
Abstract
Cancer genome sequencing has created an opportunity for precision medicine. Thus far, genetic alterations can only be used to guide treatment for small subsets of certain cancer types with these key alterations. Similar to mutations, epigenetic events are equally suitable for personalized medicine. DNA methylation alterations have been used to identify tumor-specific drug responsive markers. Methylation of MGMT sensitizes gliomas to alkylating agents is an example of epigenetic personalized medicine. Recent studies have revealed that 5-azacytidine and decitabine show activity in myelodysplasia, lung and other cancers. There are currently at least 20 kinds of histone deacetylase inhibitors in clinical testing. Inhibitors targeting other epigenetic regulators are being clinically tested, such as EZH2 inhibitor EPZ-6438.
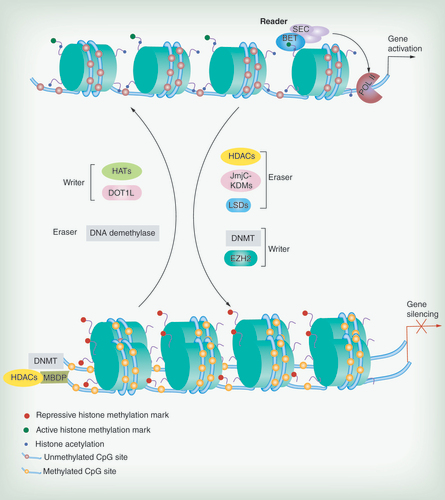
BET, SEC: representative reader (proteins that bind modifications and facilitate epigenetic effects); HATs, DOT1L, DNMT, EZH2: representative writers (enzymes that establish DNA methylation or histone modifications); histone deacetylases, JmjC–KDMs, LSDs, DNA demethylase: representative erasers (proteins that remove DNA methylation or histone modification marks).
Accumulation of genetic and epigenetic alterations is regarded as a major factor for cancer initiation and progression [Citation1–3]. The genetic structure is similar to the ‘hard drive’ of a computer, and epigenetic modifications may serve as the ‘software’ [Citation4]. Precision medicine refers to individualized prevention and treatment strategies that take individual variability into account based on the underlying molecular causes and other factors, defined by geneticist [Citation5,Citation6]. The perfect ‘precision medicine’ is coupling established clinical–pathological indices with state-of-the-art molecular profiling to create diagnostic, prognostic and therapeutic strategies precisely tailored to each patient’ requirements [Citation5–7]. Pharmacogeneticists regard ‘precision medicine’ as an approach to discovering and developing medicines and vaccines by integrating clinical and molecular information [Citation8,Citation9]. Precision medicine is more about identifying individual therapies than causes. Data from the Human Genome Project have advanced the practice of personalized medicine. Knowledge of a patient’s genetic profile can help doctors select the proper medication or therapy and administer it using the proper dose or regimen [Citation5]. Targeting therapy based on aberrant genomic changes has already had an impact on the clinical care of cancer patients. Treatment of cancers with EGFR gene mutations by EGFR kinase inhibitors is a successful example [Citation10]. A total of 138 ‘mutation (Mut)-driver’ genes were discovered by cancer genome landscape in medulloblastoma, pancreatic cancer, glioblastoma, colorectal cancer and breast cancer. Although there are many mutations found in most tumors, it has been suggested that the average tumor contains only two to eight ‘driver gene’ mutations which are involved in 12 signaling pathways that regulate cell fate, cell survival and genome maintenance [Citation11]. Aberrant epigenetic changes occur more frequently than gene mutations in human cancers [Citation12]. For example, epigenetic silencing of CDK2NA and MLH1 is much more common than mutational inactivation of either of these two well-recognized driver genes [Citation12].
It is clear that disruption of the epigenetic ‘machinery’ plays an important role in cancer development, while the value of epigenetic modifications in personalized medicine is still not extensively studied. Epigenome-based personalized medicine may be suitable for human cancer patients with the recognition of cancer epigenome landscapes. Understanding the causative epigenetic changes in cancer has led to the development of novel therapeutic agents that target epigenetic ‘machinery’, such as ORY-1001, a LSD1 inhibitor, is being tested in clinical trial (Oryzon Genomics SA, Eudra CT Number: 2013-002447-29). Recent studies may pave the way for newly emerging drugs and novel drug combinations in cancer treatment, which will be described in this review.
Epigenetic markers as predictors of response to chemotherapy
CpG island is defined as a region of DNA > 200 bp with a GC content ≥50%, and the ratio of observed/expected CpG >0.6 [Citation13]. There are about 28 million CpG sites in the genome, around 7% of CpGs reside within CpG islands (CGIs), and the majority of CpG sites exist outside of CGIs [Citation14,Citation15]. Depending on the genomic location, DNA methylation may have different biological functions. Promoter region methylation is a repressive mark, while the gene body methylation is usually associated with active gene expression. Increasing evidence showed that the intergenic regions contain many regulatory elements, such as enhancers, silencers and noncoding RNAs, and their function may also be affected by DNA methylation [Citation15,Citation16]. Methylation of CpG island ‘shore’, a region outside the CpG island and within 2 kb from transcriptional start site, was regarded as tissue specific methylation [Citation14,Citation17–18]. The most widely studied epigenetic change is DNA hypermethylation in the promoter region of tumor suppressor genes in human cancer.
The identification of biomarkers that predict response to chemotherapy is also a part of personalized medicine. Methylation patterns can be useful to assess clinical outcomes or response to chemotherapeutic agents. DNA methylation profiling has identified tumor-specific drug responsive markers in different cancers. For example, MGMT removes mutagenic alkyl groups from the O6 position of guanine, which otherwise would lead to G-A transitions and the formation of lethal cross-links in DNA after DNA damage [Citation19]. In glioma, MGMT is frequently silenced by methylation, and methylation of MGMT was found to be associated with responsiveness to alkylator-based chemotherapy and an increase in overall survival and time to progression of disease [Citation20]. The CHFR gene is an early mitotic checkpoint gene that functions as a key player in controlling chromosomal integrity [Citation21]. CHFR controls cell cycle progression at the G2/M checkpoint. Increased expression of CHFR leads to a mitotic arrest. CHFR methylation is a sensitive marker for taxanes in human esophageal, gastric, cervical, lung, endometrial cancers and oral squamous cell carcinoma [Citation22,Citation23]. In docetaxel-treated gastric cancer patients, the overall survival was longer in the CHFR methylated group compared with the CHFR unmethylated group [Citation23,Citation24]. FANCF is a member of the Fanconi anemia gene family. FANCF methylation induces disruption of the FA-BRCA pathway and leads to malignant tumors sensitive to cisplatin [Citation25,Citation26]. BRCA1 methylation was associated with an improved response to cisplatin in triple-negative breast cancer (HER2, estrogen and progesterone receptors) [Citation27]. MLH1 methylation is a marker of resistance to oxaliplatin. In oxaliplatin-treated gastric cancer patients, overall survival was longer in the MLH1 unmethylated group compared with the MLH1 methylated group [Citation24]. Many epigenetic chemosensitive markers have been found in different cancer types (). In the future, the combination of multiple epigenetic markers may effectively predict the chemosensitivity of various cancers.
DNA demethylating agents
DNA methylation patterns are established and maintained by three DNA methyltransferases (DNMTs): DNMT1, DNMT3A and DNMT3B. DNMT1 maintains existing methylation patterns, while DNMT3A and DNMT3B are responsible for de novo methylation [Citation42]. The DNA demethylating agents 5-azacytidine and 5-aza-2′deoxycytidine (decitabine) are inhibitors of DNMT1 and DNMT3B. These demethylating drugs were initially developed as chemotherapeutic agents and were used at cytotoxic doses producing significant toxicity [Citation43]. Decitabine has been approved by US FDA for myelodysplasia and acute myeloid leukemia (AML) treatment, and 5-azacytidine has also been approved for myelodysplasia therapy. They are also being tested in human patients with solid tumors [Citation44]. Severe toxicities and off-target effects induced by high dosages precluded true efficacy assessment and limited their application, especially in solid tumors [Citation45]. Reduced dosage to nanomolar drug level permits prolonged exposure times and exposing patient tumor cells. Patients with myelodysplastic syndrome (MDS)/AML generally take months to respond, suggesting the possibility of mechanism is by exhausting tumor stem-like cell renewal [Citation46]. Recent studies found that decitabine and 5-azacytidine suppress tumor growth by impeding cell cycle, inducing apoptosis, promoting cellular differentiation, reversing immune evasion signatures and changing cell adhesion properties [Citation46].
More specific demethylating agents are desirable. Zebularine is a cytidine analog that lacks an amino group in the 4 position of the pyrimidine ring. Zebularine is a stable, antitumor agent that preferentially targets cancer cells and shows activity both in vitro and in experimental animals [Citation47]. Zebularine induces demethylation by complete depleting DNMT1 and partially depleting DNMT3A and DNMT3B in cancer cells [Citation48]. Guadecitabine (SGI-110) is a second-generation demethylating agent, which is more stable in aqueous solution [Citation49]. It is now being tested in clinical trials [Citation4,Citation50]. Some other small non-nucleoside DNMT inhibitors are also being tested, including epigallocatechin-3-gallate, hydralazine, procainamide, procaine and RG108 [Citation51].
DNMT1, DNMT3A and DNMT3B can directly repress transcription partially through the activity of histone deacetylase (HDAC), independent of their methylating capacities. DNMT1 forms a stable complex with transcriptional factors (Rb, E2F1 and HDAC1) to repress transcription. DNMT3A and DNMT3B repress transcription by interacting with HDAC through the ATRX-like PHD domain, which is not found in DNMT1. In addition to re-expression of tumor suppressor genes silenced by promoter hypermethylation, 5-azacytidine and decitabine may regulate gene expression in a DNA methylation-independent manner by separating these complex protein interactions through the inhibition and removal of DNMTs from the nucleus. These effects are accompanied by partial genome-wide DNA demethylation and altered gene expression events that are involved in multiple signaling pathways thought to drive carcinogenesis [Citation46,Citation52–54].
HDAC inhibitors
There are various types of histone tail modifications, such as acetylation, methylation and ubiquitination. These modifications regulate gene expression through their interactions with chromatin-associated proteins in marking regions of transcriptionally active euchromatin and inactive heterochromatin [Citation50]. The chromatin configurations and functions may be dictated by DNA sequence. The dynamics of histone modifications in the overlaid sequence features, such as promoters, enhancers and gene bodies, may induce transcriptional activation or repression. For example, histone marks such as acetylated histone H3, and especially di- or tri-methylated histone H3 lysine 4 (H3K4me2, H3K4me3) marked promoters are often hypomethylated at DNA level. Thus, these promoter regions are also enriched for RNA polymerase II (RNAPII) and subject to transcriptional initiation [Citation55]. Repressed promoters are usually marked with H3K27me3 and H3K9me3, which correlates with constitutive heterochromatin and DNA methylation. When the DNA-hypermethylated genes are reactivated by 5-aza-2′-deoxycytidine, the repressive chromatin does not fully return to an active euchromatic state. Rather, the gene promoters are left in a ‘semiheterochromatic’ state, in which the gene promoters have restored levels, to a variable degree, of the active H3K4me mark but retained some levels of the repressive mark, H3K27me3 [Citation50]. Enhancers are preferentially occupied by sequence-specific DNA-binding proteins and co-activators such as p300 [Citation56]. The chromatin patterns at enhancers were much more variable and cell type specific than chromatin patterns at promoters [Citation55]. In the gene bodies, expressed exons have particularly strong enrichment for H3K36me3 compared with introns. Several studies have confirmed that histone modifications in gene bodies can regulate alternative splicing patterns [Citation57,Citation58]. Histone is modified by different enzymes, including histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methytransferases (HMTs) and demethylases (HDMs) [Citation59]. Inhibitors targeting these enzymes have been tested in clinic.
HDACs are enzymes responsible for removing the acetyl group from lysine residues in histones, inducing a condensed state of chromatin (heterochromatin) and transcriptional repression to balance the activity of HATs [Citation60]. HDAC enzymes are divided into four classes according to their structures and functions: class I (HDAC 1–3 and 8), II (HDAC 4–7, 9 and 10), III (Sir-2 related – SIRT1–7) and IV (HDAC 11) [Citation61]. Class I, II and IV share identity in sequence and structure, while, class III HDACs are different in sequence or structure and require nicotinamide adenine dinucleotide (NAD+) for the catalytic activity [Citation62]. HDACs play an important role in carcinogenesis by silencing tumor suppressor genes. Trichostatin A (TSA) is the first natural product discovered to inhibit HDACs. There are currently at least 20 HDAC inhibitors in clinical testing [Citation63]. Vorinostat (also known as suberoylanilide hydroxamic acid [SAHA]) and romidepsin (also known as depsipeptide or FK228) were approved by the FDA for treatment of cutaneous T-cell lymphoma [Citation52]. Recently, belinostat (PXD-101) obtained FDA approval for the treatment of refractory or relapsed peripheral T-cell lymphoma [Citation64]. Other HDAC inhibitors include abexinostat (PCI-24781), givinostat (ITF-2357), entinostat (MS-275), mocetinostat (MGCD0103), panobinostat (LBH-589) and valproic acid, which are used as alternatives or adjuvants to traditional chemotherapeutics [Citation65]. HDAC inhibitors can affect acetylation of nonhistone proteins as well as histones, potentially leading to more global effects. Furthermore, nonisoform selective HDAC inhibitors target only approximately 10% of all acetylation sites. To date, specific inhibitors of HDAC6 (class II) and HDAC8 (I) have been developed [Citation66]. For example, the HDAC inhibitor PCI-34051 was recently shown to selectively inhibit HDAC8 and induce apoptosis specifically in T-cell lymphomas and not in other tumor types or normal cells [Citation67].
Targeting the key epigenetic regulators
The epigenetic regulation ‘machinery’ is shown in . Epigenetic regulators are divided into ‘writers’ (enzymes that establish DNA methylation or histone modifications), ‘erasers’ (proteins that remove these marks) and ‘readers’ (proteins that bind to modifications and facilitate epigenetic effects). Protein complexes that position the nucleosomes across the genome are called ‘movers’ [Citation43]. The major goal of epigenetic therapy is to target the specific individual proteins that regulate the cancer epigenome. New ‘players’ related to chromatin remodeling are continuously emerging. These key ‘players’, which are related to the ‘driver events’ of the cancer epigenome, are regarded as therapeutic targets. The representative inhibitors of key epigenetic regulators are listed in .
Unlike acetylation, which generally correlates with transcriptional activation, histone lysine methylation can signal either activation or repression depending on the particular lysine residue that is methylated [Citation76]. Even within the same lysine residue, the biological consequence of methylation can differ depending on whether the lysine residue contains mono-, di- or trimethylation [Citation77]. In general, methylation at H3K4, H3K36 and H3K79 is associated with gene active genes, whereas methylation at H3K9, H3K27 and H3K20 correlates with repressed genes [Citation78,Citation79]. Histone methyltransferase (HMT) and demethylase (HDM) enzymes are relatively more specific than HDACs to target limited residues [Citation80]. A great deal of effort is underway to find drugs able to revert specific histone methylation marks or target HMTs or HDMs.
EZH2 inhibitor
Histone lysine methylation has emerged as a critical player in the regulation of gene expression, cell cycle, genome stability and nuclear architecture. Over the past decade, a tremendous amount of progress has led to the characterization of methyl modifications and the lysine methyltransferases (KMTs) and lysine demethylases (KDMs) that regulate them [Citation81]. EZH2 is the catalytic core subunit of the PRC2. It is responsible for catalyzing trimethylation of histone H3 at lysine 27, which serves as a docking site for DNA methyltransferases and HDAC. EZH2 and the whole PRC2 are critical for silencing a large number of genes involved in the development and differentiation processes. EZH2 is reported to be upregulated in various types of solid tumors, including prostate, breast, kidney and lung cancers, and it has been shown to induce cancer cell migration, colony formation and genomic instability [Citation82]. Pharmacological inhibition of PRC2 by 3-deazaneplanocin A (DZNep), an S-adenosylhomocysteine (SAH) hydrolase inhibitor, has been demonstrated to reduce H3K27 methylation and inhibit cancer cell growth. DZNep interferes with S-adenosylmethionine and SAH metabolism and can indirectly inhibit the methylation reaction [Citation83]. As the C-terminal SET domain of EZH2 exhibit methyltransferase activity, specific inhibitor has been designed by targeting the conserved SET domain [Citation84,Citation85]. Because more than 50 SET domain proteins have been identified in humans thus far, the selectivity of the SET domain inhibitors is crucial for minimizing off-target effects [Citation86]. The SAM-competitive inhibitor EPZ005687 can inhibit H3K27 methylation by the EZH2 mutants Y641 and A677, and it has been shown to selectively kill lymphoma cells that are heterozygous for one of these EZH2 mutations [Citation87]. The most potent inhibitor of EZH2 thus far is GSK126. The selectivity of GSK126 for EZH2 is more than 1000-fold higher than its selectivity for 20 other human methyltransferases containing SET or non-SET domains [Citation88]. It was found that point mutations within the catalytic domain of EZH2 occur in subsets of non-Hodgkin lymphoma (NHL) patients. GSK126 effectively inhibited the proliferation of EZH2 mutants in diffuse large B-cell lymphoma cell lines and the growth of EZH2-mutant diffuse large B-cell lymphoma xenografts in mice [Citation88]. EPZ-6438, another EZH2 inhibitor, selectively kills NHL cells bearing mutations within EZH2, and it has minimal effects on the proliferation of EZH2 wild-type NHL cells. It is currently undergoing a Phase I trial in patients with advanced solid tumors or with relapsed or refractory B-cell lymphoma [Citation69,Citation89]. Recent studies find that EZH2 is a multifaceted molecule which could switch to ‘transcriptional activator’ function and act independently of PRC2 complex on nonhistone substrate. It may limit its application in different circumstances [Citation90].
DOT1L inhibitor
DOT1L is a HMT that produces methylated H3K79 [Citation91]. The MLL gene encodes a large multidomain protein which regulates transcription of developmental genes, including the HOX genes [Citation92]. Translocations involving breakage of the MLL gene result in the loss of the carboxy-terminal methyltransferase domain, which is replaced by sequences derived from AF4, AF9, AF10 and ENL to form fusion proteins [Citation93]. DOT1L has been found to be assembled as a member of several large transcription protein complexes containing transcription factors AF4, AF9, AF10 and ENL [Citation94–101]. MLL-fusion proteins gain the ability to recruit DOT1L to MLL target genes, leading to aberrant expression of these genes by methylating H3K79 [Citation86,Citation102–104]. DOT1L deficiency or siRNA knocking down inhibits lung cancer cells proliferation [Citation105]. DOT1L–H3K79 methylation inhibition selectively inhibited proliferation, induced differentiation and reduced cell migration and invasiveness of breast cancer cells with a relatively high DOT1L [Citation106]. EPZ004777 and EPZ-5676 are small molecules, competitive inhibitors of the human DOT1L [Citation106,Citation107]. EPZ-5676 was the result of structure-guided design and optimization of a series of amino nucleoside compounds that includes EPZ004777. The x-ray crystal structure of EPZ-5676 in complex with the human DOT1L methyltransferase domain reveals that EPZ-5676 occupies the S-adenosyl methionine (SAM) binding pocket and induces conformational changes in DOT1L leading to the opening of a hydrophobic pocket beyond the amino acid portion of SAM as described for other members of this series [Citation108,Citation109]. EPZ-5676 is superior to EPZ004777, with >37,000-fold selectivity against all of the tested protein methyltransferases [Citation93]. The use of EPZ004777 in leukemic cells results in selective killing of those cells bearing the MLL gene translocation, with little effect on non-MLL-translocated cells [Citation68]. The preclinical efficacy of DOT1L inhibition on reversing MLL-rearranged leukemiain in vitro and in animal models led to the development of Phase I clinical trial of the DOT1L inhibitor EPZ-5676 [Citation107].
Inhibitors of LSD1 & JmjC domain-containing demethylases family
The methylation status of histone lysine is controlled by lysine demethylases (KDMs) and their counterparts lysine methyltransferases (KMTs). LSD1 (also known as KDM1) was the first discovered histone demethylase and belongs to the superfamily of the flavin adenine dinucleotide (FAD)-dependent amine oxidases [Citation110]. LSD1 catalyzes the demethylation of mono- and di-methylated lysines, but not tri-methylated lysines from H3K4 and H3K9 [Citation110]. LSD1 and HDAC1/2 are common components of several transcriptional corepressor complexes, such as REST corepressor (CoREST) and nucleosome remodeling and deacetylation complex (NuRD). They can cooperate with each other to repress transcription [Citation111–113]. LSD1 was found to be highly expressed in neuroblastoma, prostate, estrogen-negative breast, bladder and colorectal cancers, and the expression of LSD1 positively correlates with some of these malignancies [Citation114–118]. Because of the high structural and mechanistic similarities between LSD1 and conventional amine oxidases, monoamine oxidase inhibitors can also inhibit LSD1, such as pargyline and tranylcypromine [Citation114,Citation119]. Given that nonselective amine oxidase inhibitors have several side effects, derivatives of tranylcypromine and other molecules are being developed as more selective and potent inhibitors. GSK2879552 (GlaxoSmithKline, clinicaltrials.gov identifier: NCT02034123) and ORY-1001 (Oryzon Genomics S A, Eudra CT Number: 2013-002447-29) are two more specific inhibitors of LSD1. The major role of pargyline and tranylcypromine is oxidizing arylalkylamine, though they act as potential LSD1 inhibitors. GSK2879552 and ORY-1001 target LSD1 specifically and result in reduction of the side effect. They are currently in clinical trials for small-cell lung carcinoma and relapsed or refractory AML, respectively. NCL1 (N-[(1S)-3-[3-(trans-2-aminocyclopropyl) phenoxy]-1-(benzylcarbamoyl) propyl] benzamide) is selective LSD1 inhibitor, which impairs LSD1 demethylase activity and blocks cell proliferation in prostate cancer, glioma and breast cancer cell lines [Citation106–108]. In addition, NCL1 induced H3K9me2 accumulation at the promoter regions of androgen-responsive genes in prostate cancer cells and suppressed prostate cancer growth significantly without adverse events. It may serve as a potential therapeutic agent for hormone-resistant prostate cancer [Citation120].
In addition to FAD-dependent oxidases, JmjC domain-containing enzymes are the other larger class of KDMs, which were identified by Tsukada et al. in 2006 [Citation77]. In contrast to LSD1, the JmjC domain-containing KDMs catalyse demethylation through a hydroxylation reaction, using 2′-oxoglutarate (2OG) and dioxygen as cosubstrates and Fe (II) as a cofactor [Citation121]. All JmjC–KDMs contain a catalytic domain with a conserved core double-stranded β helix (DSBH) structural motif to support the catalytic site [Citation79]. The JmjC–KDMs have been categorized into seven KDM subfamilies (KDM2-8) based on the JmjC domain sequence homology and their demethylase activities [Citation121,Citation122]. For instance, KDM2/3/7 subfamilies are mono- and dimethyl demethylases, while, KDM4-6/8 can also remove trimethyl marks. Further, KDM5 and KDM6 members specifically demethylate H3K4 and H3K27 respectively, whereas all KDM4 members can demethylate H3K9. In addition to the catalytic domain, most of JmjC–KDMs contain binding domains to assist recruitment of JmjC–KDMs to certain chromatin regions and/or enable substrate specificity.
Emerging evidence suggests that many KDMs are associated with cancer initiation and progression. For example, KDM4A and B are overexpressed in prostate, bladder, breast, colorectal and gastric cancers [Citation123]. KDM4C is amplified in squamous cell carcinoma [Citation124]. KDM6B is highly expressed in T-cell acute lymphoblastic leukemia cells and is essential for the initiation and progression of T-cell acute lymphoblastic leukemia [Citation125]. The majority of JmjC–KDM inhibitors currently developed contain a metal-chelating motif and inhibit demethylase activity via chelation of the active site Fe (II) [Citation79,Citation126]. 8-hydroxyquinolines, pyridine hydrazine, 5-carboxy-8-hydroxyquinoline (IOX1) and pyridine-hydrazone (JIB-04) are JmjC–KDM inhibitors identified by different method [Citation127–130]. A major challenge in JmjC–KDM inhibitor development is achieving selectivity. EPT103182 is a new JmjC–KDM inhibitor, which specifically targeting KDM5 [Citation131].
BET bromodomain inhibitor
The bromodomain (BRD) is composed of a stretch of 110 conserved amino acids forming a hydrophobic acetyl-lysine (KAc) binding pocket that recognizes ∊-N-acetylated lysines on histone and non-histone proteins [Citation132]. BRDs may contribute to highly specific histone acetylation by tethering transcriptional HATs to specific chromosomal sites, or to the activity of multiprotein complexes in chromatin remodeling [Citation133]. The human genome encodes over 60 BRDs in 46 different proteins. BET family poteins, including BRD2, BRD3, BRD4 and BRDT, contain two BRDs and an extra-terminal (ET) domain [Citation134]. The BET proteins act as scaffolds for the recruitment of transcription factors and chromatin organizers required in transcription initiation and elongation [Citation135]. The BET family proteins have been identified in oncogenic rearrangements, leading to highly oncogenic fusion proteins, and thus play key roles in development of several types of cancer, including nuclear protein of the testis (NUT) midline carcinoma (NMC; a squamous cell carcinoma) and hematologic malignances [Citation136].
Extensive studies have explored small-molecule inhibitors of BET family proteins for cancer therapy. JQ1 was identified based on a previous observation that some novel benzodiazepines inhibit BET proteins. JQ1 displaces BRD4 from nuclear chromatin at nanomolar concentrations and was active against NMC cell lines in vitro and in a relevant xenograft model, thus establishing one of the first paradigms of successful BET inhibition in oncology [Citation135,Citation137]. I-BET762, also known as GSK525762A, was identified at the same time. It binds selectively to the acetyl-recognizing BET pocket with nanomolar affinity. It was initially applied in inflammatory disease, and the antitumor activity was found later in myeloma, acute leukemia and solid cancers. I-BET762 is being tested in early phase clinical trials (clinical trials.gov identifier: NCT01587703) [Citation135]. The exciting results from JQ1 and I-BET762 encouraged the development of similar structure BET inhibitors. Currently, there have been five registered active clinical trials investigating the targeting of BET family proteins, such as RVX-208, I-BET 762, OTX 015, CPI-0610 and TEN-010, in which OTX 015 has reported encouraging results in treating hematologic malignancies [Citation133,Citation138–139]. As the role of BET remains to be elucidated, it limits the development of domain and isoform specific inhibitors.
Combination of epigenetic & other therapies
There are usually multiple epigenetic aberrations occurred to an individual cancer patient, demethylation agents and HDAC inhibitors may synergistically reactivate gene expression and result in more effective tumor suppression. A Phase I clinical trial of patients with unresectable and refractory solid tumors showed that combinational treatment with 5-azacitidine and valproic acid resulted in stable disease lasting up to 12 months in a subset of patients [Citation140].
Combined epigenetic therapy and chemotherapy was reported more effective than chemotherapy alone [Citation46]. Loss of DNA mismatch repair by promoter region hypermethylation of MLH1 occurs frequently in varies cancers. It has been shown that loss of mismatch repair is resistant to a number of anticancer drugs in clinic. Decitabine induced MLH1 re-expression by decreasing MLH1 promoter region methylation in ovarian and colon tumor xenografts that are MLH1 negative because of gene promoter hypermethylation. Decitabine treatment alone has no effect on the growth rate of the tumors. However, decitabine treatment sensitizes the xenografts to cisplatin, carboplatin, temozolomide and epirubicin [Citation141]. In recent Phase I and II trials, combined low doses of azacytidine and entinostat increased the patients’ survival time and improved tumor responses to cytotoxic chemotherapy in non-small-cell lung cancer [Citation142]. Combined epigenetic therapy and immune therapy is another example. A Phase I/II study indicates that efficacy of gemtuzumab ozogamicin can be improved when combined with vorinostat and azacitidine in the difficult-to-treat AML patients [Citation143].
In addition to the demethylating agents and HDAC inhibitors, other epigenetic targeting agents may sensitize tumor cells to chemotherapeutic agents. A preclinical study found that combination of vorinostat and tranylcypromine (KDM1A inhibitor) can suppress glioblastoma growth in xenograft model [Citation144]. DZNep/GSK126 sensitizes lung cancer cells with BRG1 and EGFR mutations to topoisomerase II inhibitors [Citation145]. EZP-5676, a DOT1L inhibitor, can induce a synergistic and durable antiproliferative effect in acute myelocytic leukemia when combined with cytarabine and daunorubicin [Citation146].
Strategies for noncoding RNA-based cancer therapeutics
ncRNAs are functional RNA molecules that do not code for proteins. They are divided into different classes based on size, including siRNAs, miRNA, piRNAs and lncRNA [Citation63].
miRNAs are small single-stranded noncoding RNAs of about 18–25 nucleotides to degrade mRNA or block translation by targeting 3′-UTRs of mRNA. miRNAs are estimated to regulate 30–60% of human genes. Each miRNA may target different amount of mRNAs, and each mRNA may be targeted by different miRNAs. These miRNAs can be classified as tumor-suppressive miRNA and oncogenic miRNA by their function which is mainly determined by the effects of its target mRNAs on cancer growth [Citation60].
miRNA-based anticancer therapeutics are being developed, either alone or in combination with current targeted therapies. Many studies to date reported effects of miRNA in sensitization to chemotherapy, radiotherapy and immunotherapy [Citation147]. For example, p53-deficient human gastric cancer cells, restoration of functional miR-34 inhibited cell growth and induced chemosensitization and apoptosis by targeting Bcl-2, Notch and HMGA2 [Citation148]. miR-155 blockage increased the chemosensitivity to taxol in glioblastoma multiforme cells, making combined treatment an effective therapeutic strategy for controlling the growth by inhibiting EAG1 expression [Citation149].
miRNA-based anticancer therapeutics
The activity of a lost or downregulated miRNA can be restored by using miRNA mimics [Citation150,Citation151]. Recently, Wu et al. used a cationic LPs-based delivery system to efficiently deliver miR-29b in lung cancer in vitro and in vivo. miR-29 suppressed tumorigenicity by downregulating multiple oncogenes, including CDK6, DNMT3B [Citation152]. For miRNAs with oncogenic capabilities, potential therapies include anti-miRNA oligonucleotides, miRNA sponges, miRNA masking and small molecule inhibitor. The anti-miRNA oligonucleotides (AMOs) can block the binding of miRNAs to their binding targets competitively. Therefore, miRNAs can serve as novel therapeutic targets of AMOs in cancer [Citation153]. Studies targeting miR-21 represent one of the first examples of inhibiting cancer development by downregulating oncogenic miRNA expression [Citation154]. The action of AMOs is sequence-specific but not gene-specific. Thus, AMOs may elicit off-target side effects and unwanted toxicity. A miRNA sponge is defined as a synthetic mRNA containing multiple binding sites for an endogenous miRNA, therefore preventing the interaction between miRNA and its endogenous targets. In vitro experiments, these ‘sponges’ derepressed miRNA targets effectively [Citation155]. Despite the early promise and exciting potential, delivery of miRNA-targeting agents remains to be overcome before transition to clinical applications. SiRNAs are often originated from synthetic/exogenous dsRNAs and require perfect complementarity to a particular target mRNA to cleave that target, miRNAs are derived from genome encoded hairpin-shaped precursors and only need partial sequence match to repress target gene expression. Therefore, therapeutic approaches based on miRNA and siRNA have intrinsic similarities and differences [Citation156].
Long noncoding RNA-based anticancer therapeutics
The use of lncRNA as therapeutic agents is only beginning to be explored [Citation157]. Single-stranded oligonucleotides can be designed to strand-specifically block the interaction of the antisense transcript with the sense gene mRNA and/or degrade the antisense transcript, causing transcriptional derepression of the gene. Double-stranded siRNAs can also be utilized to induce natural antisense transcript related derepression and upregulation, provided that the antisense transcript is targeted in a region that does not directly overlap with the sense transcript [Citation158,Citation159]. Oligonucleotides have the potential to hybridize both on- or off-target, which can result in unwanted and unanticipated events. Most of the toxic effects could be observed with all oligonucleotide antisense technologies [Citation160].
Conclusion
The recognition of activating mutations in driver genes, which encode protein kinases in tumors, has led to the development of small-molecule inhibitors. Thus far, genetic alterations can only be used to guide treatment for small subsets of certain cancer types with these key alterations. Similar to mutations, epigenetic events are equally suitable for personalized medicine. DNA methylation alterations have been used to identify tumor-specific drug responsive markers. Unlike genetic events, epigenetic changes are reversible. Demethylating agents, 5-azacytidine and decitabine, have been found effective in myelodysplasia, lung and other cancers. Recent studies revealed that zebularine induces demethylation by complete depleting DNMT1 and partially depleting DNMT3A and DNMT3B in cancer cells. More small non-nucleoside DNMT inhibitors are also being tested. There are currently at least 20 kinds of HDAC inhibitors in clinical testing. Inhibitors targeting other epigenetic regulators, such as EZH2, DOT1L, LSD1 and BET, are also being clinical tested. In addition, some strategies for noncoding RNA based cancer therapeutics have also emerged. Activities of epigenetic therapies offer a unique opprtunity to alter the cancer phenotype. The combination of epigenetic therapy with chemotherapy may increase activity in cancer patients.
Future perspective
It is now a well-established concept that epigenetic alterations are driver events in the pathogenesis of human cancers. In contrast to ‘mut-driver genes’, a greater number of ‘epi-driver genes’ are abnormally expressed by aberrant epigenetic changes in human cancers [Citation9]. A growing number of tyrosine and serine-threonine kinase inhibitors have been developed based on the human cancer genome. These agents have been shown to possess antitumor activity and they have reached the pharmacy. Accumulating studies of the epigenetic profile in different cancer has already found a number of ‘epi-driver genes’. However, the landscape of human epigenome and human cancer epigenome is waiting to be characterized due to the technique limitations. The whole picture of ‘epi-driver genes’ is eagerly needed in the epigenome-based precision medicine. In the past twenty years, researchers mainly focused on promoter region regulation. More complete DNA methylomes in different cancers will help to better understand the disease mechanisms and to develop improved therapies. Currently, epigenetic therapies are successfully used in the clinic to treat hematological malignancies. However, little success has been achieved in treating solid tumors. Deeper understanding of the normal physiological functions of the epigenetic regulators such as writers, readers and erasers will help identify more potential epigenetic targets. The combination of genetic and epigenetic markers may improve the efficiency of chemotherapy by selecting specific reagents for different individuals.
Table 1 Hypermethylated genes as predictors of chemosensitivity.
Table 2 Representative inhibitors of key epigenetic regulators.
Opportunity of precision medicine in genetics
The recognition of activating mutations in driver genes, which encode protein kinases in tumors, has led to the development of small-molecule inhibitors. Genetic alterations can only be used to guide treatment for small subsets of certain cancer types.
Epigenetic events suitable for personalized medicine
DNA methylation alterations have been used to identify tumor-specific drug responsive markers.
Epigenetic changes are reversible.
Small molecules targeting epigenetic regulators are being clinically tested
The combination of epigenetic therapy with chemotherapy
DZNep/GSK126 sensitizes lung cancer cells with BRG1 and EGFR mutations to Topoisomerase II inhibitors.
EZP-5676 can induce a synergistic and durable antiproliferative effect in acute myelocytic leukemia when combined with cytarabine and daunorubicin.
Financial & competing interests disclosure
This work was supported by the following grants: National Basic Research Program of China (973 Program No. 2012CB934002, 2015CB553904); National High-tech R & D Program of China (863 Program No. SS2012AA020314, SS2012AA020821, SS2012AA020303); National Key Scientific instrument Special Programme of China (Grant No. 2011YQ03013405); National Science Foundation of China (Grant No. 81490753). JG Herman is a consultant to MDxHealth. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Guo M , RenJ , BrockMV , HermanJG , CarrawayHE . Promoter methylation of HIN-1 in the progression to esophageal squamous cancer . Epigenetics3 ( 6 ), 336 – 341 ( 2008 ).
- Fearon ER , VogelsteinB . A genetic model for colorectal tumorigenesis . Cell61 ( 5 ), 759 – 767 ( 1990 ).
- Guo M , RenJ , HouseMG , QiY , BrockMV , HermanJG . Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus . Clin. Cancer Res.12 ( 15 ), 4515 – 4522 ( 2006 ).
- Azad N , ZahnowCA , RudinCM , BaylinSB . The future of epigenetic therapy in solid tumours – lessons from the past . Nat. Rev. Clin. Oncol.10 ( 5 ), 256 – 266 ( 2013 ).
- Hamburg MA , CollinsFS . The path to personalized medicine . N. Engl. J. Med.363 ( 4 ), 301 – 304 ( 2010 ).
- Collins FS , VarmusH . A new initiative on precision medicine . N. Engl. J. Med.372 ( 9 ), 793 – 795 ( 2015 ).
- Mirnezami R , NicholsonJ , DarziA . Preparing for precision medicine . N. Engl. J. Med.366 ( 6 ), 489 – 491 ( 2012 ).
- Katsnelson A . Momentum grows to make ‘personalized’ medicine more ‘precise’ . Nat. Med.19 ( 3 ), 249 ( 2013 ).
- Dolsten M , SogaardM . Precision medicine: an approach to R & D for delivering superior medicines to patients . Clin. Transl. Med.1 ( 1 ), 7 ( 2012 ).
- Sharma SV , BellDW , SettlemanJ , HaberDA . Epidermal growth factor receptor mutations in lung cancer . Nat. Rev. Cancer7 ( 3 ), 169 – 181 ( 2007 ).
- Vogelstein B , PapadopoulosN , VelculescuVE , ZhouS , DiazLAJr , KinzlerKW . Cancer genome landscapes . Science339 ( 6127 ), 1546 – 1558 ( 2013 ).
- Beggs AD , JonesA , El-BahrawyM , AbulafiM , HodgsonSV , TomlinsonIP . Whole-genome methylation analysis of benign and malignant colorectal tumours . J. Pathol.229 ( 5 ), 697 – 704 ( 2013 ).
- Takai D , JonesPA . Comprehensive analysis of CpG islands in human chromosomes 21 and 22 . Proc. Natl Acad. Sci. USA99 ( 6 ), 3740 – 3745 ( 2002 ).
- Stirzaker C , TaberlayPC , StathamAL , ClarkSJ . Mining cancer methylomes: prospects and challenges . Trends Genet.30 ( 2 ), 75 – 84 ( 2014 ).
- Jeong M , GoodellMA . New answers to old questions from genome-wide maps of DNA methylation in hematopoietic cells . Exp. Hematol.42 ( 8 ), 609 – 617 ( 2014 ).
- Jeschke J , CollignonE , FuksF . DNA methylome profiling beyond promoters – taking an epigenetic snapshot of the breast tumor microenvironment . FEBS J.282 ( 9 ), 1801 – 1814 ( 2015 ).
- Irizarry RA , Ladd-AcostaC , WenBet al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores . Nat. Genet.41 ( 2 ), 178 – 186 ( 2009 ).
- Doi A , ParkIH , WenBet al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts . Nat. Genet.41 ( 12 ), 1350 – 1353 ( 2009 ).
- Gerson SL . MGMT: its role in cancer aetiology and cancer therapeutics . Nat. Rev. Cancer4 ( 4 ), 296 – 307 ( 2004 ).
- Esteller M , Garcia-FoncillasJ , AndionEet al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents . N. Engl. J. Med.343 ( 19 ), 1350 – 1354 ( 2000 ).
- Scolnick DM , HalazonetisTD . Chfr defines a mitotic stress checkpoint that delays entry into metaphase . Nature406 ( 6794 ), 430 – 435 ( 2000 ).
- Yun T , LiuY , GaoDet al. Methylation of CHFR sensitizes esophageal squamous cell cancer to docetaxel and paclitaxel . Genes Cancer6 ( 1–2 ), 38 – 48 ( 2015 ).
- Derks S , ClevenAH , MelotteVet al. Emerging evidence for CHFR as a cancer biomarker: from tumor biology to precision medicine . Cancer Metastasis Rev.33 ( 1 ), 161 – 171 ( 2014 ).
- Li Y , YangY , LuYet al. Predictive value of CHFR and MLH1 methylation in human gastric cancer . Gastric Cancer18 ( 2 ), 280 – 287 ( 2015 ).
- Taniguchi T , TischkowitzM , AmezianeNet al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors . Nat. Med.9 ( 5 ), 568 – 574 ( 2003 ).
- Swisher EM , GonzalezRM , TaniguchiTet al. Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas . Mol. Cancer8 , 48 ( 2009 ).
- Heyn H , EstellerM . DNA methylation profiling in the clinic: applications and challenges . Nat. Rev. Genet.13 ( 10 ), 679 – 692 ( 2012 ).
- Tanaka M , ChangP , LiYet al. Association of CHFR promoter methylation with disease recurrence in locally advanced colon cancer . Clin. Cancer Res.17 ( 13 ), 4531 – 4540 ( 2011 ).
- Veeck J , RoperoS , SetienFet al. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors . J. Clin. Oncol.28 ( 29 ), e563 – e564 ( 2010 ).
- Strathdee G , MackeanMJ , IllandM , BrownR . A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer . Oncogene18 ( 14 ), 2335 – 2341 ( 1999 ).
- Dejeux E , RonnebergJA , SolvangHet al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response . Mol. Cancer9 , 68 ( 2010 ).
- Lee JH , KangMJ , HanHYet al. Epigenetic alteration of PRKCDBP in colorectal cancers and its implication in tumor cell resistance to TNFα-induced apoptosis . Clin. Cancer Res.17 ( 24 ), 7551 – 7562 ( 2011 ).
- Ramirez JL , RosellR , TaronMet al. 14-3-3sigma methylation in pretreatment serum circulating DNA of cisplatin-plus-gemcitabine-treated advanced non-small-cell lung cancer patients predicts survival: The Spanish Lung Cancer Group . J. Clin. Oncol.23 ( 36 ), 9105 – 9112 ( 2005 ).
- Ebert MP , TanzerM , BalluffBet al. TFAP2E–DKK4 and chemoresistance in colorectal cancer . N. Engl. J. Med.366 ( 1 ), 44 – 53 ( 2012 ).
- Chekhun VF , KulikGI , YurchenkoOVet al. Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells . Cancer Lett.231 ( 1 ), 87 – 93 ( 2006 ).
- Soengas MS , CapodieciP , PolskyDet al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma . Nature409 ( 6817 ), 207 – 211 ( 2001 ).
- Iorns E , TurnerNC , ElliottRet al. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer . Cancer Cell13 ( 2 ), 91 – 104 ( 2008 ).
- Ibanez De Caceres I , Cortes-SempereM , MoratillaCet al. IGFBP–3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer . Oncogene29 ( 11 ), 1681 – 1690 ( 2010 ).
- Faller WJ , RaffertyM , HegartySet al. Metallothionein 1E is methylated in malignant melanoma and increases sensitivity to cisplatin-induced apoptosis . Melanoma Res.20 ( 5 ), 392 – 400 ( 2010 ).
- Ai L , KimWJ , DemircanBet al. The transglutaminase 2 gene (TGM2), a potential molecular marker for chemotherapeutic drug sensitivity, is epigenetically silenced in breast cancer . Carcinogenesis29 ( 3 ), 510 – 518 ( 2008 ).
- Shen L , KondoY , AhmedSet al. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel . Cancer Res.67 ( 23 ), 11335 – 11343 ( 2007 ).
- Kim H , ParkJ , JungYet al. DNA methyltransferase 3-like affects promoter methylation of thymine DNA glycosylase independently of DNMT1 and DNMT3B in cancer cells . Int. J. Oncol.36 ( 6 ), 1563 – 1572 ( 2010 ).
- Ahuja N , EaswaranH , BaylinSB . Harnessing the potential of epigenetic therapy to target solid tumors . J. Clin. Invest.124 ( 1 ), 56 – 63 ( 2014 ).
- Sekeres MA , TiuRV , KomrokjiRet al. Phase 2 study of the lenalidomide and azacitidine combination in patients with higher-risk myelodysplastic syndromes . Blood120 ( 25 ), 4945 – 4951 ( 2012 ).
- Aparicio A , WeberJS . Review of the clinical experience with 5-azacytidine and 5-aza-2′-deoxycytidine in solid tumors . Curr. Opin. Invest. Drugs3 ( 4 ), 627 – 633 ( 2002 ).
- Tsai HC , LiH , Van NesteLet al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells . Cancer Cell21 ( 3 ), 430 – 446 ( 2012 ).
- Marquez VE , BarchiJJJr , KelleyJAet al. Zebularine: a unique molecule for an epigenetically based strategy in cancer chemotherapy. The magic of its chemistry and biology . Nucleosides, Nucleotides Nucleic Acids24 ( 5–7 ), 305 – 318 ( 2005 ).
- Cheng JC , WeisenbergerDJ , GonzalesFAet al. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells . Mol. Cell. Biol.24 ( 3 ), 1270 – 1278 ( 2004 ).
- Kuang Y , El-KhoueiryA , TavernaP , LjungmanM , NeamatiN . Guadecitabine (SGI–110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin . Mol. Oncol. doi:10.1016/j.molonc.2015.06.002 ( 2015 ) ( Epub ahead of print ).
- Laille E , SavonaMR , ScottBL , BoydTE , DongQ , SkikneB . Pharmacokinetics of different formulations of oral azacitidine (CC-486) and the effect of food and modified gastric pH on pharmacokinetics in subjects with hematologic malignancies . J. Clin. Pharmacol.54 ( 6 ), 630 – 639 ( 2014 ).
- Ren J , SinghBN , HuangQet al. DNA hypermethylation as a chemotherapy target . Cell. Signal.23 ( 7 ), 1082 – 1093 ( 2011 ).
- Tsai HC , BaylinSB . Cancer epigenetics: linking basic biology to clinical medicine . Cell Res.21 ( 3 ), 502 – 517 ( 2011 ).
- Bachman KE , RountreeMR , BaylinSB . DNMT3A and DNMT3B are transcriptional repressors that exhibit unique localization properties to heterochromatin . J. Biol. Chem.276 ( 34 ), 32282 – 32287 ( 2001 ).
- Robertson KD , Ait-Si-AliS , YokochiT , WadePA , JonesPL , WolffeAP . DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters . Nat. Genet.25 ( 3 ), 338 – 342 ( 2000 ).
- Zhou VW , GorenA , BernsteinBE . Charting histone modifications and the functional organization of mammalian genomes . Nat. Rev. Genet.12 ( 1 ), 7 – 18 ( 2011 ).
- Visel A , BlowMJ , LiZet al. ChIP-seq accurately predicts tissue-specific activity of enhancers . Nature457 ( 7231 ), 854 – 858 ( 2009 ).
- Kornblihtt AR , SchorIE , AlloM , BlencoweBJ . When chromatin meets splicing . Nat. Struct. Mol. Biol.16 ( 9 ), 902 – 903 ( 2009 ).
- Luco RF , PanQ , TominagaK , BlencoweBJ , Pereira-SmithOM , MisteliT . Regulation of alternative splicing by histone modifications . Science327 ( 5968 ), 996 – 1000 ( 2010 ).
- You JS , JonesPA . Cancer genetics and epigenetics: two sides of the same coin?Cancer Cell22 ( 1 ), 9 – 20 ( 2012 ).
- Guo M , YanW . Epigenetics of gastric cancer . Methods Mol. Biol.1238 , 783 – 799 ( 2015 ).
- Gigek CO , ChenES , CalcagnoDQ , WisnieskiF , BurbanoRR , SmithMA . Epigenetic mechanisms in gastric cancer . Epigenomics4 ( 3 ), 279 – 294 ( 2012 ).
- Bolden JE , PeartMJ , JohnstoneRW . Anticancer activities of histone deacetylase inhibitors . Nat. Rev. Drug Discov.5 ( 9 ), 769 – 784 ( 2006 ).
- Yan W , GuoM . Epigenetics of colorectal cancer . Methods Mol. Biol.1238 , 405 – 424 ( 2015 ).
- Lee HZ , KwitkowskiVE , Del VallePLet al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma . Clin. Cancer Res.21 ( 12 ), 2666 – 2670 ( 2015 ).
- De Souza C , ChatterjiBP . HDAC inhibitors as novel anti-cancer therapeutics . Recent Pat. Anticancer Drug Discov.10 ( 2 ), 145 – 162 ( 2015 ).
- Kelly TK , De CarvalhoDD , JonesPA . Epigenetic modifications as therapeutic targets . Nat. Biotechnol.28 ( 10 ), 1069 – 1078 ( 2010 ).
- Balasubramanian S , RamosJ , LuoW , SirisawadM , VernerE , BuggyJJ . A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI–34051 induces apoptosis in T-cell lymphomas . Leukemia22 ( 5 ), 1026 – 1034 ( 2008 ).
- Daigle SR , OlhavaEJ , TherkelsenCAet al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor . Cancer Cell20 ( 1 ), 53 – 65 ( 2011 ).
- Wee S , DhanakD , LiHet al. Targeting epigenetic regulators for cancer therapy . Ann. NY Acad. Sci.1309 , 30 – 36 ( 2014 ).
- Tan J , YangX , ZhuangLet al. Pharmacologic disruption of Polycomb-repressive complex 2–mediated gene repression selectively induces apoptosis in cancer cells . Genes Dev.21 ( 9 ), 1050 – 1063 ( 2007 ).
- EU Clinical Trials Register . www.clinicaltrialsregister.eu/ctr-search/trial/2013-002447-29/ES .
- Investigation of GSK2879552 in Subjects With Relapsed/Refractory Small Cell Lung Carcinoma . https://clinicaltrials.gov/ct2/show/NCT02034123 .
- Lucas X , GuntherS . Targeting the BET family for the treatment of leukemia . Epigenomics6 ( 2 ), 153 – 155 ( 2014 ).
- Dawson MA , PrinjhaRK , DittmannAet al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia . Nature478 ( 7370 ), 529 – 533 ( 2011 ).
- Delmore JE , IssaGC , LemieuxMEet al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc . Cell146 ( 6 ), 904 – 917 ( 2011 ).
- Martin C , ZhangY . The diverse functions of histone lysine methylation . Nat. Rev. Mol. Cell Biol.6 ( 11 ), 838 – 849 ( 2005 ).
- Tsukada Y , FangJ , Erdjument-BromageHet al. Histone demethylation by a family of JmjC domain-containing proteins . Nature439 ( 7078 ), 811 – 816 ( 2006 ).
- Barski A , CuddapahS , CuiKet al. High-resolution profiling of histone methylations in the human genome . Cell129 ( 4 ), 823 – 837 ( 2007 ).
- Hancock RL , DunneK , WalportLJ , FlashmanE , KawamuraA . Epigenetic regulation by histone demethylases in hypoxia . Epigenomics doi:10.2217/epi.15.24 ( 2015 ) ( Epub ahead of print ).
- Lall S . Primers on chromatin . Nat. Struct. Mol. Biol.14 ( 11 ), 1110 – 1115 ( 2007 ).
- Black JC , Van RechemC , WhetstineJR . Histone lysine methylation dynamics: establishment, regulation, and biological impact . Mol. Cell48 ( 4 ), 491 – 507 ( 2012 ).
- Takawa M , MasudaK , KunizakiMet al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker . Cancer Sci.102 ( 7 ), 1298 – 1305 ( 2011 ).
- Kondo Y . Targeting histone methyltransferase EZH2 as cancer treatment . J. Biochem.156 ( 5 ), 249 – 257 ( 2014 ).
- Tan JZ , YanY , WangXX , JiangY , XuHE . EZH2: biology, disease, and structure-based drug discovery . Acta Pharmacol. Sin.35 ( 2 ), 161 – 174 ( 2014 ).
- Simon JA , LangeCA . Roles of the EZH2 histone methyltransferase in cancer epigenetics . Mutat. Res.647 ( 1–2 ), 21 – 29 ( 2008 ).
- Copeland RA , SolomonME , RichonVM . Protein methyltransferases as a target class for drug discovery . Nat. Rev. Drug Discov.8 ( 9 ), 724 – 732 ( 2009 ).
- Knutson SK , WigleTJ , WarholicNMet al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells . Nat. Chem. Biol.8 ( 11 ), 890 – 896 ( 2012 ).
- Mccabe MT , OttHM , GanjiGet al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations . Nature492 ( 7427 ), 108 – 112 ( 2012 ).
- Knutson SK , KawanoS , MinoshimaYet al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma . Mol. Cancer Ther.13 ( 4 ), 842 – 854 ( 2014 ).
- Deb G , SinghAK , GuptaS . EZH2: not EZHY (easy) to deal . Mol. Cancer Res.12 ( 5 ), 639 – 653 ( 2014 ).
- Nguyen AT , ZhangY . The diverse functions of DOT1 and H3K79 methylation . Genes Dev.25 ( 13 ), 1345 – 1358 ( 2011 ).
- Hess JL . MLL: a histone methyltransferase disrupted in leukemia . Trends Mol. Med.10 ( 10 ), 500 – 507 ( 2004 ).
- Daigle SR , OlhavaEJ , TherkelsenCAet al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia . Blood122 ( 6 ), 1017 – 1025 ( 2013 ).
- Bitoun E , OliverPL , DaviesKE . The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling . Hum. Mol. Genet.16 ( 1 ), 92 – 106 ( 2007 ).
- Biswas D , MilneTA , BasrurVet al. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes . Proc. Natl Acad. Sci. USA108 ( 38 ), 15751 – 15756 ( 2011 ).
- Mueller D , BachC , ZeisigDet al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification . Blood110 ( 13 ), 4445 – 4454 ( 2007 ).
- Mueller D , Garcia-CuellarMP , BachC , BuhlS , MaethnerE , SlanyRK . Misguided transcriptional elongation causes mixed lineage leukemia . PLoS Biol.7 ( 11 ), e1000249 ( 2009 ).
- Yokoyama A , LinM , NareshA , KitabayashiI , ClearyML . A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription . Cancer Cell17 ( 2 ), 198 – 212 ( 2010 ).
- Park G , GongZ , ChenJ , KimJE . Characterization of the DOT1L network: implications of diverse roles for DOT1L . Protein J.29 ( 3 ), 213 – 223 ( 2010 ).
- Zhang W , XiaX , ReisenauerMR , HemenwayCS , KoneBC . DOT1A–AF9 complex mediates histone H3 Lys–79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner . J. Biol. Chem.281 ( 26 ), 18059 – 18068 ( 2006 ).
- Mohan M , HerzHM , TakahashiYHet al. Linking H3K79 trimethylation to Wnt signaling through a novel DOT1-containing complex (DotCom) . Genes Dev.24 ( 6 ), 574 – 589 ( 2010 ).
- Nguyen AT , TaranovaO , HeJ , ZhangY . DOT1L, the H3K79 methyltransferase, is required for MLL–AF9-mediated leukemogenesis . Blood117 ( 25 ), 6912 – 6922 ( 2011 ).
- Monroe SC , JoSY , SandersDSet al. MLL–AF9 and MLL–ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia . Exp. Hematol.39 ( 1 ), 77 – 86e71 – 75 ( 2011 ).
- Milne TA , MartinME , BrockHW , SlanyRK , HessJL . Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications . Cancer Res.65 ( 24 ), 11367 – 11374 ( 2005 ).
- Kim W , KimR , ParkG , ParkJW , KimJE . Deficiency of H3K79 histone methyltransferase DOT-like protein (DOT1L) inhibits cell proliferation . J. Biol. Chem.287 ( 8 ), 5588 – 5599 ( 2012 ).
- Zhang L , DengL , ChenFet al. Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer . Oncotarget5 ( 21 ), 10665 – 10677 ( 2014 ).
- Stein EM , TallmanMS . Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L . Curr. Opin. Hematol.22 ( 2 ), 92 – 96 ( 2015 ).
- Yu W , ChoryEJ , WernimontAKet al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors . Nat. Commun.3 , 1288 ( 2012 ).
- Basavapathruni A , JinL , DaigleSRet al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L . Chem. Biol. Drug Des.80 ( 6 ), 971 – 980 ( 2012 ).
- Shi Y , LanF , MatsonCet al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1 . Cell119 ( 7 ), 941 – 953 ( 2004 ).
- You A , TongJK , GrozingerCM , SchreiberSL . CoREST is an integral component of the CoREST – human histone deacetylase complex . Proc. Natl Acad. Sci. USA98 ( 4 ), 1454 – 1458 ( 2001 ).
- Hou H , YuH . Structural insights into histone lysine demethylation . Curr. Opin. Struct. Biol.20 ( 6 ), 739 – 748 ( 2010 ).
- Wang Y , ZhangH , ChenYet al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer . Cell138 ( 4 ), 660 – 672 ( 2009 ).
- Metzger E , WissmannM , YinNet al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription . Nature437 ( 7057 ), 436 – 439 ( 2005 ).
- Kahl P , GullottiL , HeukampLCet al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence . Cancer Res.66 ( 23 ), 11341 – 11347 ( 2006 ).
- Hayami S , KellyJD , ChoHSet al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers . Int. J. Cancer128 ( 3 ), 574 – 586 ( 2011 ).
- Kauffman EC , RobinsonBD , DownesMJet al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer . Mol. Carcinog.50 ( 12 ), 931 – 944 ( 2011 ).
- Schulte JH , LimS , SchrammAet al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy . Cancer Res.69 ( 5 ), 2065 – 2071 ( 2009 ).
- Lee MG , WynderC , SchmidtDM , McCaffertyDG , ShiekhattarR . Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications . Chem. Biol.13 ( 6 ), 563 – 567 ( 2006 ).
- Etani T , SuzukiT , NaikiTet al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses prostate cancer without adverse effect . Oncotarget6 ( 5 ), 2865 – 2878 ( 2015 ).
- Klose RJ , KallinEM , ZhangY . JmjC-domain-containing proteins and histone demethylation . Nat. Rev. Genet.7 ( 9 ), 715 – 727 ( 2006 ).
- Hojfeldt JW , AggerK , HelinK . Histone lysine demethylases as targets for anticancer therapy . Nat. Rev. Drug Discov.12 ( 12 ), 917 – 930 ( 2013 ).
- Berry WL , JanknechtR . KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells . Cancer Res.73 ( 10 ), 2936 – 2942 ( 2013 ).
- Cloos PA , ChristensenJ , AggerKet al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3 . Nature442 ( 7100 ), 307 – 311 ( 2006 ).
- Ntziachristos P , TsirigosA , WelsteadGGet al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia . Nature514 ( 7523 ), 513 – 517 ( 2014 ).
- Lohse B , NielsenAL , KristensenJBet al. Targeting histone lysine demethylases by truncating the histone 3 tail to obtain selective substrate-based inhibitors . Angew. Chem. Int. Ed. Engl.50 ( 39 ), 9100 – 9103 ( 2011 ).
- Wang L , ChangJ , VargheseDet al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth . Nat. Commun.4 , 2035 ( 2013 ).
- King ON , LiXS , SakuraiMet al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors . PLoS ONE5 ( 11 ), e15535 ( 2010 ).
- Chang KH , KingON , TumberAet al. Inhibition of histone demethylases by 4-carboxy-2,2′-bipyridyl compounds . ChemMedChem6 ( 5 ), 759 – 764 ( 2011 ).
- Schiller R , ScozzafavaG , TumberAet al. A cell-permeable ester derivative of the JmjC histone demethylase inhibitor IOX1 . ChemMedChem9 ( 3 ), 566 – 571 ( 2014 ).
- Steller P . [Radiographic diagnosis of sliding hiatal hernias in children] . Bratisl. Lek. Listy54 ( 2 ), 224 – 229 ( 1970 ).
- Filippakopoulos P , KnappS . Targeting bromodomains: epigenetic readers of lysine acetylation . Nat. Rev. Drug Discov.13 ( 5 ), 337 – 356 ( 2014 ).
- Barbieri I , CannizzaroE , DawsonMA . Bromodomains as therapeutic targets in cancer . Brief. Funct. Genom.12 ( 3 ), 219 – 230 ( 2013 ).
- Bayarsaihan D , ShinDG . Epigenetic drug therapy based on bromodomain inhibition . Epigenomics6 ( 5 ), 473 – 476 ( 2014 ).
- Chaidos A , CaputoV , KaradimitrisA . Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence . Ther. Adv. Hematol.6 ( 3 ), 128 – 141 ( 2015 ).
- Papavassiliou KA , PapavassiliouAG . Bromodomains: pockets with therapeutic potential . Trends Mol. Med.20 ( 9 ), 477 – 478 ( 2014 ).
- Filippakopoulos P , QiJ , PicaudSet al. Selective inhibition of BET bromodomains . Nature468 ( 7327 ), 1067 – 1073 ( 2010 ).
- Chung CW , CosteH , WhiteJHet al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains . J. Med. Chem.54 ( 11 ), 3827 – 3838 ( 2011 ).
- Zeng L , LiJ , MullerMet al. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association . J. Am. Chem. Soc.127 ( 8 ), 2376 – 2377 ( 2005 ).
- Braiteh F , SorianoAO , Garcia-ManeroGet al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers . Clin. Cancer Res.14 ( 19 ), 6296 – 6301 ( 2008 ).
- Plumb JA , StrathdeeG , SluddenJ , KayeSB , BrownR . Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter . Cancer Res.60 ( 21 ), 6039 – 6044 ( 2000 ).
- Juergens RA , WrangleJ , VendettiFPet al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer . Cancer Discov.1 ( 7 ), 598 – 607 ( 2011 ).
- Walter RB , MedeirosBC , GardnerKMet al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a Phase I/II study . Haematologica99 ( 1 ), 54 – 59 ( 2014 ).
- Singh MM , JohnsonB , VenkatarayanAet al. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma . Neuro Oncol. doi:10.1093/neuonc/nov041 ( 2015 ) ( Epub ahead of print ).
- Fillmore CM , XuC , DesaiPTet al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors . Nature520 ( 7546 ), 239 – 242 ( 2015 ).
- Klaus CR , IwanowiczD , JohnstonDet al. DOT1L inhibitor EPZ–5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells . J. Pharmacol. Exp. Ther.350 ( 3 ), 646 – 656 ( 2014 ).
- Garofalo M , LevaGD , CroceCM . MicroRNAs as anti-cancer therapy . Curr. Pharm. Des.20 ( 33 ), 5328 – 5335 ( 2014 ).
- Ji Q , HaoX , MengYet al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres . BMC Cancer8 , 266 ( 2008 ).
- Meng W , JiangL , LuLet al. Anti-miR-155 oligonucleotide enhances chemosensitivity of U251 cell to taxol by inducing apoptosis . Cell Biol. Int.36 ( 7 ), 653 – 659 ( 2012 ).
- Garzon R , MarcucciG , CroceCM . Targeting microRNAs in cancer: rationale, strategies and challenges . Nat. Rev. Drug Discov.9 ( 10 ), 775 – 789 ( 2010 ).
- Bader AG , BrownD , StoudemireJ , LammersP . Developing therapeutic microRNAs for cancer . Gene Ther.18 ( 12 ), 1121 – 1126 ( 2011 ).
- Wu Y , CrawfordM , MaoYet al. Therapeutic delivery of microRNA–29b by cationic lipoplexes for lung cancer. Molecular therapy . Nucleic Acids2 , e84 ( 2013 ).
- Weiler J , HunzikerJ , HallJ . Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease?Gene Ther.13 ( 6 ), 496 – 502 ( 2006 ).
- Krichevsky AM , GabrielyG . miR-21: a small multi-faceted RNA . J. Cell. Mol. Med.13 ( 1 ), 39 – 53 ( 2009 ).
- Ebert MS , NeilsonJR , SharpPA . MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells . Nat. Methods4 ( 9 ), 721 – 726 ( 2007 ).
- Li C , FengY , CoukosG , ZhangL . Therapeutic microRNA strategies in human cancer . AAPS J.11 ( 4 ), 747 – 757 ( 2009 ).
- Qi P , DuX . The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine . Mod. Pathol.26 ( 2 ), 155 – 165 ( 2013 ).
- Faghihi MA , KocerhaJ , ModarresiFet al. RNAi screen indicates widespread biological function for human natural antisense transcripts . PLoS ONE doi:10.1371/journal.pone.0013177 ( 2010 ) ( Epub ahead of print ).
- Modarresi F , FaghihiMA , Lopez-ToledanoMAet al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation . Nat. Biotechnol.30 ( 5 ), 453 – 459 ( 2012 ).
- Bennett CF , SwayzeEE . RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform . Annu. Rev. Pharmacol. Toxicol.50 , 259 – 293 ( 2010 ).