
Abstract
Paroxysmal dyskinesias (PxD) comprise a group of heterogeneous syndromes characterized by recurrent attacks of mainly dystonia and/or chorea, without loss of consciousness. PxD have been classified according to their triggers and duration as paroxysmal kinesigenic dyskinesia, paroxysmal nonkinesigenic dyskinesia and paroxysmal exertion-induced dyskinesia. Of note, the spectrum of genetic and nongenetic conditions underlying PxD is continuously increasing, but not always a phenotype–etiology correlation exists. This creates a challenge in the diagnostic work-up, increased by the fact that most of these episodes are unwitnessed. Furthermore, other paroxysmal disorders, included those of psychogenic origin, should be considered in the differential diagnosis. In this review, some key points for the diagnosis are provided, as well as the appropriate treatment and future approaches discussed.
Paroxysmal dyskinesias: redefining concepts & classifications
Paroxysmal dyskinesias (PxD) encompass a group of heterogeneous syndromes characterized by recurrent attacks of involuntary movements, intermittent or episodic in nature and abrupt in onset, without loss of consciousness. The abnormal movements consist of dystonia and/or chorea, with ballism or athetosis being less possible, but do not include tremor or myoclonus [Citation1].
The classification of PxD has changed over the years. First, the episodes were cataloged depending on the duration (short, <5 min; long, >5 min) [Citation2]. Following this former classification, Dermikian and Jankovic proposed three different subtypes based on triggers: paroxysmal kinesigenic (PKD), nonkinesigenic (PNKD) and exercise-induced (PED) dyskinesias [Citation3]. A fourth subtype, paroxysmal hypnogenic dyskinesia (PHD), has been subsequently considered to be a form of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) in most cases [Citation4]. Noteworthy, in recent years, the differentiation of subgroups depending on the etiology (primary [familial or sporadic] and secondary forms) has gained relevance. Primary forms comprise cases without definite cause for PxD, labeled as idiopathic, and carriers of specific genetic mutations (i.e., PRRT2-PKD or SLC2A1-PED) [Citation1,Citation5]. In secondary forms, an acquired condition underlying PxD has been identified, such as autoimmune, vascular or metabolic disorders [Citation6].
The prevalence of PxD is not clearly defined, but some authors have reported to be lesser than 1% [Citation6]. However, PxD are probably underdiagnosed as the episodes of PxD are often unwitnessed due to its short duration. In addition, the common lack of abnormal signs between the attacks, especially in primary forms, increases the diagnosis challenge [Citation7]. Therefore, recognition of triggers and clinical features of attacks could lead us to conduct the appropriate investigations to reach a definite diagnosis, on which treatment is highly dependent [Citation1].
Clinical features: role of triggers
Demographic & clinical features in paroxysmal dyskinesias are summarized in .
Table 1. Demographic and clinical features in paroxysmal dyskinesias.
Paroxysmal kinesigenic dyskinesia (PKD)
In 1967, Kertesz described an episodic disorder which was termed ‘paroxysmal kinesigenic choreoathetosis’ because the attacks were triggered by sudden movements [Citation8]. As new cases are detected, clinical features of this disorder are better detailed, emphasizing that the attacks are relatively brief and respond well to antiepileptic drugs (AEDs) [Citation9,Citation10].
Mean age at onset is 5–15 years (range: 6 months–33 years), with a male:female ratio 4:1 (even up to 8:1) in sporadic but not familial forms. Familial cases typically experience other paroxysmal neurological disorders (e.g., seizures or migraine) [Citation7].
The attacks are triggered by an abrupt movement or an increase of speed, amplitude or strength, the sudden addition of new actions during ongoing steady movements or a change in direction. Light, sound and vestibular stimulation, startle, hyperventilation and stress can also induce attacks [Citation7,Citation9]. Almost 70% of patients presented a premonitory aura referred as numbness, tingling or tightness in the affected limb, or a vague sensation in head, epigastrium or abdomen [Citation9]. Those patients aware of these warning signs can prevent the attacks by slowing down or holding the affected limb firmly [Citation7].
Patients manifest any form of dyskinesia: dystonia, chorea, ballism or a combination of them. Therefore, the PKD term was proposed. Episodes are focal, multifocal or generalized and involve limbs, trunk and face, although unilateral dystonia is observed in most cases [Citation11]. A total of 30% of patients experienced slurred speech (dysarthria or anarthria) possibly due to facial weakness [Citation10]. The episodes with hemiballismus, tongue movements and freezing of gait are rare [Citation12]. New attacks cannot be induced for a refractory period of 20 min approximately [Citation7].
The duration of attacks is brief, lasting mostly less than a minute; and frequency is variable, on average 1–20 attacks per day, although coexistence of depression and anxiety worsen both aspects [Citation5,Citation13]. The majority of affected women improve during pregnancy. The frequency of episodes is maximum in puberty, with up to 30–100 attacks per day, often diminishes after the 20s; and up to 50% of cases reach complete or partial remission in adulthood [Citation9].
Paroxysmal nonkinesigenic dyskinesia (PNKD)
In 1940, Mount and Reback made the first clear description of the disorder, which they called ‘paroxysmal choreoathetosis’, into a large family with 28 members affected in five generations. The attacks lasted several hours, were precipitated by alcohol or coffee and the use of AEDs was ineffective [Citation14]. Since this initial description, the phenotypes of PNKD have evolved into a broad clinical spectrum [Citation12,Citation15,Citation16].
The primary PNKD forms usually have a childhood onset (mean age, 8 years) [Citation7], albeit attacks that began at 20 years have been reported. The starting range is extended in secondary forms (2.5–79 years), with two peaks in the twenties and sixties due to trauma and cerebrovascular events, respectively [Citation6]. Sporadic cases have shown a male:female ratio 2:1 and genetic cases 1:1 [Citation7].
Attacks occur at rest or are provoked by alcohol, coffee, tea, cola, tobacco, emotional excitement, hunger, fever, concentration and fatigue [Citation5,Citation15]. Exacerbation periods have been identified in relation to menstruation, ovulation and postpartum; as well as remissions during pregnancy and sleep [Citation7,Citation15]. Premonitory sensations are described in up to 80% of familial cases as a feeling of numbness or stiffness in a limb and involuntary movements of the mouth [Citation15]. Other prodromal symptoms comprise headache, hyperventilation and restlessness [Citation12]. Exposure to cold water palliates some episodes [Citation17].
Movements may involve face, trunk and limbs, initially on one side and tend to spread or even generalize. Dystonia and chorea predominate, which explains the use of the term PNKD nowadays, although the phenomenology is heterogeneous and variable over time [Citation15]. Blepharospasm, sardonic laugh and diplopia are rare [Citation7,Citation17]. Dysarthria or anarthria with normal breath and laryngeal dystonia with respiratory failure occur during severe episodes [Citation15,Citation18]. Interictal myokymia has been observed in several familial PNKD cases [Citation19,Citation20].
In PNKD, attacks are longer lasting (10 min–12 h, generally less than an hour) and less frequent (range between three-times a day and two-times per year, most often once a week) than in PKD [Citation15,Citation16]. PNKD has a variable prognosis, and some patients have shown remissions in the elderly and with menopause [Citation7,Citation12].
Paroxysmal exercise-induced dyskinesia
In 1977, Lance described a third ‘intermediate’ category of PxD in a family presenting attacks after physical exhaustion that lasted between 5 and 30 min [Citation2]. Paroxysmal exercise-induced dyskinesia (PED) presents a male:female ratio 2:3. Mean age at onset is 5 years (range 2–30 years) in familial forms. PED appears isolated or coexists with migraine, epilepsy, alternating hemiplegia of childhood (AHC) and ataxia [Citation7].
By definition, a prolonged or maintained exercise normally triggers the episodes [Citation2]. However, in some cases, muscle vibration, passive movements, electrical stimulation of the nerves, stress, hunger, sleep deprivation and exposure to cold may also cause attacks [Citation21,Citation22]. The most common presentation is dystonia in feet (79%) followed by hemidystonia [Citation23]. An exercise with the upper limb may induce an attack limited to the upper limb [Citation21]. Painful dystonia in hands and dancing-like postures mimicking psychogenic attacks have also been reported [Citation24,Citation25]. Attacks usually last between 2 and 5 min and up to 2 h, are not preceded by prodromes and disappear in 10 min after discontinuing the exercise. The episodes vary in frequency, from one per day to two in a month, depending on the routine level of exercise [Citation12].
A sporadic variant of PED is ‘runner’s dystonia’, reported as episodes of leg or foot dystonia after variable forms of exercise (long-distance running, cycling, hiking…). This condition shares with primary focal dystonia the relief by means of sensory tricks; with PKD, the favorable response to AEDs; and with peripherally induced dystonia, prior injury to the affected limb [Citation26].
Genetics: an ever-increasing number of mutations identified
Genetic mutations associated with PxD are listed in .
Table 2. Genetic mutations associated with paroxysmal dyskinesias.
PRRT2 (16p11.2)
In 2011, PRRT2 gene was first reported in eight families with PKD (MIM#128200) [Citation27]. To date, more than 1500 individuals with 70 different mutations in PRRT2, 95% nonsense or frameshift, have been described. c.649dupC/p.Arg217Profs*8 mutation, which leads to a premature stop codon, accounts for 82% of all these [Citation28,Citation29].
Prevalence of PRRT2 mutations in PKD oscillates between 40 and 90% [Citation5,Citation30], with autosomal dominant (AD) transmission and 60–90% penetrance in family forms [Citation31]. The estimation of penetrance may be difficult because some cases present transient or mild phenotypes, and even family members sharing the same mutation express variable phenotypes [Citation31]. Almost all PRRT2-related PxD cases have attacks induced by a kinesigenic trigger (PKD), albeit in up to 40% anxiety/startle, coffee intake or sleep deprivation can also provoke attacks [Citation5]. PRRT2-associated PKD is characterized by young age at onset; brief duration of attacks; both dystonia and chorea that tend to generalize; premonitory feelings; many attacks per day that decrease after puberty and effectivity of AEDs at low doses [Citation5,Citation30,Citation32]. PRRT2-related PNKD and PED have been identified in about 2% of cases [Citation5,Citation33]. In addition, Liu et al. reported two of 11 patients with PRRT2 mutations in PHD, suggesting re-inclusion of PHD as a subtype of PxD [Citation34].
Overlap with other syndromes has been identified. The PRRT2 gene is the leading cause of benign familial infantile seizures (BFIS; MIM#605751) and familial infantile convulsions with paroxysmal choreoathetosis (ICCA) (MIM#602066). PKD, BFIS and ICCA are considered a continuous disease spectrum [Citation29,Citation30]. A total of 5% of patients experience other disorders of early onset with paroxysmal stereotypical manifestations, such as familial hemiplegic migraine (FHM) and other types of migraine, benign paroxysmal torticollis of infancy (BPTI), episodic ataxia, childhood absence epilepsy and febrile seizures [Citation35,Citation36]. Biallelic (homozygous or compound heterozygous) PRRT2 mutations have been linked to a more severe phenotype characterized by a combination of different forms of PxD with persistence of paroxysmal attacks, prolonged episodes of ataxia and developmental delays [Citation37,Citation38]. Moreover, 16p11.2 (micro)deletions have been identified in PKD, especially for sporadic cases, generally with intellectual disability [Citation39,Citation40].
PRRT2 protein is highly expressed in neocortex, hippocampus, basal ganglia and cerebellum, which is consistent with the varied clinical manifestations of PRRT2-related disorders. PRRT2 interacts in glutamatergic synapses with synaptosomal-associated protein (SNAP25), a presynaptic membrane protein that facilitates fusion of synaptic vesicles and calcium-mediated neuronal exocytosis, in order to modulate glutamate release [Citation41]. In PRRT2 mutations, PRRT2 protein changes its usual location from the membrane to the cytoplasm, interacts with SNAP25, alters the properties of the calcium voltage-dependent channel Cav2.1, and thus, enhances intracellular glutamate levels leading to neuronal hyperexcitability [Citation16]. Otherwise, PRRT2 expression increases during the development and is correlated to neuronal migration and synaptic density. This explains that biallelic PPRT2 mutations can result in neurodevelopmental disorders [Citation42].
MR-1/PNKD (2q35)
In 2004, missense mutations in the PNKD gene, also the so-called regulatory gene of the miofibrilogenesis (MR-1) was implicated for the first time in PNKD (MIM#118800) [Citation43]. The most common mutation is p.Ala7Val followed by p.Ala9Val [Citation44]. The mutation alters the amino-terminal α-helix of two MR-1 isoforms: MR-1L, located in cell membranes of the brain and MR-1S, with diffuse cytoplasmic and nuclear expression [Citation45]. The MR-1/PNKD mutations have been identified in families of different ethnicities – the majority of European origin – with PNKD and result from an AD pattern of inheritance with 95% penetrance. No sporadic cases have been reported so far [Citation1,Citation5].
Prevalence of MR-1/PNKD mutations in PNKD has been estimated over 70% [Citation5]. MR-1/PNKD-associated PNKD shows a characteristic phenotype: childhood onset; mixture of chorea and dystonia in limbs, face and trunk; typical duration of attacks between 10 min and 1 h; triggered by caffeine, tea, alcohol or emotional stress; normal interictal examination and favorable response to benzodiazepines. Dysarthria, dysphagia, inability to move, oculogyric crises, blepharospasm and pain are rare [Citation5]. Unlike KCNMA1 mutations discussed below, MR-1/PNKD carriers do not have epilepsy [Citation46]. Nevertheless, 50% of those with MR-1/PNKD mutations experience migraine [Citation47]. Regardless of any treatment, the attacks tend to decrease or disappear in adulthood [Citation5]. In addition to the PNKD phenotype, isolated cases of MR-1/PNKD-related PKD and PED have been discovered [Citation1].
Although the exact MR-1/PNKD function is unknown, this gene is homologous with the HAGH gene, which encodes an enzyme that is involved in the pathway of methylglyoxal – a compound present in coffee and alcoholic beverages – detoxification, producing a byproduct of oxidative stress [Citation45]. This suggests a possible causal mechanism by which alcohol or coffee may induce PNKD attacks, although in vivo studies have dismissed a key role for MR-1L in gluthation metabolism, because MR-1L could not improve HAGH activity [Citation42]. Recently, the fact that MR-1 binds to Rab-3 interacting molecules (RIM) 1 and 2, which interact with vesicle and presynaptic membrane proteins, have raised the hypothesis that the absence of MR-1 displays a calcium-dependent abnormal neurotransmitter release. As caffeine is a ryanodine receptor agonist at presynaptic terminals, favors high concentration of calcium in striatal neurons and enhances the hyperexcitability due to lack of MR-1 [Citation48].
SLC2A1 (1p34.2)
Several mutations in the GLUT1 SLC2A1 had been reported in PED as an isolated syndrome (about 33% of SLC2A1 cases) or accompanied by other features of the GLUT deficiency syndrome (GLUT1-DS MIM#138140), especially epilepsy [Citation49]. Despite the fact that there are cases of autosomal recessive (AR) inheritance in the GLUT1-DS 2, this type of inheritance has not been described in PED associated to SLC2A1, in which the majority (90%) corresponds to sporadic cases and 10% to familial cases with AD transmission [Citation50,Citation51]. In addition, a German family with PED and progressive spastic paraparesis (paroxysmal choreoathetosis/spasticity [CSE, DYT9]) (MIM #601042) has been reported as a result of a heterozygous mutation (p.Arg232Cys) in SCL2A1 [Citation52]. Another mutation (p.Arg126Cys) in this gene has been found in Australian monozygotic twins with PED and progressive spastic paraparesis, but not in 139 patients with hereditary spastic paraparesis without PED [Citation53].
A correlation phenotype–genotype has been observed for SLC2A1: splicing and nonsense mutations, insertions and deletions (mutations with loss-of-function) are linked to a more severe GLUT-SD clinical phenotype (drug-resistant epilepsy, hypotonia, microcephaly, dysarthria, spasticity, ataxia, complex movement disorders, hemolytic anemia and developmental delay), whereas missense mutations are more common in PED [Citation1,Citation9,Citation49]. Other atypical paroxysmal manifestations have been identified in GLUT1-DS, such as parkinsonism, eye-head movements or AHC [Citation51,Citation54,Citation55]. The diagnosis of GLUT-DS relies mainly on the identification of pathogenic SLC2A1 mutations and a cerebrospinal fluid (CSF)/serum glucose ratio <0.60 in a fasting sample of at least 4 h [Citation56]. The glucose transporter facilitates glucose to cross the blood–brain barrier, which explains low concentrations of glucose in CSF of patients with SLC2A1 mutations. Recently, the detection of a 20% decrease in GLUT expression on the surface of erythrocytes by means of flow cytometry analysis has been proposed as a less-invasive and inexpensive alternative diagnostic method [Citation57].
Prevalence of SLC2A1 mutations in PED is less than 20% [Citation9]. PED cases carrying SLC2A1 mutations involve: choreoathetosis episodes, with tightness or cramps and focal or unilateral distribution, principally in the lower limbs; typical duration of attacks between 15 and 45 min; triggered by prolonged exercise, fasting, stress and anxiety; favorable response to ketogenic diet; and frequent comorbidity [Citation9,Citation58,Citation59]. Additional features observed in 10% of cases include migraine, clumsiness, gait disturbances, oculogyric crises and weakness. About 5% are precipitated by nonkinesigenic triggers (PNKD) or experience episodic ataxia, whereas isolated cases correspond to PKD [Citation9,Citation47,Citation60,Citation61].
KCNMA1 (10q22.3)
In 2005, Du et al. communicated a family with generalized epilepsy and PNKD inherited in an AD with incomplete penetrance pattern carrying a missense mutation (p.Asp434Gly) in the α subunit of the calcium-sensitive potassium channel (KCNMA1). Mean age at onset was 5 years (range: 1–15 years). Clinically, PNKD presented alcohol as a possible (but not constant) trigger [Citation46]. Later on, KCNMA1-related PNKD mutations (p.Glu884Lys, p.Asn1053Ser) have been reported with epilepsy and/or neurodevelopmental delay, unlike MR-1/PNKD carriers (MIM#609446) [Citation62]. One case with PKD, migraine and infantile convulsions has also been reported [Citation61]. Recent research has attributed to homozygous KCNMA1 mutations (c.2026dupT) a phenotype of cerebellar atrophy, developmental delay and seizures (MIM#617643) [Citation63]. KCNMA1 mutations enhance neuronal excitability due to rapid repolarization of the action potential [Citation42].
ECHS1 (10q26.3)
Mutations in the ECHS1 (enoyl CoA hydratase, short chain, 1 or crotonase, mitochondrial) gene which show AR inheritance convey a variable phenotype – even within families – ranging from early-onset Leigh syndrome to atypical forms often milder with later onset (Leigh-like syndrome), with different combinations of psychomotor retardation, epilepsy, spasticity, optic atrophy, sensorineural deafness or PxD (MIM#616277). PxD may be superimposed to theses syndromes or isolated [Citation64].
ECHS1 carriers of missense (p.Ala173Val, p.Lys273Glu) and nonsense mutations (p.Glu78Ter) displayed intermittent opisthotonus or generalized dystonia of long duration (30–50 min) without any clear trigger in more severe phenotypes, whereas those with Leigh-like syndrome or isolated PxD revealed PED in legs of shorter duration (5–40 min). Interestingly, all patients had pallidal hyperintensity on T2-MRI sequences, regardless of clinical severity [Citation64–66]. Crotonase is involved in the catabolism of fatty acids and amino acids such as valine. ECHS1 mutations have been linked to a diminished pyruvate oxidation, ATP-production and activity of complexes I and IV of the respiratory mitochondrial chain, favoring the accumulation of toxic metabolites (e.g., methylacrylyl-Co) and, thus, the lesions in the basal ganglia [Citation64,Citation67]. Early treatment based on mitochondrial cocktail, as described below, has showed a possible benefit in PxD [Citation64–66].
PDHA1 (Xp22.12), DLAT (11q23.1), PDHX (11p13)
The mitochondrial pyruvate dehydrogenase complex (PDC) catalyzes decarboxylation of pyruvate to acetyl-CoA, a key-step in aerobic oxidation of glucose, and therefore, in brain metabolism. PDC includes three enzymes pyruvate dehydrogenase (PDC-E1, encoded by PDHA1 gene), dihydrolipoamide acyltransferase (PDC-E2, encoded by DLAT gene) and dihydrolipoyl dehydrogenase (PDC-E3) as well as one structural E2/E3 binding protein (both encoded by PDHX gene) [Citation1,Citation68].
Mutations in PDHA1 (most common), DLAT and PDHX genes – with an X-linked transmission (except for PDHX, AR) – can cause PxD usually in addition to other manifestations, ranging from early-onset severe encephalopathy with lactic acidosis to Leigh syndrome, hypotonia, epilepsy, neurodevelopmental delay, episodic ataxia or intermittent flaccid paralysis (MIM#312170, 245348 and 245349) [Citation1,Citation69,Citation70]. Episodic dystonia are sometimes triggered by exercise (PED) and others by stress, a warm bath or no identifiable trigger (PNKD) [Citation1,Citation68–71]. Noteworthy, PxD can be an isolated symptom in cases with PDC deficiency, even in absence of classical hallmarks such as high serum or CSF lactate or piruvate or pallidal hyperintensity on T2-MRI sequences due to striatal necrosis [Citation1,Citation70].
GCH1 (14q22.2)
GCH1 gene codes for the GTP cyclohydrolase I, a rate-limiting enzyme in the metabolism of tetrahydrobiopterin, which is required for dopamine synthesis. Accordingly, GCH1 carriers present a reduction in striatal dopamine levels. Heterozygous GCH1 mutations account for 50% of dopa-responsive dystonia (DRD: MIM#128230), which typically presents in childhood, involves lower limbs and benefits from levodopa at low doses [Citation1,Citation72]. Besides dopa-responsive dystonia, GCH1 mutations have been reported as a cause of PED, either in isolation or embedded in a more complex phenotype. Dale et al. found a family with a nonsense mutation leading to a premature stop codon (p.Glu84X) in an AD pattern that suffered PED (attacks of dystonia lasting 5 min), restless legs syndrome, depression and adulthood onset parkinsonism, which ameliorate with levodopa [Citation73]. Erro et al. have estimated that GCH1 mutations occur in 12.5% of those with PED and hence a levodopa trial should be considered in PED patients [Citation74].
SCN8A (12q13.3)
SCN8A gene encodes the α-subunit NaV1.6 of voltage-gated sodium channel, which is involved in the initiation and propagation of action potentials [Citation42]. Mutations in SCN8A have been suggested as an alternative cause to PRRT2 in some cases of PKD, BFIS, and their combination ICCA. Gardella et al. described three families with 16 members carrying the same heterozygous mutation (p.Glu1483Lys) in the SCN8A gene, which experienced seizures within the first 2 years and five of them added choreo-dystonic attacks precipitated by stretching, movement initiation or emotions at older age. However, a cortical involvement was documented in one case of ‘PKD’ spell recorded by video-electroencephalography (EEG), suggesting an epileptic origin [Citation1,Citation75].
Interestingly, the presence of episodic dystonia has been described in other SCN8A carriers (p.Ala1214Thr) [Citation61]. Therefore, SCNA8 mutations should be considered in the differential diagnosis of PKD, particularly in those cases with seizures refractory to AEDs, neurodevelopment delay and co-existence with nonepisodic dystonia and ataxia, all of them different from the features associated with PRRT2 [Citation1,Citation76]. Functional studies have proposed that SCN8A mutations can result in either gain-of-function (i.e., persistent sodium current) promoting seizures and loss-of-function (i.e., an unstable protein) provoking cognitive dysfunction, although no clear genotype–phenotype correlation have been defined so far [Citation42].
ADCY5 (3q21.1)
ADCY5 gene encodes for adenylate cyclase 5, which is strongly expressed in the striatum and converts adenosine triphosphate to 3′, 5′-cyclic adenosine monophosphate and pyrophosphate. ADCY5 integrates signals from adenosine A2A, D1 and D2 dopamine receptors, among others [Citation72]. ADCY5 mutation carriers display a broad phenotypic spectrum (MIM#606703) with a genotype–phenotype correlation: the most common mutation, p.Arg418Trp, presents with a more severe phenotype than p.Arg418Gln and p.Ala726Thr [Citation77]. These mutations likely increase adenylate cyclase activity to a lesser or greater degree, thereby distinct affecting signal transduction in the striatum [Citation72].
ADCY5 patients typically have a childhood onset of mixed persistent hyperkinetic movements (chorea, dystonia or myoclonus) characterized by the following features: axial hypotonia, orofacial jerks, not true myokymia, sleep-related PxD, painful PxD, marked fluctuations in frequency and severity of movements, no seizures, ataxia or marked cognitive impairment, normal MRI and stable or very slow progression [Citation78–80]. Attacks of PxD last for min with discrete on and off set and wax and wane over weeks to months [Citation79]. This pattern makes difficult to distinguish triggers or response to medications [Citation78]. In fact, the combination of multiple PxD subtypes (e.g., PKD, PNKD and PHD) or PxD that do not fit clearly into previous defined PxD categories are clues to suspect ADCY5 mutations [Citation79]. Recently, Westenberg et al. reported two unrelated ADCY5 patients with episodes similar to those of AHC [Citation77].
ATP1A3 (19q13.2)
Mutations in ATP1A3, which encodes the α3-subunit of the Na+-K+-ATPase pump, cause different clinical syndromes, including AHC (MIM#614820) (50% missense mutations p.Asp801Asn and p.Glu815Lys, with a high prevalence of de novo mutations), rapid-onset dystonia-parkinsonism (RDP) (MIM#128235) and the cerebellar ataxia, areflexia, pes cavus, optic atrophy and sensorineural hearing loss (CAPOS) syndrome (MIM #601338) (with AD transmission and mainly mutations in exons 8, 14 and 17), the relapsing encephalopathy with cerebellar ataxia (RECA) syndrome and intermediate syndromes with clinical overlap [Citation72,Citation81]. There is a growing notion of genotype–phenotype correlations, albeit the pathophysiology is poorly understood. However, Na+-K+-ATPase enzyme is determinant for resting membrane potentials; and its α3-subunit is mainly expressed in GABAergic neurons of basal ganglia and cerebellum [Citation72,Citation82].
PNKD and/or PED are described in most AHC patients and in a few rapid-onset dystonia-parkinsonism cases. AHC manifest within 18 months of age as recurrent episodes of hemiplegia involving either side of the body, tetraplegia, tonic or dystonic attacks, ocular abnormalities (especially monocular nystagmus) and autonomic phenomena. The duration and frequency of the episodes is variable, even in the same patient, lasting from minutes to days and occurring up to several times a day. Attacks are triggered by emotional or physical stressors (physical activity, exposure to hot water, light and sounds, etc) and disappear falling asleep. AHC usually evolves with epilepsy, developmental delay, dysarthria, ataxia and choreoathetosis with a rostrocaudal gradient of severity [Citation81–84].
CACNA1A (19p13.13)
Mutations in the CACNA1A gene, which encodes the pore-forming subunit of a voltage-dependent P/Q-type calcium channel, have been linked to familial hemiplegic migraine (MIM#141500), episodic ataxia type 2 (MIM#148500), SCA6 (MIM#183086) and BPTI [Citation72,Citation85]. In BPTI: episodes of head tilt last from 10 min to 2 months; the shorter episodes are followed by vomiting, apathy and unsteadiness; BPTI begin within the first 12 months and resolve by 5 years, but can be replaced by vertigo and/or migraine [Citation85–87].
SLC16A2 (Xq13.2)
SLC16A2 gene encodes the monocarboxylate transporter type 8 (MCT8), which carry out neural uptake of triiodothyronine (T3). SLC16A2 mutations are the cause of Allan–Herndon–Dudley syndrome (MIM#300523), characterized by proximal hypotonia, PxD with spasticity, marked development delay, ataxia, dysarthria and craniofacial dysmorphic appearance [Citation88]. PxD in SLC16A2 carriers classically fit into PKD category, because attacks are provoked by passive movements (e.g., by changing their clothes or nappies or by lifting them). Nevertheless, episodes can be precipitated by emotional stressors (e.g., laughing, crying). Attacks usually are dystonic with duration from seconds to few minutes. Thyroid hormone function tests evidence elevated serum T3 and fT3 levels and decreased T4 and fT4 levels with TSH levels in the upper normal range or elevated [Citation88,Citation89].
Secondary causes: an extensive list
Approximately 22% of PxD presented an acquired cause [Citation6]. Secondary PxD should be considered, especially in adulthood onset and episodes not fitting in a specific trigger-based category [Citation1]. Development of PxD does not always occur immediately after the injury [Citation90]. ‘Secondary’ causes of PxD are summarized in [Citation1,Citation6,Citation72,Citation91].
Table 3. Causes of secondary paroxysmal dyskinesia.
Multiple sclerosis (MS)-related PxD imply brief (seconds to minutes) hemidystonic painful postures preceded by aura and triggered by hyperventilation, fatigue or startle reaction. PxD appear during the course of the disease or rarely as the presenting symptom, and a self-limiting course independent of disease activity predominates. PxD in MS are associated with lesions in any part of the CNS and are attributed to alterations of ephaptic transmission [Citation92].
LGI1-associated limbic encephalitis presents late in life with prototypical PxD so-called ‘faciobrachial dystonic seizures’. These are brief hemidystonic postures, up to several hundred per day, which occur spontaneously or precipitated by emotions, auditory stimuli or movement, although longer, bilateral and drop attacks have been reported. A total of 40% of them show basal ganglia hyperintensities on T1- or T2-MRI. Brief dystonic episodes and PED have been linked to NMDAR encephalitis. And painful tonic spasms are evidenced more frequently in neuromyelitis optica with AQP4 antibodies than in MS [Citation93].
In metabolic disorders, the identification of isolated PxD in members of two families carrying mutations in SLC20A2 and PDGFB genes, related to familial idiopathic basal ganglia calcification, raises controversy about the role of brain calcification as a lesional mechanism, since clinical expression of brain calcification is not constant [Citation1,Citation94,Citation95]. Otherwise, PxD related to stroke often occur after resolution of the initial deficits and contralateral to the infarct, mainly located in thalamus or putamen. PxD in form of ‘limb-shaking’ appear as a rare manifestation of carotid-occlusive disease in transient ischemic attacks [Citation6,Citation96].
PxD associated with HIV may be the result of opportunistic infections or reveal in advanced stages encephalitis due to severe HIV and loss of positive neurons to calbindin in the basal ganglia [Citation97]. Peripheral trauma has been suggested to promote PxD by means of aberrant cortical and spinal reorganization [Citation98]. Finally, an indirect antidopaminergic effect for the development of PxD has been postulated in most drug-induced PxD, such as selective serotonin reuptake inhibitors (SSRIs) [Citation99].
Diagnostic work up
The first step in the diagnosis of PxD is to exclude other paroxysmal disorders: seizures, tetany (carpopedal spasm), neuromyotonia, motor stereotypies, cataplexy, periodic paralyses, episodic ataxias, etc () [Citation1]. For clinical characterization of the episodes to be useful, ask the patient that imitates the attacks or that someone records them using a video mobile device [Citation100]. Those doubtful cases may require appropriate investigations (e.g., focal seizures arising from the supplementary motor area that often mimics PKD and in which an interictal EEG or even a video EEG may be necessary to rule out epilepsy) [Citation91,Citation101].
Asterisks denote main genes to consider within each category.
PED: Paroxysmal exercise-induced dyskinesia; PHD: Paroxysmal hypnogenic dyskinesia; PKD: Paroxysmal kinesigenic dyskinesia; PNKD: Paroxysmal nonkinesigenic dyskinesia.
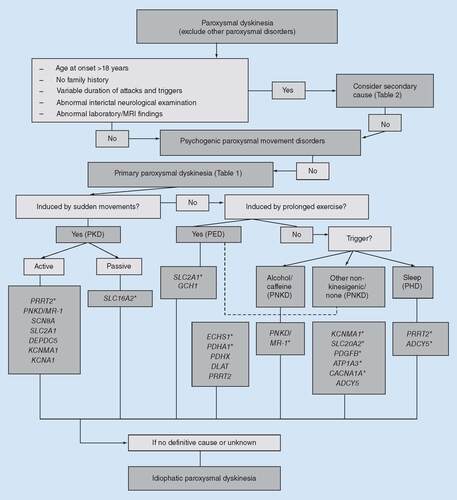
Whenever attacks have variable duration and no specific triggers, adulthood onset and absence of family history, a ‘secondary’ cause should be considered [Citation6]. There are no specific patterns for a certain etiology, but painful PxD, fluctuating levels of consciousness and dysautonomic symptoms are rarely observed in genetic forms. Focal neurological signs in the interictal examination are discarded as a differential criterion between primary and secondary cases by some authors, because additional clinical features have been reported in several mutations [Citation1]. An initial investigation including blood tests with metabolic and autoimmune panels as well as brain imaging will reveal the acquired cause in most cases.
A psychogenic paroxysmal movement disorder (PPMD) accounts for 10% of PxD cases. Though the presentation of PPMD is diverse, Ganos et al. established some positive signs that support a psychogenic origin instead of a primary form of PxD. The list of red flags include: adulthood onset; paroxysmal tremor; high within-subject phenomenological variability; marked increases in attack frequency and severity during examination; highly variable attack duration; numerous and unusual triggers; alteration of responsiveness during attacks; odd precipitating factors and relieving manoeuvres; medically unexplained somatic symptoms and atypical response to medication [Citation102,Citation103]. Nevertheless, PPMD can occur in the context of previously documented ‘organic’ PxD [Citation104]. Physical and cognitive-behavioural therapies are useful in a number of patients [Citation102].
Once a ‘primary’ PxD is suspected, a trigger-based approach (PKD, PNKD, PED) with a further distinction in precipitating factors between alcohol/caffeine, sleep and other nonkinesigenic triggers in PNKD or active and passive movements in PKD should be helpful to aim genetic studies. Additional diagnostic clues are supported by the determination of duration and distribution of attacks, the presence of suggestive associated clinical features and inheritance pattern [Citation1]. Notwithstanding, genetic diagnosis is sometimes hindered by a lack of accurate genotype–phenotype correlation, because certain phenotypes may be caused by mutations in different genes [Citation100].
Treatment
Treatment for PxD is based on anecdotal evidence, observational studies and expert consensus. The fact that effective treatments were reported before the knowledge of underlying genotypes support a therapeutic trial directed to the phenotypes [Citation100].
PKD responds well to low doses of AEDs, particularly carbamazepine [Citation5]. Oxcarbazepine, phenytoin, phenobarbital, lamotrigine, levetiracetam, gabapentin, valproate and topiramate can also be useful [Citation105–107]. A dramatic reduction in attacks with low doses of carbamazepine or other AEDs modulating voltage-gated sodium channels is expected in PRRT2 carriers as compared with PRRT2 negative-associated PKD cases (e.g., SCN8A) [Citation1,Citation76]. In PRRT2 mutations, a gradual tapering of AEDs may be indicated because of the propensity to remissions beyond adolescence [Citation1,Citation5]. Thalamotomy has been discovered to be beneficial in four members of a PRRT2 family suggesting a thalamic involvement in PKD [Citation108]. To date, no treatment is available for SLC16A2 carriers [Citation88,Citation89].
For patients with PNKD, identification and avoidance of triggering factors (e.g., caffeine, alcohol, stress…) is recommended. Benzodiazepines such as clonazepam may be effective in treating or preventing attacks, particularly in PNKD/MR-1 – the principal causative gene in isolated forms [Citation5,Citation105]. Anecdotal responses to levetiracetam, valproate, acetazolamide, gabapentin and oxcarbazepine have also been reported [Citation12]. Other treatments have shown inconsistent results, including haloperidol, oxazepam and anticholinergics [Citation105]. A patient with painful PNKD has been successfully treated with deep brain stimulation targeting ventral intermediate (Vim) nucleus of the thalamus, while internal globus pallidus (Gpi) stimulation proved to be useful in a couple of cases with refractory severe PNKD [Citation109–111].
Apart from PNKD/MR-1, other mutations manifest mainly with PNKD. KCNMA1 carriers variably respond to AEDs [Citation46]. In ADCY5 mutations, clonazepam reduced sleep myoclonic episodes in several patients and levetiracetam improved movements in a few; other isolated cases obtained benefits from tetrabenazine and deep brain stimulation [Citation1,Citation78]. Management in ATP1A3-related AHC includes a double approach: in acute treatment, fast-acting benzodiazepines or AEDs (e.g., phenobarbital), which often relieve attacks directly – increasing activity of the inhibitory neurotransmitter GABA – or by inducing sleep; and in preventive treatment, flunarizine reduced duration and frequency of episodes in 50% and intensity in 32% of 30 patients included in a retrospective study, and topiramate was also used in anecdotal reports [Citation112,Citation113]. For CACNA1A usually no treatment is required due to its self-limiting nature [Citation1,Citation86].
Prolonged exercise abstinence has been proposed in PED [Citation107]. Those patients tend to respond poorly to medication, albeit a trial with any of the available options (levodopa, AEDs, benzodiazepines, acetazolamide, ketogenic diet) is recommended [Citation100]. Ketogenic diet in SLC1A2 mutations results effective on seizures control but less on PED. This diet, which is calorie-reduced and high-fat, produces ketone bodies as an alternative energy source for brain metabolism, but often side effects (e.g., diarrhoea, fatigue) are intolerable [Citation114]. A less restrictive modified Atkins diet shows similar effectivity [Citation115]. Recently, an open-label study with triheptanoin, an odd-chain triglyceride that replenishes metabolic intermediates in the Krebs circle, resulted in a 90% reduction in nonepileptic paroxysmal manifestations in a cohort of GLUT1-DS with PED [Citation116]. GCH1 carriers dramatically benefit from levodopa supplementation [Citation74]. In ECHS1 mutations, PED is possible to benefit from ketogenic diet or a mitochondrial cocktail (vitamins B1, B2, B6 and C plus carnitine and coenzyme Q-10) [Citation65,Citation66]. Similarly, in PDHA1, DLAT and PDHX carriers, ketogenic diet and especially thiamine supplementation have been reported to improve clinical, biochemical and MRI parameters [Citation69,Citation70].
In patients with secondary PxD, the identification and treatment of the underlying cause is essential [Citation6]. PxD in the context of MS responds to AEDs; acetazolamide is useful as an alternative or a complement [Citation92]. Benzodiazepines seem to be beneficial in patients with HIV infection [Citation97]. PxD secondary to traumatic brain injury sometimes respond to AEDs alone or in combination with trihexyphenidyl. Botulinum toxin has been used in focal paroxysmal dystonia. In metabolic cases, the underlying disorder should be treated (e.g., hypoglycemia, hyperglycemia, thyrotoxicosis…). Treatment of hypocalcemia with vitamin D is crucial in PKD associated with hypoparathyroidism [Citation6,Citation105] as well as immunotherapy in limbic encephalitis [Citation93].
Conclusion
PxD should be considered in the differential diagnosis of paroxysmal disorders, in order to avoid a delay in diagnosis and treatment, undergoing unnecessary additional investigations. The ever-expanding list of genetic and acquired causes and lack of reliable phenotype–etiology correlations in some cases deserve a more updated classification system for PxD.
Future perspective
First of all, a more precise nomenclature globally agreed in the future for these disorders should be argued, since the term ‘paroxysmal dyskinesias’ is ambiguous per se. Etymologically the concept ‘paroxysmal’ indicates sudden attacks, recurrence or intensification of a disease, while ‘dyskinesias’ refers to involuntary jerky movements with a fixed pattern or an impairment of the ability to execute voluntary movements. In practice, conditions that do not match with the meaning that movement disorders experts pretend give to PxD, such as tic-syndromes, action-myoclonus or action-tremor, would fit into this category [Citation1]. Moreover, the term ‘paroxysmal’ can be mistaken as it somehow implies normal interictal examination, which is not always found [Citation100]. In fact, the stratification of PxD into ‘pure’ and ‘complicated’ PxD, depending on the absence or presence of interictal neurological signs, respectively, has been proposed [Citation1].
Likewise, a more up-to-date classification system for PxD should be implemented. For instance, the vagueness of nonkinesigenic category would justify distinguish alcohol/caffeine sensitivity and sleep as distinctive triggers (as referred in the diagnostic work-up), or consider other clinical features such as body distribution (i.e., AHC or BPTI) [Citation1]. Also, the inclusion of new terms like ‘status dyskineticus’ or ‘dyskinetic storms’ (coined by Silveira-Moriyama et al. to report life-threatening exacerbations associated to GNAO1 gene mutations) can be helpful [Citation100,Citation117]. Besides, the use of the eponyms ‘DYT’ for PxD is strongly discouraged, because the initial allocation of PxD to loci DYT generated confusion. In this sense, PKD was initially assigned DYT 10, but the c.649dupC mutation in the PRRT2 gene was also found in the family used to define DYT19, a supposedly distinct form of PKD, suggesting that the first linkage in this family was wrong. Similarly, DYT9 (paroxysmal choreoathetosis with spasticity) and DYT 18 (PED) were attributable to SLCA1 mutations [Citation5].
In addition, both the lack of a specific phenotype associated to a single gene mutation and the ever-expanding list of causative genes involved in PxD creates a challenge in diagnosis work-up [Citation1,Citation100]. The application of novel technologies (next generation sequencing) instead of candidate gene studies has been endorsed not only for a more rapid, successful and cost-effective genetic screening but also to uncover novel pathogenic variants in familial and sporadic cases [Citation118,Citation119]. Moreover, replication is required for those genes in which real nature of paroxysmal episodes remains to be elucidated (e.g., DEPDC5 and CHRNA1 mutations observed in PKD plus epilepsy in some families but also in autosomal dominant nocturnal frontal lobe epilepsy) [Citation1,Citation61,Citation120]. Future functional studies in animal models might shed light into the role of identified genes [Citation121].
Finally, the usefulness of a two-pronged method, in which the underlying etiology (genetic or not) and the clinical phenotype are specified (i.e., PRRT2-PKD, HIV encephalitis-PNKD) has been suggested. And the label ‘idiophatic’ PxD should be reserved for those patients who remain unclassified because no definite cause has been found till the date [Citation1,Citation100].
Paroxysmal dyskinesias: redefining concepts & classifications
Paroxysmal dyskinesias (PxD) are characterized by intermittent attacks of abnormal involuntary movements, without loss of consciousness.
PxD are classified by triggers (paroxysmal kinesigenic [PKD], nonkinesigenic [PNKD] and exercise-induced [PED] dyskinesias) and etiology (primary [genetic or idiopathic] and secondary).
Clinical features: role of triggers
PKD are brief attacks induced by sudden movements, PNKD are longer-lasting attacks provoked by alcohol, caffeine or other nonkinesigenic triggers, whereas PED are intermediate attacks triggered by prolonged exercise.
Genetics: an ever-increasing number of mutations identified
PKD is most commonly due to mutations in the PRTT2 gene, PNKD in the PNKD/MR-1 gene and PED in the SLC2A1 gene.
There is an ever-expanding list of causative genes involved in PxD, which lack complete phenotypic–genotypic correlation, with pure and complicated forms.
Secondary causes: an extensive list
Secondary PxD should be considered in adulthood onset of attacks, not fitting in a specific trigger-based category.
The acquired causes include immune-mediated, metabolic, vascular, infectious and neurodegenerative disorders, as well as trauma, tumors and drugs.
Diagnostic work-up
The exclusion of other paroxysmal disorders, the identification of secondary forms with appropriate investigations and the recognition of psychogenic paroxysmal movement disorders constitute the first steps in diagnostic work-up.
Whenever primary PxD is considered, an approach based on triggers, duration and distribution of attacks, and associated clinical features should be helpful.
Treatment
PKD responds well to antiepileptics, benzodiazepines are useful in PNKD and in PED ketogenic diet and levodopa can be tried.
As genotypic spectrum of PxD evolves, so does the range of therapeutic options.
Conclusion
An updated classification system for PxD is required in order to avoid delay in diagnosis and treatment.
Future perspective
Improvements in nomenclature and classification should be implemented, with both ethiological and phenotypic specifications recommended.
The role of novel pathogenic variants uncovered deserves replication and further functional studies.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- ErroR, BhatiaKP. Unravelling of the paroxysmal dyskinesias. J. Neurol. Neurosurg. Psychiatr.0, 1–8 (2018). doi:10.1136/jnnp-2018-318932.
- LanceJW. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann. Neurol.2, 285–293 (1977).
- DemirkiranM, JankovicJ. Paroxysmal dyskinesias: clinical features and classification. Ann. Neurol.38, 571–579 (1995).
- TinuperP, CerulloA, CirignottaF, CortelliP, LugaresiE, MontagnaP. Nocturnal paroxysmal dystonia with short lasting attacks. Three cases with evidence for an epileptic frontal lobe origin of seizures. Epilepsia31, 549–556 (1990).
- ErroR, SheerinUM, BhatiaK. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov. Disord.29, 1108–1116 (2014).
- BlakeleyJ, JankovicJ. Secondary paroxysmal dyskinesias. Mov. Disord.17, 726–734 (2002).
- BhatiaKP. Paroxysmal dyskinesias. Mov. Disord.26, 1157–1165 (2011).
- KerteszA. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology17, 680–690 (1967).
- BrunoMK, HallettM, Gwinn-HardyKet al.Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology63, 2280–2287 (2004).
- HouserMK, SolandVL, BhatiaKP, QuinnNP, MarsdenCD. Paroxysmal kinesigenic choreoathetosis: a report of 26 patients. J. Neurol.246, 120–126 (1999).
- SunW, LiJ, ZhuY, YanX, WangW. Clinical features of paroxysmal kinesigenic dyskinesia: report of 24 cases. Epilepsy Behav.25, 695–699 (2012).
- McGovernEM, RozeE, CounihanTJ. The expanding spectrum of paroxysmal movement disorders: update from clinical features to therapeutics. Curr. Opin. Neurol.31, 491–497 (2018).
- HuangX-J, WangT, WangJ-Let al.Paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 110 patients. Neurology85, 1546–1553 (2015).
- MountLA, RebackS. Familial paroxysmal choreoathetosis. Arch. Neurol. Psychiatr.44, 841–847 (1940).
- BrunoMK, LeeHY, AuburgerGWet al.Genotype–phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology68, 1782–1789 (2007).
- FinkJK, RainerS, WilkowskiJet al.Paroxysmal dystonic choreoathetosis: tight linkage to chromosome 2q. Am. J. Hum. Genet.59, 140–145 (1996).
- GhezziD, ViscomiC, FerliniAet al.Paroxysmal non-kinesigenic dyskinesia is caused by mutations of the MR-1 mitochondrial targeting sequence. Hum. Mol. Genet.18, 1058–1064 (2009).
- ZittelS, GanosC, MünchauA. Fatal paroxysmal non-kinesigenic dyskinesia. Eur. J. Neurol.22, e30–e31 (2015).
- StefanoE, DjarmatiA, MomcilovicDet al.Clinical characteristics of paroxysmal nonkinesigenic dyskinesia in Serbian family with Myofibrillogenesis regulator 1 gene mutation. Mov. Disord.21, 2010–2015 (2006).
- ByrneE, WhiteO, CookM. Familial dystonic choreoathetosis with myokymia; a sleep responsive disorder. J. Neurol. Neurosurg. Psychiatr.54, 1090–1092 (1991).
- PlantGT, WilliamsAC, EarlCJ, MarsdenCD. Familial paroxysmal dystonia induced by exercise. J. Neurol. Neurosurg. Psychiatr.47, 275–279 (1984).
- WaliGM. Paroxysmal hemidystonia induced by prolonged exercise and cold. J. Neurol. Neurosurg. Psychiatr.55, 236–237 (1992).
- BhatiaKP, SolandVL, BhattMH, QuinnNP, MarsdenCD. Paroxysmal exercise-induced dystonia: eight new sporadic cases and a review of the literature. Mov. Disord.12, 1007–1012 (1997).
- ClarkCN, WeberYW, LercheHet al.Paroxysmal exercise-induced dyskinesia of the hands. Mov. Disord.27, 1579–1580 (2012).
- TacikP, LoensS, SchraderCet al.Severe familial paroxysmal exercise-induced dyskinesia. J. Neurol.261, 2009–2015 (2014).
- WuLJ, JankovicJ. Runner’s dystonia. J. Neurol. Sci.251, 73–76 (2006).
- ChenWJ, LinY, XiongZQet al.Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet.43, 1252–1255 (2011).
- FruscioneF, ValenteP, SterliniBet al.PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain141, 1000–1016 (2018).
- MeneretA, GaudeboutC, RiantF, VidailhetM, DepienneC, RozeE. PRRT2 mutations and paroxysmal disorders. Eur. J. Neurol.20, 872–878 (2013).
- Ebrahimi-FakhariD, SaffariA, WestenbergerAet al.The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain138, 3476–3495 (2015).
- van VlietR, BreedveldG, deRijk-van Andel Jet al.PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology79, 777–784 (2012).
- LiHF, ChenWJ, NiWet al.PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology80, 1534–1535 (2013).
- WangK, ZhaoX, DuYet al.Phenotypic overlap among paroxysmal dyskinesia subtypes: lesson from a family with PRRT2 gene mutation. Brain Dev.35, 664–666 (2013).
- LiXR, HuangD, WangJet al.Paroxysmal hypnogenic dyskinesia is associated with mutations in the PRRT2 gene. Neurol. Genet.2, 66 (2016).
- GardinerAR, BhatiaKP, StamelouMet al.PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology79, 2115–2121 (2012).
- DaleRC, GardinerA, AntonyJet al.Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine. Dev. Med. Child Neurol.54, 958–960 (2012).
- DelcourtM, RiantF, ManciniJet al.Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J. Neurol. Neurosurg. Psychiatr.86, 782–785 (2015).
- LabateA, TarantinoP, ViriMet al.Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype wtih mental retardation, episodic ataxia, and absences. Epilepsia53, 196–199 (2012).
- WeberA, KöhlerA, HahnAet al.Benign infantile convulsions (IC) and subsequent paroxysmal kinesigenic dyskinesia (PKD) in a patient with 16p11.2 microdeletion syndrome. Neurogenetics14, 251–253 (2013).
- LiW, WangY, LiB, TangBet al.16p11.2 deletion in patients with paroxysmal kinesigenic dyskinesia but without intellectual disability. Brain Behav.8, e01134 (2018).
- ValtortaF, BenfenatiF, ZaraFet al.PRRT2: from paroxysmal disorders to regulation of synaptic function. Trends Neurosci.39, 668–679 (2016).
- ErroR, BhatiaKP, EspayAJet al.The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: channelopathies, synaptopathies, and transportopathies. Mov. Disord.32, 310–318 (2017).
- RainierS, ThomasD, TokarzDet al.Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch. Neurol.61, 1025–1029 (2004).
- YehTH, LinJJ, LaiSCet al.Familial paroxysmal nonkinesigenic dyskinesia: clinical and genetic analysis of a Taiwanese family. J. Neurol. Sci.323, 80–84 (2012).
- LeeHY, XuY, HuangYet al.The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum. Mol. Genet.13, 3161–3170 (2004).
- DuW, BautistaJF, YangHet al.Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet.37, 733–738 (2005).
- GardinerAR, JafferF, DaleRCet al.The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain138, 3567–3580 (2015).
- ShenY, GeWP, LiYet al.Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc. Natl Acad. Sci. USA112, 2935–2941 (2015).
- LeenWG, KlepperJ, VerbeekMMet al.Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain133, 655–670 (2010).
- WeberYG, StorchA, WuttkeTVet al.GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J. Clin. Invest.118, 2157–2168 (2008).
- SchneiderSA, Paisan-RuizC, Garcia-GorostiagaIet al.GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov. Disord.24, 1684–1688 (2009).
- WeberYG, KammC, SulsAet al.Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology77, 959–964 (2011).
- AuburgerG, RatzlaffT, LunkesAet al.A gene for autosomal dominant paroxysmal choreoathetosis/spasticity (CSE) maps to the vicinity of a potassium channel gene cluster on chromosome 1p, probably within 2 cM between D1S443 and D1S197. Genomics31, 90–94 (1996).
- PearsonTS, PonsR, EngelstadK, KaneSA, GoldbergME, DeVivo DC. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology88, 1666–1673 (2017).
- PellegrinS, CantalupoG, OpriRet al.EEG findings during ‘paroxysmal hemiplegia’ in a patient with GLUT1-deficiency. Eur. J. Paediatr. Neurol.21, 580–582 (2017).
- KochH, WeberYG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav.91, 90–93 (2018).
- GrasD, CousinC, KappelerCet al.A simple blood test expedites the diagnosis of glucose transporter type 1 deficiency syndrome. Ann. Neurol.82, 133–138 (2017).
- SulsA, DedekenP, GoffinKet al.Paroxysmal exercise induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain131, 1831–1844 (2008).
- PearsonTS, AkmanC, HintonVJet al.Phenotypic spectrum of glucose transporter type1 deficiency syndrome(Glut1DS). Curr. Neurol. Neurosci. Rep.13, 342 (2013).
- WangHX, LiHF, LiuGL, WenXD, WuZY. Mutation analysis of MR-1, SLC2A1, and CLCN1 in 28 PRRT2-negative paroxysmal kinesigenic dyskinesia patients. Chin. Med. J.129, 1017–1021 (2016).
- TianWT, HuangXJ, MaoXet al.Proline-rich transmembrane protein 2-negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov. Disord.33, 459–467 (2018).
- ZhangZ, TianM, JiangYet al.De novo KCNMA1mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov. Disord.30, 1290–1292 (2015).
- YeşilG, AralaşmakA, AkyüzE, İçağasıoğluD, UygurŞahin T, BayramY. Expanding the phenotype of homozygous KCNMA1 mutations; dyskinesia, epilepsy, intellectual disability, cerebellar and corticospinal tract atrophy. Balkan. Med. J.35, 336–339 (2018).
- OlgiatiS, SkorvanekM, QuadriMet al.Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord.31, 1041–1048 (2016).
- KorenkeG, NuofferJ-M, AlhaddadBet al.Paroxysmal dyskinesia in ECHS1 defect with globus pallidus lesions. Neuropediatrics47, S01–10 (2016).
- MahajanA, ConstantinouJ, SidiropoulosC. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J. Neurol.264, 185–187 (2017).
- PetersH, FerdinandusseS, RuiterJP, WandersRJ, BonehA, PittJ. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol. Genet. Metab.115, 168–173 (2015).
- McWilliamCA, RidoutCK, BrownRMet al.Pyruvate dehydrogenase E2 deficiency: a potentially treatable cause of episodic dystonia. Eur. J. Paediatr. Neurol.14, 349–353 (2010).
- HeadRA, BrownRM, ZolkipliZet al.Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann. Neurol.58, 234–241 (2005).
- CastiglioniC, VerrigniD, OkumaCet al.Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation: case report and mini-review. Eur. J. Paediatr. Neurol.19, 497–503 (2015).
- FriedmanJ, FeigenbaumA, ChuangNet al.Pyruvate dehydrogenase complex-E2 deficiency causes paroxysmal exercise-induced dyskinesia. Neurology89, 2297–2298 (2017).
- MéneretA, RozeE. Paroxysmal movement disorders: an update. Rev Neurol.172, 433–445 (2016).
- DaleRC, MelchersA, FungVS, Grattan-SmithP, HouldenH, EarlJ. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev. Med. Child Neurol.52, 583–586 (2010).
- ErroR, StamelouM, GanosCet al.The clinical syndrome of paroxysmal exercise induced dystonia: diagnostic outcomes and an algorithm. Mov. Disord. Clin. Pract.1, 57–61 (2014).
- GardellaE, BeckerF, MøllerRSet al.Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol.79, 428–436 (2016).
- LarsenJ, CarvillGL, GardellaEet al.The phenotypic spectrum of SCN8A encephalopathy. Neurology84, 480–489 (2015).
- WestenbergerA, MaxC, BrüggemannNet al.Alternating hemiplegia of childhood as a new presentation of adenylate cyclase 5-mutation-associated disease: a report of two cases. J. Pediatr.181, 306–308 (2017).
- ChenDH, MéneretA, FriedmanJRet al.ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology85, 2026–2035 (2015).
- FriedmanJR, MéneretA, ChenDHet al.ADCY5 mutation carriers display pleiotropic paroxysmal day and nighttime dyskinesias. Mov. Disord.31, 147–148 (2016).
- MencacciNE, ErroR, WiethoffSet al.ADCY5 mutations are another cause of benign hereditary chorea. Neurology85, 80–88 (2015).
- CarecchioM, ZorziG, RagonaF, ZibordiF, NardocciN. ATP1A3-related disorders: an update. Eur. J. Paediatr. Neurol.22, 257–263 (2018).
- HeinzenEL, ArzimanoglouA, BrashearAet al.Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol.13, 503–514 (2014).
- RosewichH, OhlenbuschA, HuppkePet al.The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology82, 945–955 (2014).
- SweneyMT, NewcombTM, SwobodaKJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-Parkinsonism, CAPOS and beyond. Pediatr. Neurol.52, 56e64 (2015).
- GiffinNJ, BentonS, GoadsbyPJ. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev. Med. Child Neurol.44, 490–493 (2002).
- Vila-PueyoM, GenéGG, Flotats-BastardesMet al.A loss-of-function CACNA1A mutation causing benign paroxysmal torticollis of infancy. Eur. J. Paediatr. Neurol.18, 430–433 (2014).
- ShinM, DouglassLM, MilunskyJMet al.The genetics of benign paroxysmal torticollis of infancy: is there an association with mutations in the CACNA1A gene?J. Child. Neurol.31, 1057–1061 (2016).
- FuchsO, PfarrN, PohlenzJet al.Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev. Med. Child. Neurol.51, 240–244 (2009).
- BrockmannK, DumitrescuAM, BestTTet al.X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J. Neurol.252, 663–666 (2005).
- ScottBL, JankovicJ. Delayed-onset progressive movement disorders after static brain lesions. Neurology46, 68–74 (1996).
- WalnO, JankovicJ. Paroxysmal movement disorders. Neurol. Clin.33, 137–152 (2015).
- CiampiE, Uribe-San-MartínR, Godoy-SantínJ, CruzJP, Cárcamo-RodríguezC, JuriC. Secondary paroxysmal dyskinesia in multiple sclerosis: Clinical–radiological features and treatment. Case report of seven patients. Mult. Scler.23, 1791–1795 (2017).
- BalintB, VincentA, MeinckHM, IraniSR, BhatiaKP. Movement disorders with neuronal antibodies: syndromic approach, genetic parallels and pathophysiology. Brain141, 13–36 (2018).
- ZhuM, ZhuX, WanH, HongD. Familial IBGC caused by SLC20A2 mutation presenting as paroxysmal kinesigenic dyskinesia. Parkinsonism Relat. Disord.20, 353–354 (2013).
- WangC, MaX, XuXet al.A PDGFB mutation causes paroxysmal nonkinesigenic dyskinesia with brain calcification. Mov. Disord.32, 1104–1106 (2017).
- PersoonS, KappelleLJ, KlijnCJ. Limb-shaking transient ischaemic attacks in patients with internal carotid artery occlusion: a case-control study. Brain133, 915–922 (2010).
- MirsattariSM, BerryME, HoldenJK, NiW, NathA, PowerC. Paroxysmal dyskinesias in patients with HIV infection. Neurology52, 109–114 (1999).
- JankovicJ. Can peripheral trauma induce distonia and other movement disorders? Yes!Mov. Disord.16, 7–12 (2001).
- García-RuizPJ, CaboI, García-BermejoP, CarnalP. Escitalopram-induced paroxysmal dystonia. Clin. Neuropharmacol.30, 124–126 (2007).
- Silveira-MoriyamaL, KovacS, KurianMAet al.Phenotypes, genotypes, and the management of paroxysmal movement disorders. Dev. Med. Child Neurol.60, 559–565 (2018).
- MadhusudananM, BhatiaKP, BalintB. Paroxysmal craniocervical dyskinesia as manifestation of frontal lobe epilepsy. Mov. Disord.26, 2580–2582 (2011).
- GanosC, AguirregomozcortaM, BatlaAet al.Psychogenic paroxysmal movement disorders--clinical features and diagnostic clues. Parkinsonism Relat. Disord.20, 41–46 (2014).
- MorganJC, HughesM, FigueroaRE, SethiKD. Psychogenic paroxysmal dyskinesia following paroxysmal hemidystonia in multiple sclerosis. Neurology65, E12 (2005).
- ErroR, BrigoF, TrinkaEet al.Psychogenic nonepileptic seizures and movement disorders: a comparative review. Neurol. Clin. Pract.6, 138–149 (2016).
- MehtaSH, MorganJC, SethiKD. Paroxysmal dyskinesias. Curr. Treat Options Neurol.11, 170–178 (2009).
- MinkJW. Treatment of paroxysmal dyskinesias in children. Curr. Treat Options Neurol.17, 350 (2015).
- McGuireS, ChanchaniS, KhuranaDS. Paroxysmal dyskinesias. Semin. Pediatr. Neurol.25, 75–81 (2018).
- HorisawaS, SumiM, AkagawaH, KawamataT, TairaT. Thalamotomy for paroxysmal kinesigenic dyskinesias in a multiplex family. Eur. J. Neurol.24, e71–e72 (2017).
- LoherTJ, KraussJK, BurgunderJM, TaubE, SiegfriedJ. Chronic thalamic stimulation for treatment fo dystonic paroxysmal nonkinesigenic dyskinesia. Neurology56, 268–270 (2001).
- van CollerR, SlabbertP, VaidyanathanJet al.Successful treatment of disabling paroxysmal nonkinesigenic dyskinesia with deep brain stimulation of the globus pallidus internus. Stereotact. Funct. Neurosurg.92, 388–392 (2014).
- YamadaK, GotoS, SoyamaNet al.Complete suppression of paroxysmal nonkinesigenic dyskinesia by globus pallidus internus pallidal stimulation. Mov. Disord.21, 576–579 (2006).
- PisciottaL, GherziM, StagnaroMet al.Alternating hemiplegia of childhood: pharmacological treatment of 30 Italian patients. Brain Dev.39, 521e8 (2017).
- ChiLY, ZhaoXH, LiuXWet al.Alternating hemiplegia of childhood in Chinese following long-term treatment with flunarizine or topiramate. Int. J. Neurosci.122, 506e10 (2012).
- KlepperJ. Glusoe-transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia49, 46–49 (2008).
- LeenWG, MewasinghL, VerbeekMMet al.Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov. Disord.28, 1439–1442 (2013).
- MochelF, HainqueE, GrasDet al.Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J. Neurol. Neurosurg. Psychiatr.87, 550–553 (2016).
- WaakM, MohammadSS, ComanDet al.GNAO1-related movement disorder with life-threatening exacerbations: movement phenomenology and response to DBS. J. Neurol. Neurosurg Psychiatr.89, 221–222 (2018).
- DiFonzo A, MonfriniE, ErroR. Genetics of movement disorders and the practicing clinician; who and what to test for?Curr. Neurol. Neurosci. Rep.18, 37 (2018).
- van EgmondME, LugtenbergCHA, BrouwerOFet al.A post hoc study on gene panel analysis for the diagnosis of dystonia. Mov. Disord.32, 569–575 (2017).
- JiangYL, YuanF, YangYet al.CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus?Seizure56, 88–91 (2018).
- LuJG, BishopJ, CheyetteSet al.A novel PRRT2 pathogenic variant in a family with paroxysmal kinesigenic dyskinesia and benign familial infantile seizures. Cold Spring Harb. Mol. Case Stud. (2018). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5793775/