
Abstract
Aim: To gain greater knowledge regarding the natural history of aromatic L-amino acid decarboxylase (AADC) deficiency, a genetic disorder that causes severe deficits in motor and cognitive development. Materials & methods: A systematic literature review was performed of all case reports and clinical studies published through December 2019 of patients with AADC deficiency. The data were summarized descriptively. Results: The search identified 94 publications that described 237 unique patients. Mean (standard deviation) age at diagnosis was 3.2 (±5.7) years and 16 deaths were reported. Most patients (57%) received the standard of care therapies, which showed limited efficacy in this patient population. Conclusion: AADC deficiency is a devastating disease and prospectively defined natural history studies are warranted to further understand this disease.
Plain language summary
What is this article about?
The article describes an analysis of the published medical literature that summarized the characteristics of patients with aromatic L-amino acid decarboxylase (AADC) deficiency. AADC deficiency is a very rare genetic disorder that results in loss of AADC enzyme activity and consequently the lack of the neurotransmitters dopamine and serotonin resulting in patients having severe loss of muscle and cognitive function. Data from publications were used to evaluate the frequency of specific symptoms of the disease, muscle function, underlying genotypes and therapies used to treat AADC deficiency.
What were the results?
The results show that AADC deficiency is present worldwide and is due to a variety of underlying genetic mutations in the gene that produces the AADC protein. Diagnosis of the disease is often delayed and does not usually occur until about 3 years of age. Often, these patients die within the first decade of life. The standard treatment of AADC, which does not involve gene therapy, is limited in its ability to treat the symptoms of the disease with only about 3% of patients, who at most have head control by 2 years of age, improving following treatment.
What do the results of the study mean?
This study clearly indicates that AADC deficiency is a devastating disease in which standard therapy is not effective and highlights the need for new therapies, including gene therapies, that treat the primary cause of the disease.
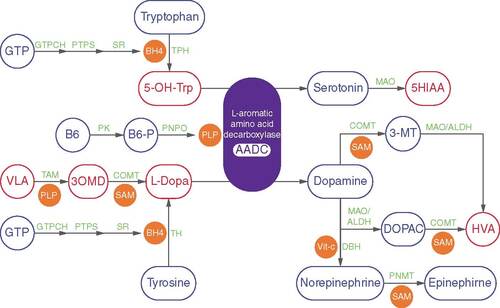
Aromatic L-amino acid decarboxylase (AADC) deficiency is an ultra-rare autosomal recessive disorder of dopaminergic and serotonergic pathways. The disease presents typically in early infancy and is due to the presence of pathological variants in the dopa decarboxylase (DDC) gene that encodes for the AADC enzyme [Citation1,Citation2]. AADC is responsible for the decarboxylation of L-3,4 dihydroxyphenylalanine (L-DOPA) and 5-hydroxytryptophan (5-HTP) to form the neurotransmitters dopamine and serotonin, respectively, and in turn, controls essential neurotransmitters such as norepinephrine and epinephrine () [Citation1–3]. AADC deficiency significantly impacts the quality of life of patients and caregivers [Citation4]. It can result in death in the first decade of life, and patients require life-long care [Citation3,Citation5].
PLP is synthesized from endogenous vitamin B6 (pyridoxine, pyridoxal or pyridoxamine), via B6-P, by PK and PNPO. Precursors of dopamine (L-dopa) and serotonin (5-hydroxytryptophan; 5-OH-Trp) are produced from amino acids tyrosine and tryptophan by tetrahydrobiopterin (BH4)-dependent TH and TPH, respectively. A 3-step reaction results in the synthesis of BH4 from GTP and catalyzed by enzymes GTPCH, PTPS and SR. Vit-c-dependent DBH converts dopamine to norepinephrine. Norepinephrine is subsequently converted to epinephrine by SAM-dependent PNMT. Dopamine is degraded by MAO and ALDH to DOPAC, which is further degraded to the final metabolite HVA by a SAM-dependent COMT. Dopamine can also be degraded to HVA via 3-MT by COMT, MAO and ALDH. Serotonin is degraded by MAO, to 5HIAA. L-dopa is methylated to 3OMD by COMT and subsequently transaminated to VLA by PLP-dependent TAMs. Abbreviated enzymes names are indicated in green, cofactors and co-substrates are shown in orange and key metabolites are indicated in blue.
3-MT: 3-methoxytyramine; 3OMD: 3-O-methyl-dopa; 5HIAA: 5-hydroxyindoleacetic acid; AADC: Aromatic L-amino acid decarboxylase; ALDH: Aldehyde dehydrogenase; B6-P: Phosphorylated B6; COMT: catechol-O-methyltransferase; DBH: Dopamine-β-hydroxylase; DOPAC: 3,4-dihydroxyphenylacetic acid; GTPCH: GTP cyclohydrolase I; HVA: Homovanillic acid; MAO: Monoamine oxidase A; PK: Pyridoxal kinase; PLP: Pyridoxal 5-phosphate; PNMT: Phenylethanolamine N-methyltransferase; PNPO: pyridox(am)ine- 5′phosphate oxidase; PTPS: 6-pyruvoyl-tetrahydropterin synthase; SAM: Sadenosyl-methionine; SR: Sepiapterin reductase; TAM: Transaminase; TH: Tyrosine hydroxylase; TPH: Tryptophan hydroxylases; Vit-c: Vitamin C; VLA: Vanillactic acid.
The neurotransmitters affected by AADC are key for control of the autonomic nervous systems [Citation5]. Patients with AADC deficiency show loss of motor function and present with autonomic symptoms such as mood disturbance, deficits and sleep disorder. Other symptoms associated with AADC deficiency include oculogyric crisis (OGC), a rare form of dystonia [Citation6,Citation7]. Patients with AADC deficiency fail to achieve motor milestones such as head control and the ability to sit, stand and walk [Citation3]. The loss of development of motor function is due to the marked or complete loss of dopamine production in the brain despite essentially preserved neurophysiology and neuroanatomy, as determined by brain imaging [Citation5].
Given that AADC deficiency is an ultra-rare disease, little is understood regarding the natural history of the disease, making it difficult to design studies, which complicates the clinical development of therapies. To gain further insight into the natural history of the disease a systematic literature review was performed of reported case studies of patients with AADC deficiency. The data from this review were used to develop a patient-level database for evaluating the natural history of AADC deficiency and to give current information on the disease.
Materials & methods
Literature search
The systematic literature review was performed in accordance with the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines [Citation8]. MEDLINE, Embase and BIOSIS Preview databases were searched using the following search terms: ((MJMESH.EXACT(‘aromatic-L-amino-acid decarboxylases’)) or (aromatic l-amino acid decarboxylase deficiency) or AADC) and (human(yes)). Additionally, the reference list from the official website of the AADC Research Trust (https://www.aadcresearch.org/cronological-order-with-links) and the reference lists of appropriate review articles were reviewed for relevant publications. No restriction was placed in the date or languages of the publications. The literature search was performed in March 2020 and included all articles published through December 2019.
Eligibility for inclusion of publications in the systematic review included case and case series reports and clinical studies of patients with diagnosed AADC deficiency. Literature reviews of publications and analyses of patients with AADC deficiency were included. Two independent reviewers screened the results of the database searches for eligibility and inclusion into the review. A third reviewer was used to adjudicate any discrepancies.
Data extraction
The following information was extracted: publication, author and year of publication; demographic data (sex, age of diagnosis, AADC variants, ethnicity, race and age of death); and medical history and disease progression (treatments administered for AADC, motor development, symptoms and symptom onset).
Data were extracted from all relevant publications, abstracts and congress presentations by a data entry team into a structured database. If necessary, additional resources were consulted to decide any uncertainties. Each member extracted information independently of the others. The data entry team performed consistency and verification checks of the data entry throughout the data entry process to minimize errors and ensure consistency. All data were quality checked by a party that was independent of the data entry team. All findings from the quality check were reviewed, and revisions were made accordingly.
It was common that a patient was reported in multiple publications. In these instances, the information from the different papers was combined into one overall summary per unique patient.
Genotypic information was updated to reflect that of the current nomenclature per the HUGO Gene Nomenclature Committee (HGNC) guidelines (https://www.genenames.org/). Ethnicity was harmonized using the US FDA’s guidance on Collection of Race and Ethnicity Data in Clinical Trials [Citation9].
Two clinicians, who were not members of the data entry team, evaluated motor function acquisition on the extracted data of each patient. The motor milestone acquisition for each patient in the database was scored based on a scoring system of 1–9 (). This assignment was retroactively performed to maximize consistency of analysis of different patients across a wide range of publications that represented heterogenous description of motor function. The score was defined by the team but was anchored to the Peabody Developmental Motor Scales; Second Edition (PDMS-2) instrument. PDMS-2 is an in-depth assessment of a child’s motor development that evaluates both gross and fine motor skills and has been used in clinical studies to assess treatment effect in children with various diseases that impact motor function [Citation10–14]. The motor milestones are known to be acquired in a sequential manner, and this fact was used to help determine the motor milestones reached by a particular patient based on the reported observations. The clinicians worked independently, the reliability of the clinicians who reviewed the data was high, and they were consistent with one another about 99% of the time. This was determined via cross-check by a third party using programmatic comparison tools available in Microsoft Word and Excel and Statistical Analysis System (SAS). Any discrepancy was resolved by a joint rereview of data by the two clinicians.
Table 1. Summary of scoring for motor milestone acquisition.
For those patients who received gene therapy, only motor milestone data prior to administration of gene therapy are included here.
Once the adjudication of the motor milestones was completed, all patients in the database were programmatically assessed to ensure a consistent approach in the determination of motor milestone acquisition, genotype, use of standard of care and demographics.
Statistical analyses
Data were summarized descriptively.
Results
Results of the literature search
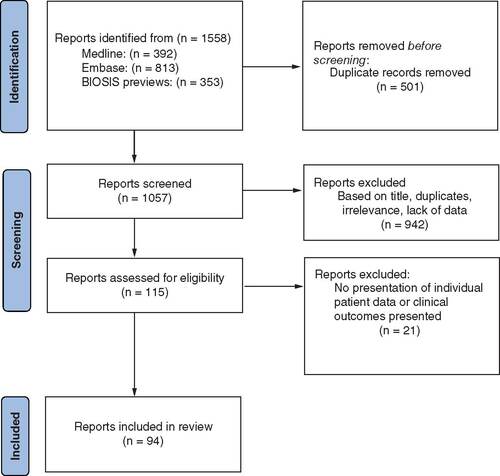
The literature search identified 1558 publications, of which 1057 were screened after the removal of duplicate records (). Following review of titles and abstracts, 115 publications were evaluated in detail, of which 94 met the inclusion criteria. The selected 94 included 55 manuscripts and 39 congress abstracts (Supplementary Table 1 for full list of included publications). Two articles were in Chinese and were translated by a certified translator; all other included publications were in English. The earliest report included was published in 1990.
Demographics & patient characteristics
Across the different reports, a total of 237 unique patients were identified, of which 185 had available high-quality data, where no ambiguities existed with respect to patient demographics and the extracted data required no clinical deductions or interpretations. The findings from these 185 are reported here.
The proportion of males and females within this population was similar, and a wide range of different ethnic backgrounds were observed (). Age at first diagnosis was approximately 3.2 years.
Table 2. Summary of patient demographics and disease characteristics.
The method of diagnosis was not reported for all case studies. However, when reported the most common methods included genetic analysis, symptomology and/or evaluation of the levels of VLA, HVA and 5HIAA in the CNS (). When available, most cases were confirmed through genetic analysis. Out of 185 subjects assessed in this systematic review, 140 (76%) had genotype confirmation of their disease.
A total of 16 deaths were reported with an average age of death of 8 years. Eight of these deaths (11%) were in patients who achieved only head control or less at 2 years of age, with the youngest being 3.5 months of age and the oldest 84.0 months (7 years) of age. Reasons for death in the 16 patients included seizure (n = 1), severe encephalopathies (n = 1), aspiration pneumonia (n = 1), asphyxia and pneumonia (n = 2), bronchoaspiration (n = 1), pulmonary infection (n = 1), influenza B encephalitis (n = 1), sepsis (n = 2) and unknown (n = 6).
Motor milestone acquisition by 2 years of age
Of the 185 patients assessed, data were available to evaluate motor milestone achievement by 2 years of age in 96 of them (). Of those with reported motor milestone, the majority (72%; n = 69) did not demonstrate any motor function, including lifting head against gravity, lifting head and push up or full head control by 2 years of age. Of the 27 patients who attained motor milestones beyond head control by 2 years of age (e.g., sitting, standing or walking), one patient could roll from side to side, two could sit with assistance and 24 could walk with assistance.
Table 3. Summary of milestone acquisition.
Genetic mutations by disease phenotype
Most patients had unique genotypes; 39 distinct genotypes were identified (). The most common genotype was either homozygous (n = 24) or heterozygous (n = 18) for the founder variant c.714 + 4A>T (). The founder mutation is common within the Chinese population and is associated with the inability of patients to obtain motor function [Citation3]. The variant c.714 + 4A>T is a splice site in intron 6 that results in the production of a truncated nonfunctional protein [Citation5].
Table 4. Summary of genotype by phenotype (n = 96).
Response to the standard care for patients with no motor function by 2 years of age
Until recently, the standard of care (SoC) for the treatment for AADC deficiency was drugs that primarily treated symptoms and not the underlying cause of the disease. However, the gene therapy eladocagene exuparvovec (Upstaza) was approved in 2022 in Europe for the treatment of AADC deficiency. As the literature search is through 2019, the data below focus on the reported outcomes with the SoC.
Of the 237 unique patients in the database, 135 had information regarding treatment using SoC. Drugs commonly reported for treating AADC deficiency as part of the SoC include pyridoxine/B6 (79%), monoamine oxidase inhibitors (MAO-I) (25%) and dopamine agonists (53%).
As the ability to interpret the effect of SoC is clearer in cases in which a patient had not achieved a motor milestone, we focused evaluating the impact of SoC on this population. Only two of the 69 patients (3%) who had not attained a motor milestone showed clinical benefit after receiving SoC. One patient, who had two different variants in the DDC gene (i.e., c.367G>A and c.876G>A), was treated at 35 months of age with pyridoxine (AADC cofactor), pramipexole (dopamine receptor agonist), tranylcypromine (MAO-I) and was able to walk with assistance at 48 months of age [Citation15]. The variant c.367G>A is predicted to reduce but not eliminate AADC enzyme activity [Citation5,Citation15]. The residual AADC enzyme activity may account for the response to SoC. The variant c.876G>A is a splice site mutation that results in a truncated nonfunctional protein [Citation5,Citation15].
The other patient (genotype unknown) was treated after diagnosis at 5 years of age with pyridoxine, tranylcypromine and bromocriptine (dopamine receptor agonist). After 4 weeks of treatment, rolling over, vocalization, increased muscle tone and head control were observed [Citation15,Citation16].
Discussion
AADC deficiency is an ultra-rare disease, and little is known about its natural history. To better understand the natural history of the disease, a systematic literature review was performed of all case reports and clinical studies published through December 2019 of patients with AADC deficiency. Wassenberg et al. previously performed a systematic review of AADC case reports of publications through 2015 as part of the development of treatment guidelines for this disease [Citation3]. The findings reported here support and extend the earlier systematic review.
The current search differs from that of Wassenburg et al. [Citation3], as this search was used to develop a patient-level database that could be used to gain insight into the frequency of outcomes such as disease phenotype, variant frequency across the AADC deficiency patient population and response to SoC. The methodology used to build the patient-level database was designed to maximize consistency of assessment of motor milestone acquisition and disease severity across the published reports. This systematic review expands upon Wassenberg et al. as it reports on a larger population of the patients with AADC deficiency, identifies additional genotypes not described in Wassenberg et al., and evaluates the relationship of genotype to motor milestone acquisition and response to standard of care.
The systematic literature search identified 94 publications that met the inclusion criteria. Evaluation of these reports identified 237 unique patients, of which high-quality data were available for 185 of them. The data show that AADC deficiency is present across a wide range of geographic populations and diagnosis is commonly delayed to about 3 years of age. Although the most common variant is the founder mutation (c.714 + 4A>T), present primarily in Chinese populations, the underlying variants in other populations are diverse with most patients having unique variants. Of the reported deaths, the mean age of death was 8 years, with patients who had gained little motor function by 2 years of age, dying within the first decade of life. Causes of death are often due to sepsis and respiratory complications including pneumonia, asphyxiation and bronchoaspiration, all of which may have resulted from poor muscle function due to AADC deficiency.
Of the 96 patients whose disease phenotype could be adjudicated, most (72%; n = 69) had not achieved a motor milestone greater than head control by the age of 2 years. In general, this phenotype was associated with variants predicted to severely reduce the activity of the AADC enzyme [Citation5,Citation17]. Only three patients who had at most head control by 2 years of age responded to the SoC. In all three cases, one of the alleles is the founder mutation that is considered not to produce an active AADC enzyme. However, two of the cases have the same second allele (c.853C>T) that is predicted to produce an AADC molecule that retains enzyme activity [Citation5,Citation17]. In the third case, the second allele was not identified and the impact of the mutation on the function of the AADC enzyme is not known. However, the phenotype suggests the unknown allele does not completely diminish AADC enzyme activity.
The data from this literature review indicate that the SoC has not changed significantly over the past 30 years and that the impact of this therapy has had no or very limited impact on the symptoms of the diseases. Available pharmacological interventions used as part of the SoC include dopamine agonists, MAO-I, pyridoxine therapies and serotonin reuptake inhibitors. Response to these therapies is particularly poor in patients with no motor development [Citation3,Citation10]. Clinical experience indicates that patients typically do not respond to available treatments because these therapies cannot replace or sufficiently increase dopamine production in the brain to adequately improve motor function and allow achievement of developmental milestones [Citation3,Citation15].
A novel gene therapy from PTC Therapeutics, called eladocagene exuparvovec, was recently approved by the EMA for the treatment of patients ≥18 months of age with genetically confirmed severe AADC deficiency, defined as not achieving motor developmental milestones including the ability to sit, stand or walk [Citation18]. Eladocagene exuparvovec is a recombinant adeno-associated virus type 2 containing the human coding sequence of the DDC gene. Eladocagene exuparvovec is delivered to the putamen where it results in the production of the AADC enzyme. Administration of the gene therapy to the putamen has the benefit that it is being administered to the site in which the AADC enzyme acts and also does not have to cross the blood–brain barrier which would be necessary if administered peripherally.
The findings from multiple clinical trials demonstrate that intraputaminal infusion of the AADC coding region is well tolerated and produces clinically meaningful and sustained improvement in motor functions in patients with AADC deficiency independent of the age of the patient at time of treatment [Citation14,Citation19–21]. These studies also found that the gene therapy improved symptoms of the disease including reduction in respiratory tract infections, a primary cause of mortality in this patient population [Citation3,Citation5]. It also reduced the duration and frequency of oculogyric crisis and improved the maintenance of or increase in body weight. Eladocagene exuparvovec also improved motor skills allowing for activities important for daily living such as self-feeding, playing and interacting with the environment [Citation14], which benefited the quality of life for both the patient and the caregiver [Citation4].
A strength of this systematic review is that it took unstructured data from the free-form articles, then structured it and analyzed the data programmatically. However, the data information collected in this systematic review was limited by what was reported. As such, it was not possible to collect all data that may be of clinical interest. For example, age of disease onset was often not clearly defined in the case study reports, and consequently, this information was not collected. Hence, this systematic review focused on age of diagnosis, which was clearly reported in most publications. Additionally, patient information, such as their death, may have been reported in multiple papers. As the analysis in this review was purely descriptive and the data were captured as reported in each publication, no formal assessment of reporting bias was performed.
Conclusion
This systematic review clearly indicates that AADC deficiency is a devastating disease that often is slow to be diagnosed, and historically the treatments available for the past 30 years have shown no or limited efficacy. Currently, AADC deficiency is not part of newborn screening. It also highlights the need for greater understanding of the natural history of the disease through prospectively designed registry studies, such as that of The International Working Group on Neurotransmitter Related Disorders [Citation22], that will also provide insights on the impact of novel gene therapies designed to treat AADC deficiency.
Aromatic L-amino acid decarboxylase (AADC) deficiency is an ultra-rare disease, and little is known about its natural history.
To better understand the natural history of the disease, a systematic literature review was performed of all case reports and clinical studies published through December 2019 of patients with AADC deficiency and used to develop a patient-level database for evaluating the natural history of the disease.
The search identified 94 publications that met the inclusion criteria. Evaluation of these reports identified 237 unique subjects of which high quality data were available for 185 of them, of which the data are described here.
The data show that AADC deficiency is present across a wide range of geographic populations and diagnosis is commonly delayed to about 3 years of age.
Although the most common variant is the founder mutation (c.714 + 4A>T), present primarily in Chinese populations, the underlying variants in other populations are diverse with most patients having unique variants.
Of the 96 patients whose disease severity could be adjudicated, most (72%) could not sit, stand or walk by ≥24 months of age.
Of the reported deaths, the mean age of death was 8 years. Causes of death included pneumonia, asphyxiation, bronchoaspiration and sepsis, all of which may have resulted from poor muscle function due to AADC deficiency.
Standard of care is designed to treat symptoms of the disease and was similar across all publications indicating that SoC has not changed significantly over the past 30 years. Only 3% of patients who at most had head control at 2 years of age responded to SoC, which may reflect the underlying mutation.
In conclusion, the systematic review clearly indicates that AADC deficiency is a devastating disease, lacking effective treatment that targets the underlying cause of the disease.
Author contributions
M Bergkvist, C Stephens and A Wang performed the systematic literature review and extracted and analyzed the data. T Schilling helped adjudicate the disease phenotypes. E Goodwin analyzed the data and drafted the manuscript. T Schilling, X Yu, A Kristensen, L Golden and M Klein critically analyzed the data and reviewed the manuscript.
Supplementary data
Download MS Word (98.1 KB)Acknowledgments
The authors would like to thank A Russel and E Leonardi of PTC Therapeutics for critical review and discussion of this manuscript.
Financial & competing interests disclosure
This work was funded by PTC Therapeutics, Inc. C Stephens, T Schilling, A Wang, X Yu, E Goodwin, L Golden, A Kristensen and M Klein are employed by PTC Therapeutics, Inc. and have received salary compensation for time and effort and hold financial interest in the company. M Bergkvist is a paid consultant for PTC Therapeutics, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- HylandK, ClaytonP. Aromatic amino acid decarboxylase deficiency in twins. J. Inherit. Metab. Dis.13(3), 301–304 (1990).
- HylandK, SurteesRA, RodeckC, ClaytonPT. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology42(10), 1980–1988 (1992).
- WassenbergT, Molero-LuisM, JeltschKet al.Consensus guideline for the diagnosis and treatment of aromatic L-amino acid decarboxylase (AADC) deficiency. Orphanet J. Rare Dis.12(1), 12 (2017).
- WilliamsK, SkrobanskiH, WernerC, O'neillS, BueschK, AcasterS. Symptoms and impact of aromatic L-amino acid decarboxylase (AADC) deficiency: a qualitative study and the development of a patient-centred conceptual model. Curr. Med. Res. Opin.37(8), 1353–1361 (2021).
- HimmelreichN, MontioliR, BertoldiMet al.Aromatic amino acid decarboxylase deficiency: molecular and metabolic basis and therapeutic outlook. Mol. Genet. Metab.127(1), 12–22 (2019).
- BoiS, Garcia-MaloC, RodríguezCI. Oculogyric crisis: a rare type of dystonia. J. Psychiatry Neurosci.46(4), E429–e430 (2021).
- MahalP, SutharN, NebhinaniN. Spotlight on oculogyric crisis: a review. Indian J. Psychol. Med.43(1), 5–9 (2021).
- MoherD, LiberatiA, TetzlaffJ, AltmanDG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Open Med.3(3), e123–130 (2009).
- FDA. Collection of race and ethnicity data in clinical trials (2016). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/collection-race-and-ethnicity-data-clinical-trials
- ChenHC, ChenCL, KangLJ, WuCY, ChenFC, HongWH. Improvement of upper extremity motor control and function after home-based constraint induced therapy in children with unilateral cerebral palsy: immediate and long-term effects. Arch. Phys. Med. Rehabil.95(8), 1423–1432 (2014).
- ChenYP, KangLJ, ChuangTYet al.Use of virtual reality to improve upper-extremity control in children with cerebral palsy: a single-subject design. Phys. Ther.87(11), 1441–1457 (2007).
- DeKegel A, MaesL, Van WaelveldeH, DhoogeI. Examining the impact of Cochlear implantation on the early gross motor development of children with a hearing loss. Ear Hear.36(3), e113–121 (2015).
- XuK, WangL, MaiJ, HeL. Efficacy of constraint-induced movement therapy and electrical stimulation on hand function of children with hemiplegic cerebral palsy: a controlled clinical trial. Disabil. Rehabil.34(4), 337–346 (2012).
- TaiCH, LeeNC, ChienYHet al.Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther.30(2), 509–518 (2022).
- BrunL, NguLH, KengWTet al.Clinical and biochemical features of aromatic l-amino acid decarboxylase deficiency. Neurology75(1), 64–71 (2010).
- MallerA, HylandK, MilstienS, BiaggioniI, ButlerIJ. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a second family. J. Child Neurol.12(6), 349–354 (1997).
- Gene therapy for the treatment of AADC deficiency. https://ncats.nih.gov/trnd/projects/active/aadc-deficiency
- First therapy to treat rare genetic nervous system disorder AADC deficiency. https://www.ema.europa.eu/en/news/first-therapy-treat-rare-genetic-nervous-system-disorder-aadc-deficiency
- ChienYH, LeeNC, TsengSHet al.Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, Phase I/II trial. Lancet Child Adolesc. Health1(4), 265–273 (2017).
- HwuW-L, MuramatsuS-I, TsengS-Het al.Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci. Transl. Med.4(134), 134ra161 (2012).
- KojimaK, NakajimaT, TagaNet al.Gene therapy improves motor and mental function of aromatic L-amino acid decarboxylase deficiency. Brain142(2), 322–333 (2019).
- KuseyriHübschmann O, HorvathG, Cortès-SaladelafontEet al.Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines. Nat. Commun.12(1), 5529 (2021).