify the methylation level of the nc886 locus in the cerebellum, dataset GSE134379, containing methylation data from the cerebellum and middle temporal gyrus (MTG), and GSE72778, containing data from the cerebellum and five other brain regions (frontal lobe, hippocampus, midbrain, occipital lobe and temporal lobe) were investigated. In both datasets, regions of the cerebrum and midbrain present a bimodal nc886 methylation pattern, whereas in the cerebellum, the median methylation level follows a unimodal distribution and is higher, close to 0.70 ( & Supplementary Figure 10).
(A) Blood, GSE55763, n = 2664, (B) middle temporal gyrus (MTG), GSE134379, n = 404, (C) cerebellum, GSE134379, n = 404 (same individuals as in panel B), (D) placenta, GSE167885, n = 411 and (E) muscle, GSE61454, n = 60. Compared with blood and MTG, cerebellum shows a unimodal distribution with elevated methylation levels in nc886 locus. Also in muscle (E), nc886 methylation showed a unimodal distribution. In placenta the methylation level at nc886 locus presented a bimodal distribution, but the overall methylation level was lower compared with blood.
The methylation level of the nc886 locus in the cerebellum was high both in individuals with an imprinted and a nonmethylated nc886 (clustered according to the methylation levels in the cerebrum or MTG) in both datasets studied ( < Supplementary Figure 11). Despite the unimodal distribution of the nc886 methylation level in the cerebellum, a difference in the methylation level of the nc886 locus was observed in the cerebellum between imprinted and nonmethylated individuals (p < 0.001 in both datasets). The six known imprinted genes analyzed did not present differences in the methylation level between the cerebellum and other brain regions (Supplementary Figure 12).
Individuals have been grouped to imprinted and nonmethylated based on data of the five brain regions where nc886 displayed a bimodal methylation pattern (frontal lobe, hippocampus, midbrain, occipital lobe and temporal lobe). Compared with other brain regions, the cerebellum shows higher levels of methylation in both groups, but there is a statistically significant difference in the methylation between individuals clustered as imprinted and nonmethylated. Similar pattern can be observed in dataset GSE134379 (Supplementary Figure 11).
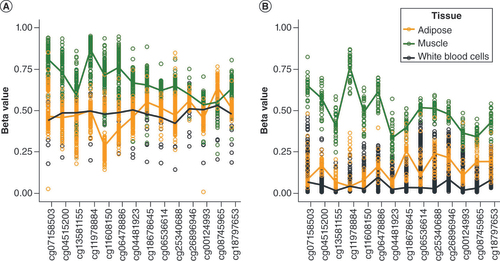
To investigate further the methylation level of the nc886 locus in muscle, six additional datasets were investigated, three of which consisted of only muscle samples (GSE142141, GSE151407 and GSE171140) and three for which other tissues were available (GSE61454 – skeletal muscle, visceral and subcutaneous fat, as well as liver; ERMA – muscle tissue and blood; FTC – muscle tissue, adipose tissue and blood). In all of the datasets, the methylation level of the nc886 locus in muscle presented a unimodal distribution, centerd slightly above 0.50 ( & Supplementary Figures 13–15). In GSE61454, ERMA and FTC, other tissues presented the expected bimodal methylation distribution at the nc886 locus (Supplementary Figures 14 & 15). Despite the unimodal methylation level observed in muscle, there was a difference in the methylation level at the nc886 locus in muscle between imprinted and nonmethylated individuals (as clustered based on the nc886 methylation levels in other tissues) in FTC and GSE61454 (Mann–Whitney U-test p < 0.001) ( & Supplementary Figure 16). Moreover, in FTC, the median methylation levels correlated well across all three tissues (adipose tissue, muscle, blood; p < 2 × 10-12). Individuals presenting intermediately methylated nc886 in blood also had intermediate methylation levels in other tissues (Supplementary Figure 17).
Methylation level of nc886 locus in FTC in (A) imprinted individuals (as clustered based on nc886 methylation levels in blood) and (B) nonmethylated individuals. Despite the unimodal methylation level in muscle (Supplementary Figure 14), a difference in the methylation level of nc886 locus in muscle was observed between imprinted and nonmethylated individuals.
Other available datasets were also investigated for potential atypical methylation patterns in the nc886 locus. Skin (GSE90124) and buccal swabs (NELLI, GSE128821) displayed the expected bimodal distribution and the expected level of methylation at the nc886 locus (Supplementary Figure 18). For the placenta ( & Supplementary Figures 19 & 20), a slight downward shift was observed in the methylation level at the nc886 locus in four datasets (GSE167885, GSE75248, GSE71678, GSE115508). Although the methylation pattern in the placenta followed a bimodal distribution, a clustering analysis could not establish a clear division between imprinted and nonmethylated individuals. In dataset GSE115508, a corresponding downward shift in the methylation values in amnion or chorion was not observed (Supplementary Figure 20). Similarly, fetal cord tissue (GSE157896) did not display a downward shift in the nc886 methylation level (Supplementary Figure 1).
In sperm, a methylation level close to 0 at the nc886 locus was observed, as is expected for a maternally imprinted gene (Supplementary Figure 21). In addition to methylation data from sperm, dataset GSE149318 also contained methylation data from blood of the same individuals. A difference in the sperm nc886 methylation level was not observed between blood-derived imprinted and nonmethylated individuals (Mann-Whitney U-test p > 0.05).
Discussion
Here, we have shown that the proportion of individuals with an imprinted nc886 locus is approximately 75% in the majority of populations and, in 32 datasets, we have shown that the variation in this proportion is limited, especially in populations consisting of White singletons. More varied proportions can be observed in populations consisting of other ethnicities and in twins. This work demonstrates that within MZ twin pairs, the methylation level of the nc886 locus is highly similar, especially in MCMA twin pairs. Finally, we confirmed that the nc886 methylation pattern is stable in the majority of somatic tissues, but we also described two exceptions: the cerebellum and skeletal muscle. These findings allow us to refine the hypotheses on timing and determinants of the polymorphically imprinted nc886 and how the methylation in this locus varies in tissues and individuals originating from the same zygote.
Variation of the nc886 methylation status group proportions is limited across populations
It has previously been reported in individual cohorts that the proportion of individuals with an imprinted nc886 locus is approximately 75%, with the remaining 25% presenting a nonmethylated nc886 locus [Citation9–11]. Here, using 32 cohorts and more than 30,000 individuals, we can confirm that the average proportion of imprinted individuals is 75%. Especially in White singletons, the variation in the proportion of individuals with an imprinted nc886 locus is limited. The lowest proportion of individuals with an imprinted nc886 is observed in cohorts of East Asian origin, whereas the highest proportion of individuals with an imprinted nc886 is observed in cohorts consisting of African–American individuals or twins. Previously, in a cohort of 82 Korean females, the proportion of individuals with methylation levels indicating an imprinted nc886 locus was reported to be 65.9% [Citation87], comparable to our findings here ( & Supplementary Table 2).
Using the GoDMC database and more than 30,000 individuals, we have previously shown that genetic variation is not associated with the establishment of the nc886 methylation pattern [Citation11,Citation88]. Similar results have been obtained in smaller studies [Citation10,Citation19]. Because these results are based mainly on White populations, we cannot rule out the possibility that other ethnic groups would present genetic variation that would affect the establishment of the nc886 imprint. Another explanation for these findings is that the lifestyle or environmental conditions of these populations are affecting the proportions of imprinted individuals. However, the East Asian cohorts included here consist of individuals born in Singapore [Citation53] and of East Asian origin, some of whom were born in the UK and others in Asia [Citation31], suggesting that, at least in this case, shared genetics, rather than shared environmental conditions, may affect the proportion of imprinted individuals. Our results again highlight the need to include more diverse populations in genetic association studies [Citation89].
The methylation status of nc886 is not associated with sex
We identified no difference in the proportion of imprinted individuals between males and females, with the exception of one dataset, GSE82273. This dataset consists of individuals born with a facial cleft and unaffected controls matched for the time of birth [Citation33,Citation90]. Because the data set did not include case–control status, we were unable to test whether the observed difference in proportions was actually due to the study setting because both nc886 methylation and sex have been associated facial clefts [Citation91,Citation92]. We observed no difference in the proportion of imprinted individuals between males and females in 26 other datasets (in a total of 27,362 individuals), and therefore we assume that the observed difference in this one dataset is due to the bias caused by the case–control setting and conclude that the nc886 methylation status is not associated with sex.
nc886 methylation is not associated with analysed case-control settings, with the exception of presence of MLID
In individuals with MLID, we identified a disturbance in the binomial methylation pattern of nc886 in blood. Among individuals with MLID, there was a greater number of people with intermediately methylated nc886 levels, indicative of hypomethylation of this site; this is in line with previously described hypomethylation of imprinted genes in MLID [Citation93].
None of the investigated adult-onset diseases or adverse life events were associated with nc886 methylation status. Previously, nc886 and other imprinted genes have been associated with cardiometabolic health [Citation11,Citation94], but none of the cohorts included a case–control study in this field.
nc886 imprint is stable across majority of somatic tissues
Previous studies have shown that the nc886 methylation status is stable within one individual in all tissues analyzed (abdominal and subcutaneous adipose tissue, bone, joint cartilage, yellow and red bone marrow, coronary and splenic artery, abdominal and thoracic aorta, gastric mucosa, lymph node, tonsils, bladder, gall bladder, medulla oblongata and ischiatic nerve) [Citation10,Citation17], and we have previously shown this in different blood cell subtypes [Citation11]. Here, we confirm these findings in more than 30 somatic tissues but also report exceptions to this pattern – namely, cerebellum and skeletal muscle. Level of nc886 methylation in these two tissues has not been previously reported in the literature. In both cerebellum and skeletal muscle, visual inspection revealed a unimodal DNA methylation pattern at the nc886 locus, with a methylation level of approximately 0.70 in the cerebellum and approximately 0.50 in skeletal muscle. Despite the unimodal methylation pattern in these tissues, there was a slight difference in the methylation level of nc886 between individuals who were imprinted and nonmethylated (according to clustering analysis on other tissues). This suggests that the methylation pattern established in early development in these tissues is not completely reset and established anew, but that the methylation level is built on the existing methylation state. This is further corroborated by our finding that the methylation level of nc886 in muscle is strongly correlated with the methylation level in other somatic tissues. It is not known what mechanism is responsible for this increase in methylation at the nc886 locus in these tissues.
In contrast to the cerebellum and skeletal muscle, in the placenta, the methylation level of the nc886 locus was slightly decreased compared with other analyzed tissues. Placenta has been previously described to have aberrant profiles of imprinted genes and also present a multitude of secondary differentially methylated regions [Citation95]. In line with previous studies [Citation9,Citation13], we show here that the nc886 locus is nonmethylated in sperm and see no difference in the methylation levels between males who present either imprinted or nonmethylated nc886 locus in other tissues. Unfortunately, no suitable dataset from oocytes were available with Illumina array data, and quality of data in the nc886 loci was not adequate in single-cell bisulphite sequencing data (e.g., GSE154762 [Citation96]) to warrant any conclusion. Previously, in a pooled sample of 202 oocytes, the average methylation of nc886 locus was reported to be close to 75% [Citation10,Citation97].
We, and others, have previously shown that the nc886 methylation is tightly associated with the level of nc886 RNAs [Citation11,Citation12,Citation98], with the imprinted individuals having lower levels of these RNAs compared with the nonmethylated individuals. Therefore, we can speculate that the cerebellum and skeletal muscle have lower levels of these RNAs compared with other tissues, whereas the placenta has higher levels of these RNAs. Because the function of these RNAs is not known [Citation15,Citation16], different regulation patterns in these specific tissues might offer possibilities to further hypothesize their role.
In addition to being stable across tissues, the methylation level of nc886 has been shown to be stable through follow-up, from childhood to adolescence [Citation17] and from adolescence to adulthood [Citation11]. However, in granulosa cells, the methylation level of the nc886 locus has been shown to be affected by age, with females over age 40 showing higher methylation values compared with females under age 30 [Citation99]. Granulosa cells are the somatic cell compartment in the follicle and are crucial for oogenesis [Citation100,Citation101]. It has been previously shown that maternal age is associated with the nc886 methylation status of the offspring [Citation9,Citation11], and thus it is interesting to speculate whether the altered methylation status of the granulosa cells is associated with this phenomenon.
Establishment of nc886 imprint
Current literature suggests that periconceptional conditions affect the establishment of the nc886 methylation pattern [Citation9–11,Citation17] and that the imprinting of nc886 is an early embryonic event happening between days 4 and 6 after fertilization [Citation13]. This notion is in slight conflict with the finding that the variation in the proportion of imprinted individuals is limited across populations. If periconceptional conditions would have a substantial role in the establishment of the methylation of the nc886 locus, one would expect to see more differences between cohorts from different countries or between different birth cohorts and, in contrast, fewer differences within DZ twin pairs. For example, we see only minor differences in the prevalence of imprinted individuals in Finnish cohorts born in the 1960s and 1970s (YFS; 73.5%), the 1950s through 1980s (FTC; 75.7%) or in 2007–2008 (NELLI; 74.1%), even though the nutritional status of Finnish expecting mothers has drastically changed during this time [Citation102,Citation103]. Furthermore, if the nonmethylated status would be caused by adverse pregnancy conditions, such as lack of energy or certain nutrients, one could expect that the proportion of nonmethylated individuals would be significantly decreased in populations with good nutritional status. For example, if the lack of folate would be causal in the establishment of the nonmethylated nc886 methylation pattern [Citation17,Citation104], the number of these individuals should have been more drastically diminished in cohorts born after folate supplementation recommendations were established [Citation103,Citation105]. For DZ twins, who share the pregnancy but originate from different zygotes, we showed that approximately one-third of pairs are discordant regarding the nc886 methylation status. Although this was slightly fewer pairs than would be expected by chance, the difference was subtle and statistically significant only in one dataset studied.
Another plausible hypothesis explaining the shown associations between nc886 and periconceptional conditions is that the methylation status is determined already in the oocyte, as suggested also by Carpenter et al. [Citation10] and that the slight variation observed in the nc886 status proportions at the population level is due to either epigenotype offering a survival advantage in specific pregnancy conditions. The establishment of the nc886 methylation status already in the oocyte is also supported by the fact that, in line with results by Carpenter et al. [Citation9], we observed no MZ twin pairs with one co-twin being imprinted and the other nonmethylated. This is also true in the subgroup of twins from FTC, who have been reported to be dichorionic and thus were separated 1–3 days after conception, indicating that the process leading to either nonmethylated or imprinted nc886 loci was completed before this time point. Notably, epigenetic similarity of MZ co-twins is not restricted to nc886 locus but is genome wide [Citation106,Citation107].
Our results also suggest that the process that leads to individuals presenting intermediate nc886 methylation pattern is over before implantation. In MZ twins reported to be separated after implantation, the median methylation in nc886 is nearly identical between the co-twins. Furthermore, individuals who present intermediate methylation levels in their blood also present very similar levels in their adipose tissue, in line with previous findings in different blood cell populations [Citation11], suggesting stability of the methylation level, including the intermediate state, through tissue differentiation. When taken together with the temporal stability of also the intermediate methylation levels [Citation11], we can suggest that the ratio of cells with an imprinted or a non-methylated maternal allele of nc886 in individuals presenting intermediately methylated nc886 status is established before implantation, concurrently to the global de- and re-methylation waves in the embryo [Citation108] and is then reflected on the individual for the rest of their life.
Previous reports on the association of periconceptional conditions and the nc886 methylation status [Citation9–11,Citation17] could be explained by selective survival in certain pregnancy conditions, instead of these conditions directly affecting the establishment of the methylation status. As shown by our results, an example of pregnancy conditions where certain nc886 status might be advantageous or disadvantageous is twin pregnancy. In the population cohorts studied, twin cohorts had high proportions of imprinted individuals, and in DZ twins, the number of pairs with discordant nc886 methylation status was slightly lower than would be expected by chance. This suggests that twin pregnancy might be favorable to fetuses with imprinted nc886 loci or, in the case of DZ twins, to pairs with concordant nc886 methylation status.
Limitations of the study
Our results are descriptive in nature and thus need to be interpreted as hypothesis-generating rather than conclusive. Clustering of individuals into nc886 methylation status groups is, to some extent, affected by different data pre-processing methods, but we have tried to mitigate this by carefully comparing different pre-processing methods. The paucity of datasets consisting of individuals of multiple ethnicities limits our possibilities to draw firm conclusions on the effect of ethnicity on nc886 methylation status proportions. The reasons for imprinting at this locus and the factors responsible for the imprinting remain to be investigated, but our results demonstrating a disturbed pattern of imprinting in the nc886 locus in individuals with MLID indicates that mechanisms responsible for the maintenance of methylation of other imprinted genes are important also in the case of nc886.
Conclusion
Current literature suggests that the polymorphic imprinting of nc886 is not due to genetic variation in White populations, but, given that our results show more variation in the proportion of individuals with an imprinted nc886 in non-White cohorts, the genetic analyses should be repeated in more diverse populations. On the basis of our results and current literature, we hypothesize that DNA methylation of the nc886 locus is established in the growing oocyte and that the variation in the proportion of imprinted individuals in a population could be due to survival advantage or disadvantage in certain pregnancy conditions, illustrated in Supplementary Figure 22. After implantation, the methylation level of nc886 is preserved across studied somatic tissues, with the exception of cerebellum and skeletal muscle. In all individuals, nc886 locus gains methylation in these tissues, even though the methylation levels still associate with the nc886 status established earlier in the development.
Variation in the proportion of individuals with an imprinted nc886 is very modest, with approximately 75% of individuals being imprinted across populations.
The observed variation is mainly limited to non-White ethnic groups and twin cohorts.
Methylation status of nc886 was not associated with sex or any of the case–control settings investigated, with the exception of presence of multilocus methylation disturbance.
Methylation level of nc886 is increased in cerebellum and in skeletal muscle but is uniform in other somatic tissues.
Placenta presents lower methylation levels than majority of somatic tissues, but a binomial methylation pattern can still be detected.
Monozygotic co-twins show highly similar nc886 methylation levels, which is even more pronounced in twins separated at a later date in development.
Approximately 30% of dizygotic twin pairs are discordant for nc886 methylation status.
We suggest that methylation status of nc886 is established in the oocyte and that the slight variation observed across populations could be due to selective survival advantage of the fetus in certain conditions.
Ethical conduct of research
YFS: approved by the first ethical committee of the Hospital District of Southwest Finland on 21 September 2010 and by local ethical committees (1st Ethical Committee of the Hospital District of Southwest Finland, Regional Ethics Committee of the Expert Responsibility area of Tampere University Hospital, Helsinki University Hospital Ethical Committee of Medicine, Research Ethics Committee of the Northern Savo Hospital District, and Ethics Committee of the Northern Ostrobothnia Hospital District).
NELLI: this follow-up study and written informed consent procedure were approved by the medical ethics committees of the Pirkanmaa hospital district (R14039)
SATSA: approved by the ethics committee at Karolinska Institutet with Dnr 2015/1729-31/5.
LURIC: study plan was approved by the ethics committee of the State Chamber of Physicians of Rhineland-Palatinate.
KORA: all study methods were approved by the ethics committee of the Bavarian Chamber of Physicians, Munich.
DILGOM: original FINRISK study has been approved by Coordinating Ethics Committee of the HUS Hospital District, decision nos. 229/E0/2006 and 332/13/03/00/2013. FINRISK and DILGOM study materials have been transferred to THL Biobank in accordance with the notification procedure permitted by the Finnish Biobank Act.
FTC: data collection and analysis were approved by the ethics committee of the Helsinki University Central Hospital (Dnro 249/E5/01, 270/13/03/01/2008, 154/13/03/00/2011).
ERMA: study was approved by the ethics committee of the central Finland health care district in 2014 (K-SSHP Dnro 8U/2014).
Supplemental document
Download MS Word (29.5 KB)Supplemental image
Download JPEG Image (344.4 KB){kind=link}
Supplemental image
Download JPEG Image (163 KB){kind=link}
Supplemental image
Download JPEG Image (341.7 KB){kind=link}
Supplemental image
Download JPEG Image (468.3 KB){kind=link}
Supplemental image
Download JPEG Image (212.2 KB){kind=link}
Supplemental image
Download JPEG Image (338.4 KB){kind=link}
Supplemental image
Download JPEG Image (139.6 KB){kind=link}
Supplemental image
Download JPEG Image (531.8 KB){kind=link}
Supplemental image
Download JPEG Image (256.2 KB){kind=link}
Supplemental image
Download JPEG Image (219.4 KB){kind=link}
Supplemental image
Download JPEG Image (138.2 KB){kind=link}
Supplemental image
Download JPEG Image (135.8 KB){kind=link}
Supplemental image
Download JPEG Image (153.1 KB){kind=link}
Supplemental image
Download JPEG Image (311.3 KB){kind=link}
Supplemental image
Download JPEG Image (937.9 KB){kind=link}
Supplemental image
Download JPEG Image (456.1 KB){kind=link}
Supplemental image
Download JPEG Image (413.5 KB){kind=link}
Supplemental image
Download JPEG Image (414.2 KB){kind=link}
Supplemental image
Download JPEG Image (468.7 KB){kind=link}
Supplemental image
Download JPEG Image (315.3 KB){kind=link}
Supplemental image
Download JPEG Image (435.7 KB){kind=link}
Supplemental image
Download JPEG Image (541.4 KB){kind=link}
Supplemental document
Download MS Excel (34.1 KB)Acknowledgments
The authors thank all the researchers who participated in the collection of the datasets included in this study and made their data publicly available. The DILGOM data used for the research were obtained from THL Biobank (study no. THLBB2021_22). The authors also thank study participants for their generous participation at THL Biobank and at National FINRISK and DILGOM Studies. In addition, the authors thank Eric Dufour and Daria Kostiniuk for their in-depth discussions of the establishment of the nc886 methylation pattern.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2217/epi-2022-0228
Financial & competing interests disclosure
This research was supported by Academy of Finland (349708 PP Mishra; 341750, 346509 E Sillanpää; 275323, 309504, 314181, 335249 EK Laakkonen; 297908, 328685 M Ollikainen; 330809, 338395 E Raitoharju), European Research Council (742927 for MULTIEPIGEN project, O Raitakari), Juho Vainio Foundation (L Kananen, E Sillanpää) Karolinska Institutet Strategic Research Program in Epidemiology (S Hägg), Laboratoriolääketieteen edistämissäätiö sr. (E Raitoharju), Pirkanmaa Regional Fund of Finnish Cultural Foundation (S Marttila), Päivikki and Sakari Sohlberg foundation (E Sillanpää), Signe och Ane Gyllenbergs stiftelse (E Raitoharju), Swedish Research Council (2015-03255; 2019-01272, 2020-06101, SNP&SEQ Technology Platform in Uppsala to S Hägg), the Sigrid Juselius Foundation (M Ollikainen), the Tampere University Hospital Medical Funds (9AC077, 9X047, 9S054, 9AB059 E Raitoharju), Yrjö Jahnsson Foundation (20207299 S Marttila; 20217416, 20197181 L Kananen; 20197212 E Raitoharju).
The Young Finns Study (YFS) was financially supported by the Academy of Finland: grants 322098, 286284, 134309 (Eye), 126925, 121584, 124282, 129378 (Salve), 117787 (Gendi), and 41071 (Skidi); the Social Insurance Institution of Finland; Competitive State Research Financing of the Expert Responsibility area of Kuopio, Tampere and Turku University Hospitals (grant X51001); Juho Vainio Foundation; Paavo Nurmi Foundation; Finnish Foundation for Cardiovascular Research; Finnish Cultural Foundation; the Sigrid Juselius Foundation; Tampere Tuberculosis Foundation; Emil Aaltonen Foundation; Yrjö Jahnsson Foundation; Signe and Ane Gyllenberg Foundation; Diabetes Research Foundation of Finnish Diabetes Association; EU Horizon 2020 (grant 755320 for TAXINOMISIS; grant 848146 for To_Aition); and Tampere University Hospital Supporting Foundation.
The DNA methylation measurement in the LURIC Study has been financially supported by the 7th Framework Program RiskyCAD (grant agreement no. 305739) of the European Union and the Competence Cluster of Nutrition and Cardiovascular Health (nutriCARD), which is funded by the German Federal Ministry of Education and Research (grant no. 01EA1411A).
The main sources of funding in NELLI follow-up study are competitive research funding from Pirkanmaa hospital district (9R030, 9S034, 9M053) and Academy of Finland (277079).
The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Reik W , WalterJ. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet.2(1), 21–32 (2001).
- Lucifero D , MannMRW , BartolomeiMS , TraslerJM. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum. Mol. Genet.13(8), 839–849 (2004).
- Smallwood SA , KelseyG. De novo DNA methylation: a germ cell perspective. Trends Genet.28(1), 33–42 (2012).
- Lees-Murdock DJ , WalshCP. DNA methylation reprogramming in the germ line. Epigenetics3(1), 5–13 (2008).
- Hudson Q , KulinskiT , HuetterS , BarlowD. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity105(1), 45–56 (2010).
- Baran Y , SubramaniamM , BitonAet al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res.25(7), 927–936 (2015).
- Sanchez-Delgado M , CourtF , VidalEet al. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PLOS Genet.12(11), e1006427 (2016).
- Carli D , RiberiE , FerreroGB , MussaA. Syndromic disorders caused by disturbed human imprinting. J. Clin. Res. Pediatr. Endocrinol.12(1), 1–16 (2020).
- Carpenter BL , ZhouW , MadajZet al. Mother–child transmission of epigenetic information by tunable polymorphic imprinting. Proc. Natl. Acad. Sci. U. S. A.115(51), E11970–E11977 (2018).
- Carpenter BL , RembaTK , ThomasSLet al. Oocyte age and preconceptual alcohol use are highly correlated with epigenetic imprinting of a noncoding RNA (nc886). Proc. Natl. Acad. Sci. U. S. A.118(12), e2026580118 (2021).
- Marttila S , ViiriLE , MishraPPet al. Methylation status of nc886 epiallele reflects periconceptional conditions and is associated with glucose metabolism through nc886 RNAs. Clin. Epigenetics13(1), 143 (2021).
- Treppendahl MB , QiuX , SøgaardAet al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood119(1), 206–216 (2012).
- Romanelli V , NakabayashiK , VizosoMet al. Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer. Epigenetics9(5), 783–790 (2014).
- Kostiniuk D , TamminenH , MishraPPet al. Methylation pattern of polymorphically imprinted nc886 is not conserved across mammalia. PLOS ONE17(3), e0261481 (2022).
- Lee YS . Are we studying non-coding RNAs correctly? Lessons from nc886. Int. J. Molec. Sci.23(8), 4251 (2022).
- Fort RS , GaratB , Sotelo-SilveiraJR , DuhagonMA. vtRNA2-1/nc886 produces a small RNA that contributes to its tumor suppression action through the microRNA pathway in prostate cancer. Non-coding RNA6, 7 (2020).
- Silver MJ , KesslerNJ , HennigBJet al. Independent genomewide screens identify the tumor suppressor VTRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Bio.16(1), 118 (2015).
- Lokk K , ModhukurV , RajashekarBet al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol.15(4), 3248 (2014).
- Dugué PA , YuC , McKayTet al. Vtrna2-1: Genetic variation, heritable methylation and disease association. Int. J. Molec. Sci.22(5), 1–18 (2021).
- van Dijk SJ , PetersTJ , BuckleyMet al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. (Lond.)42(1), 28–35 (2018).
- Shaoqing Y , RuxinZ , GuojunLet al. Microarray analysis of differentially expressed microRNAs in allergic rhinitis. Am. J. Rhinol. Aller.25(6), e242–e246 (2011).
- Suojalehto H , LindströmI , MajuriM-Let al. Altered MicroRNA expression of nasal mucosa in long-term asthma and allergic rhinitis. IAA163(3), 168–178 (2014).
- Sharbati J , LewinA , Kutz-LohroffBet al. Integrated microRNA-mRNA-analysis of human monocyte derived macrophages upon Mycobacterium avium subsp. hominissuis infection. PLOS ONE6(5), e20258 (2011).
- Asaoka T , SotolongoB , IslandERet al. MicroRNA signature of intestinal acute cellular rejection in formalin-fixed paraffin-embedded mucosal biopsies. Am. J. Transplant.12(2), 458–468 (2012).
- Lin C-H , LeeY-S , HuangY-Y , TsaiC-N. Methylation status of vault RNA 2-1 promoter is a predictor of glycemic response to glucagon-like peptide-1 analog therapy in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care.9(1), e001416 (2021).
- Barker DJP , OsmondC. Infant mortality, childhood nutrition, and ischaemic heart disease in England and in Wales. The Lancet327(8489), 1077–1081 (1986).
- Pilvar D , ReimanM , PilvarA , LaanM. Parent-of-origin-specific allelic expression in the human placenta is limited to established imprinted loci and it is stably maintained across pregnancy. Clin. Epigenetics11(1), 94 (2019).
- Wilkinson LS , DaviesW , IslesAR. Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci.8(11), 832–843 (2007).
- Edgar R , DomrachevM , LashAE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res.30(1), 207–210 (2002).
- Hannum G , GuinneyJ , ZhaoLet al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell49(2), 359–367 (2013).
- Lehne B , DrongAW , LohMet al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol.16(1), 37 (2015).
- Hannon E , DempsterE , VianaJet al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol.17(1), 176 (2016).
- Markunas CA , WilcoxAJ , XuZet al. Maternal age at delivery is associated with an epigenetic signature in both newborns and adults. PLOS ONE11(7), e0156361 (2016).
- Ventham NT , KennedyNA , AdamsATet al. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun.7(1), 13507 (2016).
- Barbosa M , JoshiRS , GargPet al. Identification of rare de novo epigenetic variations in congenital disorders. Nat. Commun.9(1), 2064 (2018).
- Li S , WongEM , JooJEet al. Genetic and environmental causes of variation in the difference between biological age based on DNA methylation and chronological age for middle-aged women. Twin Res. Hum. Genet.18(6), 720–726 (2015).
- Chuang Y-H , PaulKC , BronsteinJMet al. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med.9(1), 76 (2017).
- Curtis SW , CobbDO , KilaruVet al. Exposure to polybrominated biphenyl (PBB) associates with genome-wide DNA methylation differences in peripheral blood. Epigenetics14(1), 52–66 (2019).
- Zhang X , HuY , AouizeratBEet al. Machine learning selected smoking-associated DNA methylation signatures that predict HIV prognosis and mortality. Clin. Epigenetics10(1), 155 (2018).
- Kurushima Y , TsaiP-C , Castillo-FernandezJet al. Epigenetic findings in periodontitis in UK twins: a cross-sectional study. Clin.Epigenetics11(1), 27 (2019).
- Arloth J , EraslanG , AndlauerTFMet al. DeepWAS: multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLOS Comput. Biol.16(2), e1007616 (2020).
- Kilaru V , KnightAK , KatrinliSet al. Critical evaluation of copy number variant calling methods using DNA methylation. Genet. Epidemiol.44(2), 148–158 (2020).
- Robinson O , ChadeauHyam M , KaramanIet al. Determinants of accelerated metabolomic and epigenetic aging in a UK cohort. Aging Cell19(6), e13149 (2020).
- Nuotio M-L , PervjakovaN , JoensuuAet al. An epigenome-wide association study of metabolic syndrome and its components. Sci. Rep.10(1), 20567 (2020).
- Zeilinger S , KühnelB , KloppNet al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLOS ONE8(5), e63812 (2013).
- Laaksonen J , MishraPP , SeppäläIet al. Mitochondrial genome-wide analysis of nuclear DNA methylation quantitative trait loci. Hum. Mol. Genet.ddab339 (2021).
- Wang Y , KarlssonR , LampaEet al. Epigenetic influences on aging: a longitudinal genome-wide methylation study in old Swedish twins. Epigenetics13(9), 975–987 (2018).
- Hannon E , KnoxO , SugdenKet al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLOS Genet.14(8), e1007544 (2018).
- Liu Y , AryeeMJ , PadyukovLet al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol.31(2), 142–147 (2013).
- Reynolds LM , TaylorJR , DingJet al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun.5(1), 5366 (2014).
- McRae AF , PowellJE , HendersAKet al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol.15(5), R73 (2014).
- Everson TM , MarsitCJ , MichaelO’Shea Tet al. Epigenome-wide analysis identifies genes and pathways linked to neurobehavioral variation in preterm infants. Sci. Rep.9(1), 6322 (2019).
- Huang JY , CaiS , HuangZet al. Analyses of child cardiometabolic phenotype following assisted reproductive technologies using a pragmatic trial emulation approach. Nat. Commun.12(1), 5613 (2021).
- van Dongen J , GordonSD , McRaeAFet al. Identical twins carry a persistent epigenetic signature of early genome programming. Nat. Commun.12(1), 5618 (2021).
- Tan Q , FrostM , HeijmansBTet al. Epigenetic signature of birth weight discordance in adult twins. BMC Genomics15(1), 1062 (2014).
- Bens S , KolarovaJ , BeygoJet al. Phenotypic spectrum and extent of DNA methylation defects associated with multilocus imprinting disturbances. Epigenomics8(6), 801–816 (2016).
- Horvath S , MahV , LuATet al. The cerebellum ages slowly according to the epigenetic clock. Aging7(5), 294–306 (2015).
- Horvath S , LangfelderP , KwakSet al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging8(7), 1485–1512 (2016).
- Bonder MJ , KaselaS , KalsMet al. Genetic and epigenetic regulation of gene expression in fetal and adult human livers. BMC Genomics15(1), 860 (2014).
- Voisin S , HarveyNR , HauptLMet al. An epigenetic clock for human skeletal muscle. J. Cachexia Sarcopenia Muscle11(4), 887–898 (2020).
- Sillanpää E , HeikkinenA , KankaanpääAet al. Blood and skeletal muscle ageing determined by epigenetic clocks and their associations with physical activity and functioning. Clin. Epigenetics13(1), 110 (2021).
- Tuominen PPA , HusuP , RaitanenJ , LuotoRM. Rationale and methods for a randomized controlled trial of a movement-to-music video program for decreasing sedentary time among mother–child pairs. BMC Public Health15, 1016 (2015).
- Roos L , SandlingJK , BellCGet al. Higher nevus count exhibits a distinct DNA methylation signature in healthy human skin: implications for melanoma. J. Invest. Dermatol.137(4), 910–920 (2017).
- Åsenius F , Gorrie-StoneTJ , BrewAet al. The DNA methylome of human sperm is distinct from blood with little evidence for tissue-consistent obesity associations. PLOS Genet.16(10), e1009035 (2020).
- Jenkins TG , JamesER , AlonsoDFet al. Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology5(6), 1089–1099 (2017).
- Green BB , KaragasMR , PunshonTet al. Epigenome-wide assessment of DNA methylation in the placenta and arsenic exposure in the new hampshire birth cohort study (USA). Environ. Health Perspect.124(8), 1253–1260 (2016).
- Paquette AG , HousemanEA , GreenBBet al. Regions of variable DNA methylation in human placenta associated with newborn neurobehavior. Epigenetics11(8), 603–613 (2016).
- Konwar C , PriceEM , WangLQ , WilsonSL , TerryJ , RobinsonWP. DNA methylation profiling of acute chorioamnionitis-associated placentas and fetal membranes: insights into epigenetic variation in spontaneous preterm births. Epigenetics Chromatin11(1), 63 (2018).
- Bhattacharya A , FreedmanAN , AvulaVet al. Placental genomics mediates genetic associations with complex health traits and disease. Nat. Commun.13(1), 706 (2022).
- Borodulin K , TolonenH , JousilahtiPet al. Cohort profile: the National FINRISK Study. Int. J. Epidemiol.47(3), 696–696i (2018).
- Kaprio J . The Finnish Twin Cohort Study: an update. Twin Res. Hum. Genet.16(1), 157–162 (2013).
- Kaprio J , BollepalliS , BuchwaldJet al. The Older Finnish Twin Cohort – 45 years of follow-up. Twin Res. Hum. Genet.22(4), 240–254 (2019).
- Kaidesoja M , AaltonenS , BoglLHet al. FinnTwin16: a longitudinal study from age 16 of a population-based Finnish twin cohort. Twin Res. Hum. Genet.22(6), 530–539 (2019).
- Rose RJ , SalvatoreJE , AaltonenSet al. FinnTwin12 Cohort: an updated review. Twin Res. Hum. Genet.22(5), 302–311 (2019).
- Kovanen V , AukeeP , KokkoKet al. Design and protocol of Estrogenic Regulation of Muscle Apoptosis (ERMA) study with 47 to 55-year-old women’s cohort: novel results show menopause-related differences in blood count. Menopause25(9), 1020–1032 (2018).
- Holle R , HappichM , LowelHet al. KORA – a research platform for population based health research. Gesundheitswesen67(Suppl. 1), S19–25 (2005).
- Winkelmann BR , MärzW , BoehmBOet al. Rationale and design of the LURIC study – a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics2(1s1), S1–S73 (2001).
- Luoto RM , KinnunenTI , AittasaloMet al. Prevention of gestational diabetes: design of a cluster-randomized controlled trial and one-year follow-up. BMC Pregnancy Childbirth10, 39 (2010).
- Pedersen NL . Swedish Adoption/Twin Study on Aging (SATSA), 1984, 1987, 1990, 1993, 2004, 2007, and 2010: Version 2, ICPSR – Interuniversity Consortium for Political and Social Research. (2015).
- Raitakari OT , JuonalaM , RönnemaaTet al. Cohort profile: The Cardiovascular Risk in Young Finns Study. Int. J. Epidemiol.37(6), 1220–1226 (2008).
- Aryee MJ , JaffeAE , Corrada-BravoHet al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics30(10), 1363–1369 (2014).
- Touleimat N , TostJ. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics4(3), 325–341 (2012).
- McCartney DL , WalkerRM , MorrisSWet al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data9, 22–24 (2016).
- Pidsley R , YWong CC , VoltaMet al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics14(1), 293 (2013).
- Hernandez Mora JR , TayamaC , Sánchez-DelgadoMet al. Characterization of parent-of-origin methylation using the Illumina Infinium MethylationEPIC array platform. Epigenomics10(7), 941–954 (2018).
- Chambers JC , LohM , LehneBet al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol.3(7), 526–534 (2015).
- You Y-A , KwonEJ , HwangH-Set al. Elevated methylation of the vault RNA2-1 promoter in maternal blood is associated with preterm birth. BMC Genomics22(1), 528 (2021).
- Min JL , HemaniG , HannonEet al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat. Genet.53(9), 1311–1321 (2021).
- Sirugo G , WilliamsSM , TishkoffSA. The Missing Diversity in Human Genetic studies. Cell177(1), 26–31 (2019).
- Wilcox AJ , LieRT , SolvollKet al. Folic acid supplements and risk of facial clefts: national population based case-control study. BMJ334(7591), 464 (2007).
- Gonseth S , ShawGM , RoyRet al. Epigenomic profiling of newborns with isolated orofacial clefts reveals widespread DNA methylation changes and implicates metastable epiallele regions in disease risk. Epigenetics14(2), 198–213 (2019).
- Ahmed MK , BuiAH , TaioliE. Epidemiology of cleft lip and palate. In: Designing Strategies for Cleft Lip and Palate Care.AlmasriMA ( Ed.). IntechOpen (2017). www.intechopen.com/chapters/53918
- Sanchez-Delgado M , RiccioA , EggermannTet al. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet.32(7), 444–455 (2016).
- Smith FM , GarfieldAS , WardA. Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res.113(1-4), 279–291 (2006).
- Hanna CW , PeñaherreraMS , SaadehHet al. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res.26(6), 756–767 (2016).
- Yan R , GuC , YouDet al. Decoding dynamic epigenetic landscapes in human oocytes using single-cell multi-omics sequencing. Cell Stem Cell28(9), 1641–1656 (2021).
- Okae H , ChibaH , HiuraHet al. Genome-wide analysis of DNA methylation dynamics during early human development. PLOS Genet.10(12), e1004868 (2014).
- Cao J , SongY , BiNet al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res.73(11), 3326–3335 (2013).
- Olsen KW , Castillo-FernandezJ , ZedelerAet al. A distinctive epigenetic ageing profile in human granulosa cells. Hum. Reprod.35(6), 1332–13442020).
- Eppig JJ . Oocyte control of ovarian follicular development and function in mammals. Reproduction122(6), 829–838 (2001).
- Gilchrist RB , LaneM , ThompsonJG. Oocyte-secreted factors: regulators of cumulus cell function and oocyte quality. Hum. Reprod. Update14(2), 159–177 (2008).
- Prättälä R . Dietary changes in Finland – success stories and future challenges. Appetite41(3), 245–249 (2003).
- Erkkola M , KarppinenM , JärvinenAet al. Folate, vitamin D, and iron intakes are low among pregnant Finnish women. Eur. J. Clin. Nutr.52(10), 742–748 (1998).
- Steegers-Theunissen RPM , TwigtJ , PestingerV , SinclairKD. The periconceptional period, reproduction and long-term health of offspring: the importance of one-carbon metabolism. Hum. Reprod. Update19(6), 640–655, (2013).
- Becker W , LyhneN , PedersenANet al. Nordic Nutrition Recommendations2004 – integrating nutrition and physical activity. Food Nutr. Res.52(10), 178–187 (2004).
- van Baak TE , CoarfaC , DuguéPAet al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Biol.19(2), 2018).
- van Dongen J , NivardMG , WillemsenGet al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun.7(1), 11115 (2016).
- Zeng Y , ChenT. DNA methylation reprogramming during mammalian development. Genes10(4), 257 (2019).