
Abstract
Spinal muscular atrophy (SMA) is a childhood disorder caused by loss of the SMN gene. Pathological hallmarks are spinal cord motor neuron death, neuromuscular junction dysfunction and muscle atrophy. The first SMN genetic therapy was recently approved and other SMN-dependent treatments are not far behind. However, not all SMA patients will reap their maximal benefit due to limited accessibility, high costs and differential effects depending on timing of administration and disease severity. The repurposing of commercially available drugs is an interesting strategy to ensure more rapid and less expensive access to new treatments. In this mini-review, we will discuss the potential and relevance of repositioning drugs currently used for neurodegenerative, neuromuscular and muscle disorders for SMA.
Spinal muscular atrophy (SMA) is an inherited neuromuscular disorder characterized by motor neuron loss and muscle atrophy. SMA is the leading genetic cause of infant mortality [Citation1,Citation2]. Approximately 96% of SMA cases have been mapped to deletions and loss-of-function mutations within the ubiquitously expressed SMN1 gene, which holds several housekeeping functions, including RNA metabolism, small nuclear ribonucleoprotein assembly and spliceosomal regulation as well as numerous cell-specific roles [Citation3,Citation4]. In addition, SMN1 has an inverted duplicate gene called SMN2, which carries out similar functions, although this is limited by an 840C>T polymorphism that causes 90% of expressed proteins to be truncated and non-functional via exon 7 exclusion [Citation5]. While compensated by the fully expressed SMN1 in healthy individuals, SMN2 is clinically relevant in SMA as a disease modifier due to an association of increased copy number with reduced severity [Citation5,Citation6].
Presently, the only clinically approved treatment for SMA is nusinersen (Spinraza™), an antisense oligonucleotide that targets SMN2 to promote the production of higher levels of functional SMN protein through exon 7 inclusion [Citation7,Citation8]. However, as nusinersen is a relatively new therapy, there are no longitudinal studies to assess its long-term effects, although reports suggest its success is dependent upon the treatment being administered as early as possible [Citation9–11]. These limitations, along with others, highlight a host of issues such as limited treatment viability for type 2 and 3 patients, whose symptoms are milder and develop in later years, and elevated costs associated with both newborn screening and treatment (estimated at £450,000 for the first year and £225,000 for subsequent years) [Citation11–13]. As such, National Health Service England and Wales, for example, have only recently approved funding of nusinersen (May 2019), leaving several patients without access to any clinical treatments during a crucial period [Citation13]. While a cohort of additional novel drugs designed to enhance SMN expression are in development, none have yet passed the Phase III clinical trial stages () and similar high costs have been predicted (∼US$2 million per treatment) [Citation14,Citation15]. Therefore, more cost-efficient drugs are required to ensure accessibility of treatments for all SMA patients.
Table 1. Status of SMN-dependent drugs in clinical trials for spinal muscular atrophy.
Typically, clinical drug development is a very long overhaul with some drugs taking up to 20 years before being approved [Citation16]. Due to the limited availability of treatments for SMA and the urgency in getting them to patients, a valid alternative is the repositioning (or repurposing) of drugs originally designed to treat other conditions [Citation17]. Compared with novel drugs, most repositioned drugs have passed safety efficacy tests for human administration, thus allowing a complete omission of Phase I trials [Citation17]. Furthermore, information such as efficacy dosage, side effects and formulation costs are already known, making trials of repositioned drugs more cost and time effective [Citation17]. Drug repositioning is not a new concept in the fields of non-neurodegenerative diseases and has opened several novel avenues of research [Citation17–19]. Indeed, the Type 2 diabetes drug metformin is now being evaluated in clinical trials for breast and endometrial-based cancers, while the controversial morning sickness drug thalidomide is a possible candidate for Alzheimer’s disease (AD) [Citation20–26]. However, very little research on drug repositioning has been conducted in the context of inherited neurodegenerative diseases. This review will discuss how commercial drugs originally designed for other neurodegenerative, neuromuscular and muscular disorders have the potential to be repositioned to treat the central nervous system (CNS), neuromuscular junction (NMJ) and skeletal muscle pathologies observed in SMA.
Repositioning drugs to target CNS pathologies in SMA
The loss of motor neurons in the anterior horn of the spinal cord is the canonical feature of SMA [Citation27]. While the genetic basis behind SMA is now well understood, it has not yet been elucidated how and why loss of SMN affects and causes motor neuron degeneration [Citation27]. Besides its ubiquitous localization within nuclear Gemini of coiled bodies (Gems), SMN is also specifically abundant in the axon and synaptic regions of motor neurons where it regulates mRNA transport, actin dynamics and vesicle release [Citation28–31]. This is supported by studies showing neurite outgrowth, differentiation and synaptic defects in SMN-depleted neuronal cells, suggesting that CNS-specific functions of SMN are dysregulated in SMA [Citation31–33]. Commercial drugs that modulate similar regulatory pathways in the CNS might therefore be beneficial in SMA patients.
One plausible repositioning strategy may be through identifying drugs that are currently used to treat other motor neuron diseases such as amyotrophic lateral sclerosis (ALS). Indeed, the ALS drug riluzole (Rilutek™ or Teglutik™), a pharmacological compound thought to promote neuroprotection through Ca2+-activated K+ channel-mediated glutamate reduction, has already been evaluated in clinical trials for SMA, albeit with limited success in improving survival rates (ClinicalTrials.Gov ID: NCT00774423) [Citation34–38].
While the repositioning of riluzole for SMA is ongoing, other ALS drugs have not yet made that leap, including rasalgiline (Azilect™), an irreversible MOA-B inhibitor () [Citation39]. The MOA-B isoform is expressed in the outer membrane of mitochondria with a neuron-specific role in oxidative deamination of monoamine neurotransmitters [Citation40–43]. Based upon evidence of MOA-B upregulation in rodent models of Parkinson’s disease (PD), rasalgiline was initially evaluated in clinical trials for PD (ClinicalTrials.Gov ID: NCT00203138) and received EMA and US FDA approval in 2005 and 2006, respectively [Citation43].
Overview of pathological molecular effectors or biological pathways (in red) that could be therapeutically modulated by commercially available drugs (in blue) to treat CNS, NMJ and skeletal muscle pathologies in spinal muscular atrophy.
NMJ: Neuromuscular junction.
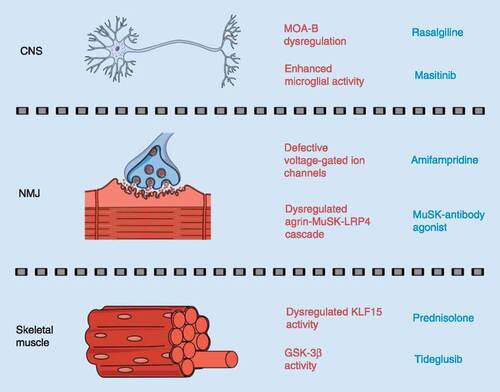
Recently, rasalgiline underwent Phase II clinical trials in ALS patients, either alone or in combination with riluzole, with the latter demonstrating moderate success, which is thought to be due to rasalgiline’s neuroprotective role through promoting mitochondrial stability (ClinicalTrials.Gov ID: NCT01786603; NCT01879241; NCT01232738) [Citation44–46]. Indeed, increased MOA-B activity could contribute to mitochondrial dysfunction and neurodegeneration via accumulation of its aldehyde and reactive oxygen species by-products [Citation47–50]. Given that mitochondrial instability has been reported in pre-symptomatic SMA motor neurons, rasalgiline could be a treatment option to improve mitochondrial stability and delay motor neuron degeneration () [Citation51].
Modulating the aberrant inflammation of nervous tissue (neuroinflammation) can also help minimize motor neuron death. The pathological impacts of neuroinflammation, predominantly mediated by astrocytes and microglia, have been associated with several neurodegenerative diseases such as AD [Citation52]. Microglia are specialized macrophage cells that regulate the removal of damaged neurons, plaques and infectious agents [Citation53]. However, the chronic release of pro-inflammatory factors by M1 microglia can result in neuroinflammation-induced neuronal loss [Citation54–57]. Studies in rodents identified the colony-stimulating factor 1 receptor, a member of the RTK family, as a key promoter of microglia proliferation and viability [Citation58–61]. The RTK inhibitor masitinib (Masivet™) has been demonstrated to reduce the levels of microglia via colony-stimulating factor 1 receptor inhibition (), thus decreasing neuronal degeneration in clinical trials for AD (ClinicalTrials.Gov ID: NCT01872598) [Citation62–64]. Masitinib has recently been evaluated in SOD1G93A ALS rats, where administration of the drug delayed disease progression as well as reduced both microglial activity and neuroinflammation [Citation65]. Furthermore, emerging results from Phase III clinical trials of masitinib in ALS patients are promising, with reports of delayed neuronal degeneration, although the extent of the delay remains unclear (ClinicalTrials.Gov ID: NCT02588677) [Citation66]. Recent reports also support a role for neuroinflammation in SMA pathogenesis [Citation67]. Indeed, increased microgliosis and release of the pro-inflammatory IL-6 cytokine by microglia have been described in SMA mice [Citation33,Citation68]. In addition, astrocytosis was observed in the spinal cord of SMA mice at early symptomatic stages, when motor neuron loss is first detected [Citation69,Citation70]. The role of astrocytes in SMA pathology was further supported by experiments demonstrating that astrocyte-specific restoration of SMN expression in SMA improves disease phenotypes [Citation70]. Recent work has also identified the Notch pathway as a mediator of neuroinflammatory activity in SMA mice [Citation71]. Thus, drugs that reduce neuroinflammation such as masitinib may provide significant therapeutic benefits for SMA ().
Preventing and/or delaying motor neuron death in SMA is a therapeutic priority and repositioning neuroprotective drugs such as rasalgiline and masitinib for SMA has the potential to rapidly expand the number of available CNS-specific treatments.
Repositioning drugs to target NMJ pathologies in SMA
SMA pathology also affects the NMJ, a specialized synaptic region between the motor nerve terminal and skeletal muscle fiber that contributes to contraction via an ACh and nicotinic AChR interaction [Citation72]. In both severe and intermediate SMA mice, NMJ defects include neurofilament accumulation, axonal sprouting, immature motor endplate development, impaired synaptic regions and dysregulated AChR clustering, the latter being a crucial step in NMJ development [Citation72,Citation73]. Thus, drugs currently aimed at modulating NMJ activity (pre- and post-synaptic) are an interesting avenue for SMA.
Lambert-Eaton myasthenic syndrome is an autoimmune disorder caused by antibodies that target motor neuron voltage-gated Ca2+ channels, resulting in a pre-synaptic NMJ pathology [Citation74]. Amifampridine (Firdapse™) has been used to treat this disorder by blocking pre-synaptic K+ channels to allow an increased duration of ACh release () [Citation75,Citation76]. Interestingly, impairments in neuronal excitation of SMN-deficient motor neurons were similarly caused by defective voltage-gated Ca2+ channels, suggesting that amifampridine could provide therapeutic benefits in SMA () [Citation77]. Furthermore, with successful use in the post-synaptic NMJ disorder muscle-specific kinase-myasthenia gravis (MuSK-MG), amifampridine could potentially modulate both pre- and post-synaptic defects observed in SMA NMJs (ClinicalTrials.Gov ID: NCT03304054) [Citation78–80].
MuSK-MG is caused by an antibody-mediated breakdown of MuSK, a specialized RTK that regulates motor endplate development, maturation and AChR clustering through an agrin-MuSK-LRP4 cascade [Citation81–84]. Although there are limited studies of MuSK regulation in SMA, agrin dysregulation contributes to impaired AChR clustering, suggesting that the agrin-MuSK-LRP4 cascade may be a suitable target for SMA treatments [Citation85,Citation86]. Although not yet in clinical trials, preliminary work is currently ongoing to evaluate the therapeutic potential of MuSK-specific antibody agonists aimed at increasing MuSK activity () [Citation87,Citation88]. Increasing MuSK activity either genetically or pharmacologically in SOD1G93A ALS mice resulted in reduced NMJ denervation and improved motor function [Citation89,Citation90]. Although the MuSK antibody agonist did not influence survival rate or disease progression in ALS mice, its ability to maintain NMJ integrity and innervation makes it an interesting candidate that could potentially be combined with amifampridine to reduce NMJ pathology in SMA ().
From a repositioning approach, one limitation in the use of NMJ-specific drugs for SMA treatment is that they were originally designed to treat disorders that display either pre- or post-synaptic NMJ defects. However, both compartments are affected in SMA and studies in ALS mice indicate that these pharmacological compounds may not be as effective when administered individually. Instead, future research endeavors in SMA could adopt a combinatorial approach when trialing NMJ targeting drugs to ensure optimal benefits.
Repositioning drugs for skeletal muscle pathologies in SMA
Reduced SMN levels also affect skeletal muscle development and function [Citation91–94]. A new avenue for SMA therapeutic development could therefore involve drugs currently used for muscular disorders. Indeed, shared treatment paradigms have recently been identified for SMA and Duchenne muscular dystrophy (DMD), an inherited X-linked recessive muscle disorder [Citation95]. Currently, the most commonly used drug to treat DMD is prednisolone, a synthetic glucocorticoid, which was originally prescribed to reduce fibrotic development in skeletal muscle via its anti-inflammatory properties [Citation96,Citation97]. Research in DMD mdx mice has recently demonstrated that when dosed intermittently, prednisolone also displayed ergogenic benefits in skeletal muscle by increasing its repair [Citation98,Citation99]. The molecular mechanism behind the prednisolone-induced muscle repair is thought to be through restoration of Klf15 expression, a transcription factor involved in muscle homeostasis via regulation of glucose, lipid and amino acid metabolism () [Citation99–102]. Subsequently, Klf15 levels were also observed to be low in skeletal muscle of pre-symptomatic SMA mice [Citation103]. Importantly, prednisolone treatment in SMA mice increased Klf15 expression and led to amelioration of muscle-specific phenotypes, weight and survival [Citation103]. Evidence of common pathological targets and treatment strategies between SMA and DMD supports further investigations into the repositioning of additional muscle-specific drugs not previously considered for SMA.
Tideglusib, a small heterocyclic thiadiazolidine-based non-ATP competitive inhibitor of GSK-3β, could potentially ameliorate SMA muscle pathology () [Citation104]. GSK-3β is a ubiquitously expressed isoform of GSK-3, which acts as a serine/threonine kinase in many cellular processes such as proliferation, survival and metabolism [Citation105,Citation106]. Tideglusib was initially developed for AD, with initial trials deeming the drug safe for human use [Citation107,Citation108]. The ubiquitous and multipurpose activities of GSK-3β thus enabled repurposing opportunities for tideglusib. In skeletal muscle, GSK-3β is a negative regulator of growth through dysregulation of the myogenic regulator factors [Citation109]. When GSK-3β was inhibited in C2C12 myoblasts and C57BL/6 mice, myogenic regulator factor activity, myotube formation and muscle growth were enhanced, confirming its usefulness as a potential drug candidate for impaired muscle development [Citation109–111]. As such, tideglusib is being evaluated in Phase II clinical trials (ClinicalTrials.gov ID: NCT03692312; NCT02858908) for congenital myotonic dystrophy, an autosomal polyQ-dominant disorder characterized by myotonia, muscle dystrophy and weakness [Citation112]. Although the trial results are not fully published, early results are promising, suggesting that a Phase III approval is in the near future [Citation113]. In addition to promotion of muscle growth, tideglusib may have a further advantage in SMA by restoring SMN levels. Indeed, chemical screens in human SMA fibroblasts exposed to GSK-3β inhibitors or GSK-3β short-hairpin RNAs revealed an increased SMN expression [Citation114]. With positive effects in congenital myotonic dystrophy and on SMN, tideglusib could be a potential therapeutic candidate for SMA, ameliorating SMN-dependent and -independent muscle atrophy.
The shared dysregulation in pathways related to muscle growth, differentiation, function and homeostasis between muscular disorders and SMA highlights the contribution of intrinsic muscle defects to SMA pathogenesis. The parallel continual development of pharmacological compounds for muscle disorders suggests that several potential muscle-specific treatments could potentially lead to significant benefits for SMA.
Conclusion
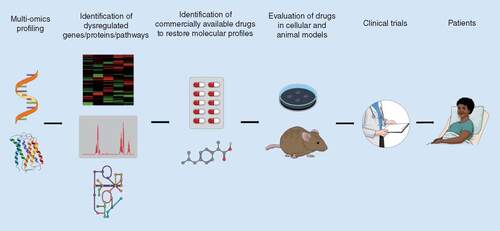
Despite being the leading cause of genetically caused infant deaths, there is presently only one clinically approved drug for SMA that is not accessible to all SMA patients and is unfortunately not a cure. Additional SMN-specific drugs may be approved in the future, thus increasing treatment options (). However, molecular and functional pathological similarities between SMA and other neurodegenerative, neuromuscular and muscular disorders highlight potential novel non-SMN treatments (). Indeed, the existence of shared dysregulated pathways between diseases offers a therapeutic solution in the form of drug repositioning. Pharmacological compounds that are either approved or in clinical trials could provide SMA patients more affordable and accessible treatment options that could be used in combination with SMN-dependent therapeutics. Furthermore, while the present review focuses on the potential of repurposing drugs currently in use for neurodegenerative, neuromuscular and muscular disease, repositioning strategies for SMA should not be limited to these groups of disorders as pathological overlaps may also exist between SMA and unrelated disorders such as cancer (e.g., RTK signaling) [Citation115]. With several published multi-omics SMA studies identifying a multitude of dysregulated pathways and molecular effectors, the opportunities for drug repositioning are many and should be a key component of future therapy development for SMA () [Citation116–118].
Proteomic and transcriptomic approaches can be used to define the differential expression patterns of genes, proteins and biological pathways between diseased and healthy states. Published literature or integrated publicly available drug databases allow for the uncovering of commercially available drugs that can restore the aberrant molecular profiles in the diseased state. The drugs then require to first be validated for safety and efficacy in relevant in vitro and in vivo models. Successful drug candidates can then be evaluated in clinical trials, with Phase I usually bypassed as information regarding safety and dosages are known. Finally, the drug is approved for patient use if it demonstrates benefits in clinical trials.
Future perspective
It is clear that the SMA therapeutic landscape has drastically changed since the approval of nusinersen/Spinraza™. In the next few years, it is likely that most, if not all, SMA patients will have been administered one form or another of an SMN-enhancing therapy (antisense oligonucleotide, gene therapy, small molecule). Our role as researchers and clinicians is to support the SMA community by anticipating additional therapeutic needs as these presently young children reach adolescence and adulthood. SMN-dependent therapies will without a doubt have to be accompanied by complementary treatments aimed at ameliorating and/or preserving neuromuscular health in SMA patients. Our goal should be to develop these drug entities well ahead of them being required, which can be ensured by understanding the pathological similarities between SMA and other diseases and by using this information for drug repositioning strategies.
Spinal muscular atrophy
Spinal muscular atrophy (SMA) is an inherited neuromuscular disorder and the leading genetic cause of infant mortality caused by homozygous loss of the SMN1 gene.
Disease severity is dependent upon the copy number of SMN2, a duplicated but mutated copy of SMN1, which only produces 10% of full-length protein.
The benefits of the only currently available treatment for SMA (nusinersen/Spinraza™), an antisense oligonucleotide that promotes expression of full-length SMN from SMN2, are limited by its accessibility and age-dependent efficacy.
Drug repositioning
There is currently a gap in treatment availability for SMA patients.
Drug repositioning provides a shorter and more cost-effective transition to the clinic as pre-clinical and clinical dosage and safety assessments have already been performed.
Molecular and functional similarities between SMA and other neurodegenerative, neuromuscular and muscle disorders present opportunities for drug repositioning strategies.
Rasalgiline
Rasalgiline (Azilect™) is an irreversible MOA-B inhibitor that promotes neuroprotection in Parkinson’s Disease through increased mitochondrial stability.
Mitochondrial instability has been reported in pre-symptomatic SMA and amyotrophic lateral sclerosis (ALS) motor neurons, which has led to rasalgiline Phase II clinical trials in the latter.
Masitinib
Masitinib (Masivet™) is a CSF1 receptor inhibitor, which reduces microglia- and neuroinflammation-induced neuronal loss.
A Phase III clinical trial of masitinib in ALS patients revealed delayed neuronal degeneration, with similar results in ALS mice that were associated with reduced microglial activity.
Although masitinib has not been trialled in SMA, neuroinflammation-induced neuronal loss has been reported, suggesting potential benefits of the drug in SMA.
Amifampridine
Amifampridine (Firdpase™) is a pre-synaptic K+ channel blocker used in neuromuscular junction disorders such as Lambert-Eaton myasthenic syndrome to promote ACh release, which is impaired due to defective voltage-gated Ca2+ channels.
Similar voltage-gated Ca2+ channel defects have been reported in SMA motor neurons, suggesting amifampridine could be effective in SMA.
Muscle-specific kinase antibody agonist
The agrin-MuSK-LRP4 cascade is important for ACh receptor remodeling at the post-synaptic motor endplate.
ACh receptor remodeling is dysregulated in a variety of neuromuscular disorders such as SMA, ALS and MuSK-myasthenia gravis.
A MuSK antibody agonist, previously designed to promote MuSK activity in MuSK-myasthenia gravis, improved motor function in ALS mice, suggesting it could have similar benefits in SMA.
Prednisolone
Prednisolone is a synthetic glucocorticoid used in Duchenne muscular dystrophy that demonstrates anti-inflammatory and ergogenic properties.
In both Duchenne muscular dystrophy and SMA mice, prednisolone improves muscle health through restoration of Klf15 activity, an important regulator of muscle metabolism.
Tideglusib
Tideglusib is a small heterocyclic thiadiazolidine-based non-ATP competitive inhibitor of GSK-3β.
In skeletal muscle, GSK-3β negatively regulates myogenic regulatory factor activity and its inhibition is associated with muscle growth.
Tideglusib is currently showing promising results in Phase II clinical trials for the muscle disorder congenital myotonic dystrophy.
Tideglusib could potentially ameliorate SMA skeletal muscle through both SMN-dependent and -independent pathways.
Second-generation SMA therapies
There is increasing evidence that future treatments need to incorporate both SMN-dependent and -independent therapies that affect both neuronal and systemic tissues.
The increasing number of multi-omic studies will allow for the identification of drug target similarities between SMA and different disorders.
Financial & competing interests disclosure
JM Hoolachan is funded by a studentship from the Keele University School of Medicine. ER Sutton is funded by a studentship from Muscular Dystrophy UK. M Bowerman receives funding from SMA Angels Charity, Association Française contre les Myopathies, Action Medical Research and Muscular Dystrophy UK for SMA research. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- KolbSJ, KisselJT. Spinal muscular atrophy. Neurol. Clin.33(4), 831–846 (2015).
- PriorTW, SnyderPJ, RinkBDet al.Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. A152A(7), 1608–1616 (2010).
- LefebvreS, BürglenL, ReboulletSet al.Identification and characterization of a spinal muscular atrophy-determining gene. Cell80(1), 155–165 (1995).
- SinghRN, HowellMD, OttesenEW, SinghNN. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech.1860(3), 299–315 (2017).
- KashimaT, ManleyJL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet.34(4), 460–463 (2003).
- MonaniUR, LorsonCL, ParsonsDWet al.A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet.8(7), 1177–1183 (1999).
- SinghNK, SinghNN, AndrophyEJ, SinghRN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol.26(4), 1333–1346 (2006).
- FinkelRS, MercuriE, DarrasBTet al.Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med.377(18), 1723–1732 (2017).
- MontesJ, McDermottMP, MirekEet al.Ambulatory function in spinal muscular atrophy: age-related patterns of progression. PLoS ONE13(6), e0199657 (2018).
- GidaroT, ServaisL. Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps. Dev. Med. Child Neurol.61(1), 19–24 (2019).
- BoardmanFK, YoungPJ, GriffithsFE. Newborn screening for spinal muscular atrophy: the views of affected families and adults. Am. J. Med. Genet. A173(6), 1546–1561 (2017).
- MousaMA, AriaDJ, SchaeferCMet al.A comprehensive institutional overview of intrathecal nusinersen injections for spinal muscular atrophy. Pediatr. Radiol.48(12), 1797–1805 (2018).
- NICE. Nusinersen for treating spinal muscular atrophy. https://www.nice.org.uk/guidance/GID-TA10281/documents/appraisal-consultation-document
- Al-ZaidyS, PickardAS, KothaKet al.Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol.54(2), 179–185 (2019).
- MaloneDC, DeanR, ArjunjiRet al.Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy7(1), 1601484 (2019).
- DicksonM, GagnonJP. The cost of new drug discovery and development. Discov. Med.4(22), 172–179 (2004).
- AshburnTT, ThorKB. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov.3(8), 673–683 (2004).
- GraulAI, CrucesE, StringerM. The year's new drugs & biologics, 2014: part I. Drugs Today51(1), 37–87 (2015).
- LiJ, ZhengS, ChenB, ButteAJ, SwamidassSJ, LuZ. A survey of current trends in computational drug repositioning. Brief Bioinform.17(1), 2–12 (2016).
- RojasLBA, GomesMB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr.5, 6 (2013).
- HeX, EstevaFJ, EnsorJ, HortobagyiGN, LeeM-H, YeungS-CJ. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol.23(7), 1771–1780 (2012).
- ChenT-M, LinC-C, HuangP-T, WenC-F. Metformin associated with lower mortality in diabetic patients with early stage hepatocellular carcinoma after radiofrequency ablation. J. Gastroenterol. Hepatol.26(5), 858–865 (2011).
- HadadS, IwamotoT, JordanLet al.Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat.128(3), 783–794 (2011).
- LaskovI, DrudiL, BeauchampM-Cet al.Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol. Oncol.134(3), 607–614 (2014).
- RidingsJE. The thalidomide disaster, lessons from the past. Methods Mol. Biol.947, 575–586 (2013).
- HeP, ChengX, StaufenbielM, LiR, ShenY. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer's disease. PLoS ONE8(2), e55091 (2013).
- BrieseM, EsmaeiliB, SattelleDB. Is spinal muscular atrophy the result of defects in motor neuron processes?Bioessays27(9), 946–957 (2005).
- BowermanM, BeckerCG, Yáñez-MuñozRJet al.Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model Mech.10(8), 943–954 (2017).
- Donlin-AspPG, BassellGJ, RossollW. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol.39, 53–61 (2016).
- KongL, WangX, ChoeDWet al.Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci.29(3), 842–851 (2009).
- HenselN, ClausP. The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration?Neuroscientist24(1), 54–72 (2018).
- van BergeijkJ, Rydel-KöneckeK, GrotheC, ClausP. The spinal muscular atrophy gene product regulates neurite outgrowth: importance of the C terminus. FASEB J.21(7), 1492–1502 (2007).
- TarabalO, Caraballo-MirallesV, Cardona-RossinyolAet al.Mechanisms involved in spinal cord central synapse loss in a mouse model of spinal muscular atrophy. J. Neuropathol. Exp. Neurol.73(6), 519–535 (2014).
- StutzmannJM, WahlF, PrattJet al.Neuroprotective profile of riluzole in in vivo models of acute neurodegenerative diseases. CNS Drug Rev.3(1), 83–101 (1997).
- DimitriadiM, KyeMJ, KallooG, YersakJM, SahinM, HartAC. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci.33(15), 6557–6562 (2013).
- FáveroFM, VoosMC, de CastroI, CaromanoFA, OliveiraASB. Epidemiological and clinical factors impact on the benefit of riluzole in the survival rates of patients with ALS. Arq. Neuropsiquiatr.75(8), 515–522 (2017).
- RussmanBS, IannacconeST, SamahaFJ. A Phase I trial of riluzole in spinal muscular atrophy. Arch. Neurol.60(11), 1601–1603 (2003).
- AbbaraC, EstournetB, LacomblezLet al.Riluzole pharmacokinetics in young patients with spinal muscular atrophy. Br. J. Clin. Pharmacol.71(3), 403–410 (2011).
- FinbergJP, LamensdorfI, CommissiongJW, YoudimMB. Pharmacology and neuroprotective properties of rasagiline. J. Neural Transm. Suppl.48, 95–101 (1996).
- StudentAK, EdwardsDJ. Subcellular localization of types A and B monoamine oxidase in rat brain. Biochem. Pharmacol.26(24), 2337–2342 (1977).
- EdmondsonDE, BindaC, WangJ, UpadhyayAK, MatteviA. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry48(20), 4220–4230 (2009).
- TabakmanR, LechtS, LazaroviciP. Neuroprotection by monoamine oxidase B inhibitors: a therapeutic strategy for Parkinson's disease?Bioessays26(1), 80–90 (2004).
- MallajosyulaJK, KaurD, ChintaSJet al.MAO-B elevation in mouse brain astrocytes results in Parkinson's pathology. PLoS ONE3(2), (2008). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2229649/
- BarohnR, StatlandJ, MooreDet al.Rasagiline for the treatment of ALS: a randomized controlled study (S27.001). Neurology88(16 Suppl.), S27.001 (2017).
- LudolphAC, SchusterJ, DorstJet al.Safety and efficacy of rasagiline as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomised, double-blind, parallel-group, placebo-controlled, Phase II trial. Lancet Neurol.17(8), 681–688 (2018).
- StatlandJM, MooreD, WangYet al.Rasagiline for amyotrophic lateral sclerosis: a randomized, controlled trial. Muscle Nerve59(2), 201–207 (2019).
- FarfánLabonne BE, GutiérrezM, Gómez-QuirozLEet al.Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol.25(6), 599–609 (2009).
- KaludercicN, CarpiA, NagayamaTet al.Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid. Redox Signal.20(2), 267–280 (2014).
- IslamMT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res.39(1), 73–82 (2017).
- WoodPL, KhanMA, KulowSR, MahmoodSA, MoskalJR. Neurotoxicity of reactive aldehydes: the concept of “aldehyde load” as demonstrated by neuroprotection with hydroxylamines. Brain Res.1095(1), 190–199 (2006).
- MillerN, ShiH, ZelikovichAS, MaY-C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet.25(16), 3395–3406 (2016).
- CalsolaroV, EdisonP. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement.12(6), 719–732 (2016).
- NayakD, RothTL, McGavernDB. Microglia development and function. Annu. Rev. Immunol.32, 367–402 (2014).
- von BernhardiR, EugenínJ. Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res.1025(1–2), 186–193 (2004).
- KrsticD, MadhusudanA, DoehnerJet al.Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation9, 151 (2012).
- ZrzavyT, HöftbergerR, BergerTet al.Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol. Appl. Neurobiol.45(3), 278–290 (2019).
- von BernhardiR, Eugenín-vonBernhardi L, EugenínJ. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci.7, 124 (2015).
- WalkerDG, TangTM, LueL-F. Studies on colony stimulating factor receptor-1 and ligands colony stimulating factor-1 and interleukin-34 in Alzheimer's disease brains and human microglia. Front. Aging Neurosci.9, 244 (2017).
- MurphyGM, ZhaoF, YangL, CordellB. Expression of macrophage colony-stimulating factor receptor is increased in the AβPPV717F transgenic mouse model of Alzheimer's disease. Am. J. Pathol.157(3), 895–904 (2000).
- ErblichB, ZhuL, EtgenAM, DobrenisK, PollardJW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE6(10), e26317 (2011).
- LemmonMA, SchlessingerJ. Cell signaling by receptor tyrosine kinases. Cell141(7), 1117–1134 (2010).
- D'allardD, GayJ, DescarpentriesCet al.Tyrosine kinase inhibitors induce down-regulation of c-Kit by targeting the ATP pocket. PLoS ONE8(4), e60961 (2013).
- DubreuilP, LetardS, CiufoliniMet al.Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE4(9), e7258 (2009).
- PietteF, BelminJ, VincentHet al.Masitinib as an adjunct therapy for mild-to-moderate Alzheimer's disease: a randomised, placebo-controlled Phase II trial. Alzheimers Res. Ther.3(2), 16 (2011).
- TriasE, IbarburuS, Barreto-NúñezRet al.Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflammation13(1), 177 (2016).
- MoraJS, HermineO. Masitinib as an add-on therapy to riluzole is safe and effective in the treatment of amyotrophic lateral sclerosis (ALS). J. Neurol. Sci.381, 183 (2017).
- PapadimitriouD, LeVerche V, JacquierA, IkizB, PrzedborskiS, ReDB. Inflammation in ALS and SMA: sorting out the good from the evil. Neurobiol. Dis.37(3), 493–502 (2010).
- WanB, FengP, GuanZ, ShengL, LiuZ, HuaY. A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet.27(23), 4061–4076 (2018).
- MartinJE, NguyenTT, GrunseichCet al.Decreased motor neuron support by SMA astrocytes due to diminished MCP1 secretion. J. Neurosci.37(21), 5309–5318 (2017).
- RindtH, FengZ, MazzasetteCet al.Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet.24(14), 4094–4102 (2015).
- OhuchiK, FunatoM, YoshinoYet al.Notch signaling mediates astrocyte abnormality in spinal muscular atrophy model systems. Sci. Rep.9(1), 3701 (2019).
- BoidoM, VercelliA. Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front. Neuroanat. (2016). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4737916/
- YoshidaM, KitaokaS, EgawaNet al.Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs. Stem Cell Rep.4(4), 561–568 (2015).
- WatermanSA, LangB, Newsom-DavisJ. Effect of Lambert-Eaton myasthenic syndrome antibodies on autonomic neurons in the mouse. Ann. Neurol.42(2), 147–156 (1997).
- MaddisonP, Newsom-DavisJ, MillsKR. Effect of 3,4-diaminopyridine on the time course of decay of compound muscle action potential augmentation in the Lambert–Eaton myasthenic syndrome. Muscle Nerve21(9), 1196–1198 (1998).
- OhSJ, ShcherbakovaN, Kostera-PruszczykAet al.Amifampridine phosphate (Firdapse(®)) is effective and safe in a Phase III clinical trial in LEMS. Muscle Nerve53(5), 717–725 (2016).
- JablonkaS, BeckM, LechnerBD, MayerC, SendtnerM. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol.179(1), 139–149 (2007).
- MorschM, ReddelSW, GhazanfariN, ToykaKV, PhillipsWD. Pyridostigmine but not 3,4-diaminopyridine exacerbates ACh receptor loss and myasthenia induced in mice by muscle-specific kinase autoantibody. J. Physiol. (Lond.)591(10), 2747–2762 (2013).
- BonannoS, PasanisiMB, FrangiamoreRet al.Amifampridine phosphate in the treatment of muscle-specific kinase myasthenia gravis: a Phase IIb, randomized, double-blind, placebo-controlled, double crossover study. SAGE Open Med.6, 2050312118819013 (2018).
- MoriS, KishiM, KuboSet al.3,4-Diaminopyridine improves neuromuscular transmission in a MuSK antibody-induced mouse model of myasthenia gravis. J. Neuroimmunol.245(1–2), 75–78 (2012).
- BowenDC, ParkJS, BodineSet al.Localization and regulation of MuSK at the neuromuscular junction. Dev. Biol.199(2), 309–319 (1998).
- HochW, McConvilleJ, HelmsS, Newsom-DavisJ, MelmsA, VincentA. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med.7(3), 365–368 (2001).
- ValenzuelaDM, StittTN, DiStefanoPSet al.Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron15(3), 573–584 (1995).
- ZongY, ZhangB, GuSet al.Structural basis of agrin-LRP4-MuSK signaling. Genes Dev.26(3), 247–258 (2012).
- BoidoM, DeAmicis E, ValsecchiVet al.Increasing agrin function antagonizes muscle atrophy and motor impairment in spinal muscular atrophy. Front. Cell Neurosci.12, 17 (2018).
- GlassDJ, BowenDC, StittTNet al.Agrin acts via a MuSK receptor complex. Cell85(4), 513–523 (1996).
- BurdenS, VanDer Maarel S. An agonist antibody to MuSK as a theraputic for MuSK myasthenia gravis. http://grantome.com/grant/NIH/R21-NS088723-01A1
- HuijbersMG, VergoossenDL, Fillié-GrijpmaYEet al.MuSK myasthenia gravis monoclonal antibodies valency dictates pathogenicity. Neurol. Neuroimmunol. Neuroinflamm.6(3), e547 (2019).
- Pérez-GarcíaMJ, BurdenSJ. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell. Rep.2(3), 497–502 (2012).
- Sengupta-GhoshA, DominguezSL, XieLet al.Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis.124, 340–352 (2019).
- ShafeyD, CôtéPD, KotharyR. Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res.311(1), 49–61 (2005).
- BoyerJG, MurrayLM, ScottK, DeRepentigny Y, RenaudJ-M, KotharyR. Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle3(1), 24 (2013).
- BriccenoKV, MartinezT, LeikinaEet al.Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Hum. Mol. Genet.23(18), 4745–4757 (2014).
- FayzullinaS, MartinLJ. Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of Spinal Muscular Atrophy (SMA). PLoS ONE9(3), e93329 (2014).
- YiuEM, KornbergAJ. Duchenne muscular dystrophy. J. Paediatrics Child Health51(8), 759–764 (2015).
- FreyFJ. Kinetics and dynamics of prednisolone. Endocr. Rev.8(4), 453–473 (1987).
- BeenakkerEAC, FockJM, Van TolMJet al.Intermittent prednisone therapy in Duchenne muscular dystrophy: a randomized controlled trial. Arch. Neurol.62(1), 128–132 (2005).
- KeelingRM, GolumbekPT, StreifEM, ConnollyAM. Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve35(1), 43–48 (2007).
- QuattrocelliM, BarefieldDY, WarnerJLet al.Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J. Clin. Invest.127(6), 2418–2432 (2017).
- GrayS, WangB, OrihuelaYet al.Regulation of gluconeogenesis by Krüppel-like factor 15. Cell. Metab.5(4), 305–312 (2007).
- HaldarSM, JeyarajD, AnandPet al.Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proc. Natl Acad. Sci. USA109(17), 6739–6744 (2012).
- LiuY, DongW, ShaoJ, WangY, ZhouM, SunH. Branched-chain amino acid negatively regulates KLF15 expression via PI3K-AKT pathway. Front. Physiol.8, 853 (2017).
- WalterLM, DeguiseM-O, MeijboomKEet al.Interventions targeting glucocorticoid-Krüppel-like factor 15-branched-chain amino acid signaling improve disease phenotypes in spinal muscular atrophy mice. EBioMedicine31, 226–242 (2018).
- MartinezA, AlonsoM, CastroA, PérezC, MorenoFJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J. Med. Chem.45(6), 1292–1299 (2002).
- WoodgettJR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J.9(8), 2431–2438 (1990).
- SutherlandC, LeightonIA, CohenP. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J.296(Pt 1), 15–19 (1993).
- OchalekA, MihalikB, AvciHXet al.Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther.9(1), 90 (2017).
- LovestoneS, BoadaM, DuboisBet al.A Phase II trial of tideglusib in Alzheimer's disease. J. Alzheimers Dis.45(1), 75–88 (2015).
- VerheesKJP, ScholsAMWJ, KeldersMCJM, Opden Kamp CMH, vander Velden JLJ, LangenRCJ. Glycogen synthase kinase-3β is required for the induction of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol.301(5), C995–C1007 (2011).
- vander Velden JLJ, LangenRCJ, KeldersMCJM, WoutersEFM, Janssen-HeiningerYMW, ScholsAMWJ. Inhibition of glycogen synthase kinase-3beta activity is sufficient to stimulate myogenic differentiation. Am. J. Physiol. Cell Physiol.290(2), C453–C462 (2006).
- vander Velden JLJ, LangenRCJ, KeldersMCJMet al.Myogenic differentiation during regrowth of atrophied skeletal muscle is associated with inactivation of GSK-3beta. Am. J. Physiol. Cell Physiol.292(5), C1636–C1644 (2007).
- ThorntonCA. Myotonic dystrophy. Neurol. Clin.32(3), 705–719 (2014).
- HorriganJ, McMornA, SnapeM, NikolenkoN, GomesT, LochmullerH. AMO-02 (tideglusib) for the treatment of congenital and childhood onset myotonic dystrophy type 1. Neuromuscul. Disord.28, S14 (2018).
- MakhortovaNR, HayhurstM, CerqueiraAet al.A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol.7(8), 544–552 (2011).
- RegadT. Targeting RTK signaling pathways in cancer. Cancers (Basel)7(3), 1758–1784 (2015).
- FinkelRS, CrawfordTO, SwobodaKJet al.Candidate proteins, metabolites and transcripts in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE7(4), e35462 (2012).
- ŠoltićD, BowermanM, StockJ, ShorrockHK, GillingwaterTH, FullerHR. Multi-study proteomic and bioinformatic identification of molecular overlap between amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). Brain Sci.8(12), (2018).
- FullerHR, GillingwaterTH, WishartTM. Commonality amid diversity: multi-study proteomic identification of conserved disease mechanisms in spinal muscular atrophy. Neuromuscul. Disord.26(9), 560–569 (2016).