
Abstract
A small percentage of patients with neuromyelitis optica spectrum disorder (NMOSD) may have radiological manifestations that completely mimic MS. Accurate diagnosis in these patients requires paying attention to patients’ history and how they respond to treatment. Here, a patient with NMOSD is introduced, in the MRI scan of whom, the diagnostic criteria of Barkhof were outlined; however, since her blurred vision did not respond to treatment with corticosteroids, she was diagnosed with NMOSD.
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory syndrome of the central nervous system associated with anti-AQP4 antibody in the majority of patients which differs from multiple sclerosis both clinically and radiologically [Citation1]. Different clinical and neuroimaging features have been defined for MS and NMOSD. In revised consensus criteria, NMOSD is categorized to NMOSD with anti-AQP4 antibody and NMOSD without or unknown anti-AQP4 antibody status [Citation1]. In this criteria, acute myelitis, optic neuritis, area postrema syndrome, acute diencephalic clinical syndrome, acute brainstem syndrome and symptomatic cerebral syndrome have been defined as the six core clinical criteria of NMOSD [Citation1]. In the anti-AQP4 antibody seropositive status, only one core criterion is enough for making the diagnosis. When one or more clinical attacks of at least two core criteria with dissemination in space present and at least one of the attacks comprise acute myelitis, optic neuritis or area postrema syndrome and anti-AQP4 antibody is negative, NMOSD with negative or unknown anti-AQP4 antibody status could be diagnosed. It is important to exclude alternative diagnoses in both NMOSD types [Citation1]. Optic neuritis and transverse myelitis are common first presentations of both multiple sclerosis (MS) and NMOSDs and these clinical overlaps may cause misdiagnosis. In seronegative NMOSD patients, myelin oligodendrocyte glycoprotein (MOG) antibody disease is supposed to be considered [Citation2]. Up to 42% of seronegative NMOSD patients might be MOG-Ab positive [Citation2]. MOG as a myelin outer membrane glycoprotein may play an important role in myelin stability. Autoimmunity against MOG presents with recurrent optic neuritis and transverse myelitis like NMOSD and MS [Citation2].
The pattern of lesions in MRI is important in making a diagnosis of NMOSD, especially for the diagnosis of seronegative patients. Some characteristic patterns have been defined.
Longitudinally extensive transverse myelitis lesions is the most specific pattern of NMOSD [Citation1]. This lesion involves the central gray matter of at least three vertebral segments length, and causes T1 hypointensity and enhances following gadolinium administration [Citation3].
Lesions involving more than half of the optic nerve length with extension to optic chiasm is another suggestive MRI characteristic of NMOSD [Citation1].
AQP4 is a water channel and it is highly expressed adjacent to the ventricular system; therefore, NMOSD brain lesions typically involve these areas [Citation4]. Lesions adjacent to the third and fourth and lateral ventricles have been reported in NMOSD patients. These lesions might be symptomatic or asymptomatic [Citation5]. Extensive and longitudinal involvement of the posterior limb of internal capsule and midbrain has been reported in NMOSD patients brain MRI [Citation5]. Tumefactive hemispheric white matter lesions without mass effect and cloud-like appearance enhancement have been defined in NMOSD patients which caused encephalopathy, headache and seizure [Citation5].
Although brain MRI lesions like MS cannot exclude NMOSD, they have been defined as red flag of NMOSD diagnosis [Citation1]. Meanwhile, up to 16% of NMOSD patients fulfill Barkhof MS criteria [Citation5]. Large edematous and heterogeneous corpus callosum lesions with marbled pattern were reported in 18.2% of Japanese NMOSD patients especially in the acute phase [Citation6].
Differentiation of multiple sclerosis (MS) from NMOSD is of great diagnostic and therapeutic importance [Citation1]. Not only the trend and prognosis of these two diseases are different, but also different treatments are used for them. It is also now known that a number of medications used in MS such as fingolimod, natalizumab, dimethyl fumarate, interferon and glatiramer can exacerbate NMOSD [Citation7]. Therefore, any diagnostic mistake can have serious consequences for the patient. Of course, understanding the patterns of magnetic resonance imaging (MRI) in each of these two diseases contributes to this differentiation. Here we report an NMOSD patient with a first MRI like multiple sclerosis.
Presentation of case
The patient was a 26-year-old woman diagnosed with optic neuritis in the right eye in April 2017. She had negative previous history of the disease and had no significant family history and no history of medication. The movements of her right eye were painful in examination. Her visual acuity was 3 m finger count (FC), and the patient’s fundoscopy test was normal. The rest of the patient’s neurological examinations were normal. In visual evoked potential P100, distal latency was increased and amplitude was preserved.
Initial diagnosis/assessment
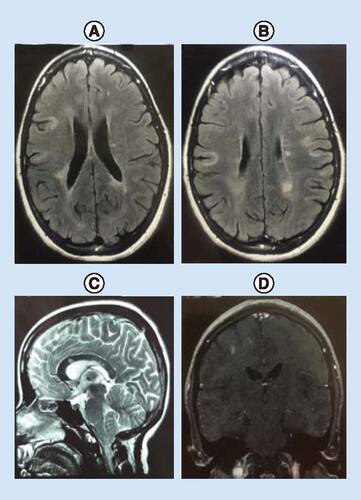
The brain MRI showed multiple periventricular plaques accompanied by U-fiber involvement antinuclear antibodies. The patient’s cervical MRI was normal. Anticardiolipin antibodies (IgG and IgM), anti-dsDNA antibodies, anti-Ro and anti-La angiotensin anticardiolipin antibodies (IgG and IgM) and angiotensin converting enzyme tests were negative (, B, C & D). Anti-MOG antibodies were also negative. The initial diagnosis was an acute inflammatory CNS disorder. Anti-AQP4 antibody reported positive with use of indirect immunofluorescence assay using HEK293 cells transfected with recombinant human full-length AQP4 gene at a 1:10 starting dilution (Euroimmun, Lübeck, Germany) at two-times apart [Citation8]. Then NMOSD with positive anti-AQP4 antibody was diagnosed.
Treatment/management
The patient underwent treatment with intravenous methylprednisolone 1 g for 5 days.
Outcome & implications
However, there was no improvement in her visual acuity after 5 days’ pulse therapy. Given that there was no visual improvement, plasma exchange (PLEX) was performed. Her blurred vision improved considerably by performing PLEX. Now PLEX is suggested as a rescue therapy in severe steroid-resistant relapses of NMOSD [Citation9]. With a diagnosis of NMOSD, the patient was treated with rituximab (1 g given IV 2 weeks apart and then repeated every 6 months). During 1 year follow-up, any new attack has been presented and her EDSS is 0 now. Now rituximab is an effective treatment option of treatment-naive NMOSD patients [Citation10].
Discussion
Here, a patient with NMOSD is introduced who was referred to the hospital with optic neuritis and her MRI showed multiple periventricular and U-fiber involvements.
A study from the UK reported U-fiber and Dawson fingers-like MRI lesions in 0% and periventricular lesions in 32% of NMOSD patients [Citation11]. In Asian countries, periventricular lesions have been reported in 40% and ovoid-shaped brain lesions in 31.6% of patients [Citation12]. In Taiwanese patients similar to Asian countries, U-fiber lesions have been reported in 48%, Dawson-like lesions in 44% and periventricular lesions in 60% of NMOSD patients [Citation13]. Although it has been reported that periventricular, U-fiber, inferior temporal and Dawson-like MRI lesions are more than 90% sensitive for distinguishing NMOSD from MS [Citation5], it seems that genetic and environmental factors should be taken into account before making this differentiation.
Brain involvement in patients with NMOSD is now a well-known issue. Studies have shown that during the course of the disease, about 60% of the patients suffer lesions in the brain which are mostly nonspecific and asymptomatic [Citation12]. Usually, brain involvements have certain patterns [Citation3] It has been found that the brain may also be involved in NMOSD which is usually associated with specific patterns in MRI [Citation3].
In research studies, patterns such as the involvement of the diencephalic region, periependymal areas in brain, dorsal medulla/area postrema or cerebral hemispheres have been reported [Citation3]; the observation of each should lead the researchers to look for more complete diagnostic tests.
Conclusion
Patients with NMOSD may have radiological manifestations similar to MS patients. Paying attention to the patients’ history and how they respond to therapeutic measures can contribute to an accurate diagnosis. Paying attention to MRI patterns can help to differentiate the disease from MS. It should be noted that in 16% of cases, it may imitate MRI scan of the patient with MS [Citation5,Citation12]. This makes it difficult to diagnose the disease. Therefore, other issues should be considered in the diagnosis. With regard to the patient of the present study, lack of improvement in her blurred vision following treatment with corticosteroids contributed, practically as a guide, to diagnose NMOSD. The differentiation of MS from NMOSD is very important because many treatments used for MS can worsen the condition of patients with NMOSD [Citation7]. Different NMOSD MRI presentations in European and Asian patients might be a future research window to define distinct subtypes of anti-AQP4 antibodies which act differently and contribute to involve different parts of the central nervous system. Altered immunological behavior of anti-AQP4 antibody in different genetic and environmental settings is a concept that warrants further investigation.
Ethical conduct of research statement
This paper has been written based on the Tehran University of Medical Science Ethical Committee. Informed consent was obtained from the participant.
Acknowledgements
The authors would like to thank the Sina Hospital Neuromyelitis Optica Spectrum Disease Cohort Study Group for providing medical records. This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Financial & competing interests disclosure
The authors would like to thank the Sina Hospital Neuromyelitis Optica Spectrum Disease Cohort Study Group for providing medical records. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- WingerchukDM, BanwellB, BennettJLet al.International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology85(2), 177–189 (2015).
- NarayanR, SimpsonA, FritscheKet al.MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord.25, 66–72 (2018).
- KimHJ, PaulF, Lana-PeixotoMAet al.MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology84(11), 1165–1173 (2015).
- PittockSJ, WeinshenkerBG, LucchinettiCFet al.Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol.63(7), 964–968 (2006).
- MatthewsL, MarascoR, JenkinsonMet al.Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology80(14), 1330–1337 (2013).
- NakamuraM, MisuT, FujiharaKet al.Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Mult. Scler.15(6), 695–700 (2009).
- KiraJI. Unexpected exacerbations following initiation of disease-modifying drugs in neuromyelitis optica spectrum disorder: which factor is responsible, anti-aquaporin 4 antibodies, B cells, Th1 cells, Th2 cells, Th17 cells, or others?Mult. Scler.23(9), 1300–1302 (2007).
- JariusS, ProbstC, BorowskiKet al.Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J. Neurol. Sci.291, 52–56 (2010).
- BonnanM, ValentinoR, DebeugnySet al.Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J. Neurol. Neurosurg. Psychiatry89(4), 346–351 (2018).
- TrebstC, JariusS, BertheleAet al.Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol.261(1), 1–16 (2014).
- MatthewsL, MarascoR, JenkinsonMet al.Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology80(14), 1330–1337 (2013).
- KimW, ParkMS, LeeSHet al.Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult. Scler.16(10), 1229–1236 (2010).
- LiaoMF, ChangKH, LyuRKet al.Comparison between the cranial magnetic resonance imaging features of neuromyelitis optica spectrum disorder versus multiple sclerosis in Taiwanese patients. BMC Neurol.14, 218 (2014).