
Abstract
Aim: Data for avelumab (anti-PD-L1 antibody) in Chinese patients are limited. Patients & methods: Phase I/Ib, open-label, dose-escalation study of Chinese patients with advanced solid tumors. Primary study objectives were to evaluate the maximum tolerated dose (MTD) and pharmacokinetics (PK) of avelumab. Results: 24 patients received avelumab 3 mg/kg every 2 weeks (Q2W; n = 3), 10 mg/kg Q2W (n = 7), 20 mg/kg Q2W (n = 6) or 10 mg/kg weekly for 12 weeks and then Q2W thereafter (n = 8). MTD was not reached. Avelumab exposure was increased in higher dose groups. Partial responses occurred in two patients (confirmed in one patient); best overall response was stable disease in nine patients. Conclusion: Data for avelumab in Chinese patients with advanced solid tumors were consistent with previous global studies.
Plain language summary
Avelumab is a form of medicine that falls under the category of immunotherapy. This means that it can help the immune system find and destroy cancer cells. In this study, researchers looked at the safety of avelumab in a small group of Chinese people with different types of cancer. Researchers also looked at blood levels of avelumab after treatment. Different doses of avelumab were given to different groups of people. Overall, study results for avelumab in Chinese people were similar to results from earlier studies in other countries.
Clinical trial registration: NCT03523390 (ClinicalTrials.gov)
Avelumab is a fully human anti-PD-L1 monoclonal antibody that has shown durable antitumor activity and an acceptable safety profile in a range of solid tumors [Citation1–7]. Avelumab is approved in various countries for the treatment of metastatic Merkel cell carcinoma, and in Europe and the USA in combination with axitinib for first-line treatment of advanced renal cell carcinoma [Citation1,Citation8–11]. It is also approved in Europe and the USA for first-line maintenance treatment of advanced urothelial carcinoma that has not progressed with first-line platinum-containing chemotherapy, on the basis of data from the phase III JAVELIN Bladder 100 study, [Citation2,Citation10,Citation11] and in the USA, Canada and Israel for treatment of advanced urothelial carcinoma with disease progression following platinum-containing chemotherapy or following disease progression within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (second line) [Citation3,Citation10].
Differences in the pharmacokinetics (PK) and pharmacodynamics of numerous oncology drugs have been observed in Asian populations compared with patients from other regions [Citation12–14]. Previous studies of avelumab were conducted in global and Japanese populations [Citation1–9,Citation15]; however, data for avelumab treatment in Chinese patients are limited.
Given the potential for differences in Asian populations, we performed a phase I dose-escalation study of avelumab to investigate safety and PK in Chinese patients with advanced solid tumors.
Materials & methods
Study design & patients
This was a phase I/Ib, open-label, dose-escalation study to evaluate the safety, PK and antitumor activity of avelumab monotherapy in Chinese patients (NCT03523390). Patients were enrolled at three sites in mainland China. Eligible patients were Chinese and had locally advanced unresectable or metastatic solid tumors for which standard therapy failed or did not exist. They also had a formalin-fixed, paraffin-embedded block containing tumor tissue (biopsy ≤6 months before the study) or ≥12 unstained tumor slides for biomarker detection; were aged ≥18 years; had an Eastern Cooperative Oncology Group performance status of 0 or 1 at study entry; had an estimated life expectancy of ≥3 months; and had adequate hematologic, liver and renal function. Exclusion criteria included prior treatment with antibodies or drugs targeting immune checkpoints (cancer vaccines were acceptable), concurrent anticancer treatment (except for palliative bone-directed radiotherapy and radiotherapy of superficial lesions) and major surgery (except for diagnostic biopsy) ≤28 days prior to the first administration of avelumab.
All patients gave informed written consent, and the informed consent form and clinical study protocol were approved by independent ethics committees/institutional review boards at each study site, according to Chinese regulations. The study was performed in accordance with the clinical study protocol, the ethical principles embodied in the Declaration of Helsinki, and the International Council for Harmonisation E6 Guideline for Good Clinical Practice (GCP; 1996), China GCP and applicable regulatory requirements.
Outcomes
The primary objective was to evaluate the maximum tolerated dose (MTD; occurrence of dose-limiting toxicity [DLT]) and PK of avelumab in Chinese patients. Secondary objectives were the safety, tolerability and immunogenicity of avelumab in Chinese patients. Exploratory objectives included assessment of antitumor activity (including best overall response [BOR], duration of response [DOR], progression-free survival [PFS] and overall survival [OS]). Data for antitumor activity in the overall study population are provided for descriptive purposes only.
Procedures & assessments
Patients received one of four avelumab dosing regimens: 3 mg/kg every 2 weeks (Q2W), 10 mg/kg Q2W, 20 mg/kg Q2W or 10 mg/kg every week (QW) for 12 weeks followed by 10 mg/kg Q2W from week 13 (QW→Q2W). The planned study enrollment was 21–24 patients. A modified 3 + 3 cohort design was used for the 3-mg and 20-mg Q2W cohorts but not for the 10-mg/kg Q2W or 10-mg/kg QW→Q2W cohort. Specifically, for the 3- and 20-mg/kg Q2W cohorts, DLT assessment was required to be completed in the first three patients before more patients were enrolled; however, because the safety of the 10-mg/kg dose had been demonstrated extensively in previous studies, [Citation1,Citation3,Citation5–8,Citation10,Citation11,Citation15] all patients in this cohort were enrolled concurrently. Escalation to the next dosing level (i.e., enrollment of the next cohort) was permitted following a review of the safety data by the safety monitoring committee. Dose increases were not permitted in individual patients. A premedication regimen of diphenhydramine 25–50 mg and acetaminophen 500–650 mg was mandatory approximately 30–60 min prior to the first four doses of avelumab to mitigate potential infusion-related reactions (IRRs).
Safety end points were the occurrence of DLTs in each cohort (excluding the 10-mg/kg QW→Q2W cohort) during the DLT observation period (defined as the initial 21 days of treatment), and occurrence of treatment-emergent adverse events (TEAEs; including adverse events [AEs] considered related or unrelated to treatment) and treatment-related TEAEs for all dose groups. The severity of TEAEs was graded by investigators using the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03 and coded using the Medical Dictionary for Regulatory Activities Version 22.0. DLTs were defined as grade ≥3 adverse drug reactions (defined as any AE suspected to be related to avelumab by the investigator and/or sponsor) that occurred during the DLT observation period and were confirmed to be related to avelumab by the safety monitoring committee. The following were exceptions to the DLT definition: grade 3 IRRs resolving within 6 h and controlled with medical management; transient (≤6 h) grade 3 flu-like symptoms or fever that were controlled with medical management; transient (≤24 h) grade 3 fatigue, local reactions, headache, nausea or emesis that resolved to grade ≤1; grade 3 skin toxicity or grade 3 liver function test (ALT, AST or GGT) value increases that resolved to grade ≤1 in <7 days after medical management (eg, immunosuppressant treatment) was initiated; grade 3 diarrhea unless treatment was discontinued according to treatment guidelines (in which case it was considered a DLT); single laboratory values out of normal range that were unlikely to be related to avelumab according to the investigator, did not have any clinical correlate, and resolved to grade ≤1 within 7 days with adequate medical management; and tumor flare phenomena defined as local pain, irritation or rash localized to sites of known or suspected tumors.
PK parameters measured were as follows: area under the concentration-time curve from zero to tlast (last sampling time; AUC0-t); area under the serum concentration-time curve from zero to tau (the respective dosing interval; AUC0-tau); area under the serum concentration-time curve from zero extrapolated to infinity (AUC0–∞); percentage of area under the serum concentration-time curve from zero to infinity obtained by extrapolation (AUCextra%); dose-normalized AUC0-t (AUC0-t/dose); dose-normalized AUC0-tau (AUC0-tau/dose); maximum serum concentration (Cmax); last quantifiable serum concentration (Clast); serum concentration observed immediately before the next dose (Ctrough); dose-normalized Cmax (Cmax/dose); time to reach maximum serum concentration (Tmax); terminal elimination half-life (t1/2); and terminal elimination rate constant (λz). Serum samples for PK testing were collected at specified time points. For patients receiving avelumab Q2W, PK serum samples were collected on day 1 within 2 h prior to and at the end of the 1-h infusion; at 0.5, 1, 2, 4, 6, 12, 24, 36, 48 and 168 h post infusion, on days 15, 29, 43, 85, 127 and 169 within 2 h prior to infusion (trough value) and immediately after infusion (peak value); then every 12 weeks beyond week 25 within 2 h prior to infusion (trough value). For patients receiving avelumab QW, PK serum samples were collected within 2 h prior to each infusion at weeks 1, 2, 3, 5, 7, 13, 15, 19 and 25, and at 12-week intervals thereafter while on treatment; samples were also collected at the end of infusion (within 15 min) at weeks 1, 7, 13 and 25. In all patients, PK samples were also collected at the end-of-treatment visit and at the 30-day safety follow-up visit.
Antidrug antibodies (ADAs) were measured in serum samples, which were collected via a specified schedule. For patients receiving avelumab Q2W, ADA serum samples were collected within 2 h prior to dosing on day 1, 15, 29, 43, 85, 127 and 169, then every 12 weeks while on treatment. For patients receiving avelumab QW, ADA serum samples were collected on day 1, 3, 5, 7, 13, 19 and 25, then every 12 weeks while on treatment. In all patients, samples were also collected at the end-of-treatment visit and at the 30-day safety follow-up visit. The presence of ADAs was analyzed by Covance® using a homogenous bridging Meso Scale Discovery electrochemiluminescence assay. Patients with treatment-emergent ADAs were considered transient positive if they had a single positive evaluation, or the duration between first and last positive result was <16 weeks apart and the last assessment was not positive; they were considered persistent positive if they had a first and last positive result ≥16 weeks apart or a positive evaluation at the last assessment.
BOR, DOR and PFS were assessed by investigators according to Response Evaluation Criteria in Solid Tumors version 1.1. Tumor assessments were performed within 14 days prior to the first administration of avelumab (baseline), every 6 weeks during the first 12 months of treatment and every 12 weeks thereafter. Patients were treated until withdrawal of consent, disease progression, significant clinical deterioration, unacceptable toxicity or occurrence of any other criterion for withdrawal.
Statistical methods
The DLT analysis set patients included in the dose-escalation schedule and excluded patients in the 10-mg/kg QW→Q2W cohort; in the 20-mg/kg Q2W cohort, if none of the first three patients enrolled had DLT, the study protocol specified that the other three patients would be enrolled for PK evaluation but not DLT assessment. All patients in the DLT analysis set received all avelumab administrations in the DLT observation period or stopped treatment because of DLTs in the DLT observation period. All patients who received ≥1 dose of avelumab were included in the safety analysis set. All patients who received ≥1 dose of avelumab and had measurable disease at baseline were included in the efficacy population set. All patients who received ≥1 dose of avelumab and provided ≥1 measurable post-dose concentration were included in the PK analysis set. All patients who received any dose of avelumab and had ≥1 valid ADA result were included in the immunogenicity analysis set.
Continuous variables were summarized using descriptive statistics, and qualitative variables were summarized by counts and percentages. The dose proportionality of the PK parameters over the administered dose range was assessed graphically on the basis of exposure data (AUC0-tau and Cmax).
Results
Patients
Between 7 May 2018 and 29 April 2019, a total of 24 patients were enrolled and treated with avelumab monotherapy: three with 3 mg/kg Q2W, seven with 10 mg/kg Q2W, six with 20 mg/kg Q2W and eight with 10 mg/kg QW→Q2W. All 24 patients comprised the safety and efficacy analysis sets. All but one patient (n = 23 [95.8%]) had received prior anticancer drug treatment and most patients (n = 15 [62.5%]) had received prior anticancer surgery ().
Table 1. Baseline characteristics.
The median duration of avelumab treatment with the 3-mg/kg, 10-mg/kg, 20-mg/kg and 10-mg/kg QW→Q2W regimens was 30.1, 30.0, 26.6 and 8.0/14.0 weeks (QW/Q2W periods), respectively (). Four patients (one patient in each cohort) remained on treatment at data cutoff (29 July 2019). The most common reasons for discontinuation were disease progression (3 mg/kg, n = 1; 10 mg/kg, n = 5; 20 mg/kg, n = 5; and 10 mg/kg QW→Q2W, n = 4]) and AEs (3 mg/kg, n = 1 and 10 mg/kg QW→Q2W, n = 2).
Table 2. Patient disposition.
Safety
In total, 13 of 24 patients (54.2%) who received avelumab were evaluable for DLTs (3 mg/kg, n = 3 [100.0%]; 10 mg/kg, n = 7 [100.0%]; and 20 mg/kg, n = 3 [50.0%]). No DLTs were reported, and the MTD was not reached.
TEAEs were reported in all 24 patients, including grade ≥3 TEAEs in 10 (41.7%; ). The most common grade ≥3 TEAEs across all cohorts were anemia (n = 4 [16.7%]), GGT increased (n = 2 [8.3%]), decreased appetite (n = 2 [8.3%]) and hyperuricemia (n = 2 [8.3%]). Avelumab-related TEAEs of any grade were reported in 20 patients (83.3%); the most common were pyrexia (n = 7 [29.2%]), proteinuria (n = 6 [25.0%]) and decreased appetite (n = 5 [20.8%]). Avelumab-related grade 3 TEAEs were reported in one of eight patients in the 10-mg/kg QW→Q2W cohort (12.5%; IRR); treatment was discontinued and not re-initiated in this patient.
Table 3. Safety.
Serious TEAEs were reported in eight patients (33.3%); no serious TEAE was considered avelumab related. TEAEs leading to death were reported in two patients (8.3%; apnea and dyspnea; both n = 1); both were considered unrelated to study treatment.
Eight cases of any-grade IRR were reported in five patients (20.8%), including a grade 3 IRR in one patient (4.2%) in the 10-mg/kg QW→Q2W cohort; all were considered treatment related (). One patient (4.2%) in the 10-mg/kg QW→Q2W cohort had two treatment-emergent immune-related AEs (autoimmune thyroiditis and hypothyroidism, both grade 2), both of which were considered treatment related.
Immunogenicity
All 24 patients were included in the immunogenicity analysis set; two were ADA positive, both in the 3-mg/kg Q2W cohort. One patient was transiently ADA positive and one was persistently ADA positive. Both patients had a BOR of stable disease (SD).
PK analyses
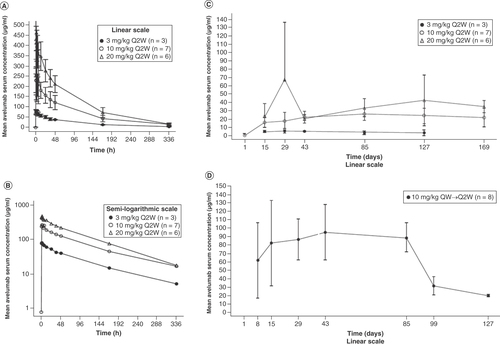
PK was assessed in all 24 patients. Avelumab exposure increased with dose across the 3-mg/kg to 20-mg/kg Q2W dose range following single intravenous administration, and 10-mg/kg QW dosing resulted in higher serum concentrations than 10-mg/kg Q2W dosing ( & ). The median Tmax for all dose levels was observed approximately 1.5–2 h following administration, and the mean observed terminal t1/2 was similar across dose levels (range: 85.5–96.5 h). Ctrough levels were numerically lower in ADA-positive patients (n = 2) than in ADA-negative patients.
Table 4. Avelumab serum pharmacokinetics on day 1 (pharmacokinetics population).
(A) Dose levels (Q2W cohorts), linear (± standard deviation) scale. (B) Dose levels (Q2W cohorts), semi-logarithmic scale. (C) Mean (± standard deviation) trough avelumab serum concentration-time profiles by study day for all dose levels (Q2W cohorts) on the linear scale. (D) Mean (± standard deviation) trough avelumab serum concentration-time profiles by study day for the 10-mg/kg QW→Q2W cohort on the linear scale.
Q2W: Every 2 weeks; QW: Every week.
Antitumor activity
Two patients had a partial response (PR): one was confirmed and one was unconfirmed at the time of data cutoff (Supplementary Table 1). The patient with confirmed PR was in the 20-mg/kg cohort. This patient was diagnosed with adenocarcinoma of the lung with metastasis in the brain, bone, adrenal gland, hilus pulmonis and inguinal lymph nodes. The DOR for the confirmed PR was 18.14 weeks. Overall, nine patients had a BOR of SD (including patients in all four dosing groups).
Discussion
This dose-escalation study sought to assess the safety, PK and antitumor activity of avelumab in a population of Chinese patients with advanced solid tumors. Avelumab was well tolerated at all dose regimens used. No DLTs were reported and the MTD was not reached. The frequency of TEAEs was not correlated with avelumab dose, and the frequency of IRRs was consistent with the known safety profile of avelumab [Citation7]. No treatment-related deaths occurred. The safety of avelumab in the Chinese population in this study was comparable to that reported in the global JAVELIN Solid Tumor study as well as in other Asian populations [Citation7,Citation15]. Immunogenicity and PK data were also consistent with findings from other phase I studies of avelumab monotherapy [Citation15,Citation16]. For example, in the 10-mg/kg Q2W cohort, the Cmax, AUC0-t, and t1/2 geometric mean values in Chinese patients in this study (294 μg/ml, 22,800 h•μg/ml, and 94.6 h, respectively) were similar to values observed in a previous study of avelumab PK with 10-mg/kg Q2W dosing in a predominantly white, US population (89%; 294 μg/ml, 20,400 h•μg/ml, and 90.3 h, respectively) [Citation15]. Only one patient (4.2%) was persistently ADA positive, which is similar to the frequency reported in a previous trial of avelumab (1 of 53 patients [1.9%] persistently ADA-positive) [Citation16]; this patient was treated with the lowest avelumab dose level assessed, which is lower than approved dosing. Clinical implications of ADAs against immune checkpoint inhibitors remain to be elucidated [Citation17]; in previous exploratory analyses for avelumab, effects of ADA development could not be determined because of low patient numbers and confounding variables [Citation10]. Indications of antitumor activity were seen with avelumab treatment, including in two patients with a PR (one confirmed prior to data cutoff) and nine patients with SD.
Limitations of the study include the small number of patients treated with each dose of avelumab, although this is appropriate for a phase I/Ib dose-escalation study. In addition, the study population included patients with a diverse range of tumor types, meaning efficacy data in the overall population cannot be interpreted.
Conclusion
In summary, findings of safety and PK of avelumab in Chinese patients were consistent with previous phase I studies in other populations, and antitumor activity was observed. Data reported support the use of avelumab in Chinese patients and support further investigation of doses up to 20 mg/kg Q2W or 10 mg/kg QW for 12 weeks and Q2W thereafter.
This phase I/Ib dose-escalation study investigated avelumab monotherapy in 24 Chinese patients with advanced solid tumors.
Avelumab dosing was 3 mg/kg every 2 weeks (Q2W), 10 mg/kg Q2W, 20 mg/kg Q2W or 10 mg/kg weekly (Q2W after 12 weeks).
No dose-limiting toxicities were reported and the maximum tolerated dose was not reached.
Safety and pharmacokinetics were consistent with earlier phase I studies in other populations; two patients had partial response (one confirmed).
Data support further investigation of avelumab at doses of up to 20 mg/kg Q2W or 10 mg/kg weekly for 12 weeks and Q2W thereafter.
Author contributions
Conception and design: Y-L Wu, H Pan and K Chin. Provision of study material or patients: Y-L Wu, Y Cheng, H Chen, H Tu, C Xu, Z Wang, Y Liu, Y Xin, H Lou, W Wang and H Pan. Collection and assembly of data: Y-L Wu, Y Cheng, H Chen, H Tu, C Xu, Z Wang, Y Liu, Y Xin, H Lou, W Wang and H Pan. Data analysis and interpretation: all authors. Manuscript writing: all authors. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors.
Financial & competing interests disclosure
This trial was sponsored by Merck (CrossRef Funder ID: 10.13039/100009945), as part of an alliance between Merck and Pfizer. Y-L Wu reports consulting or advisory roles for AstraZeneca, Boehringer Ingelheim, Novartis and Takeda; has received honoraria from AstraZeneca, Beigen, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck & Co., Pfizer, Roche and Sanofi; and has received research support from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Hengrui and Roche. C Xu has received honoraria from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Merck & Co., Novartis, Pfizer, and Roche; reports consulting or advisory roles for Boehringer Ingelheim and Pfizer; has received research support from BeiGene and Hengrui Pharmaceutical; and has received reimbursement for travel and accommodation expenses from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Merck & Co., Novartis, Pfizer and Roche. K Chin was an employee of EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA, at the time this study was conducted. D Li, D Zhao, Y Gao and W Xu are employees of Merck Serono (Beijing) Pharmaceutical R&D Co., Ltd. Beijing, China, an affiliate of Merck KGaA. All other authors declare no conflicts of interest. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing support was provided by M Holland of ClinicalThinking, and funded by Merck (CrossRef Funder ID: 10.13039/100009945), as part of an alliance between Merck and Pfizer.
Ethical conduct of research
All patients gave informed written consent, and the informed consent form and clinical study protocol were approved by independent ethics committees/institutional review boards at each study site, according to Chinese regulations. The study was performed in accordance with the clinical study protocol, the ethical principles embodied in the Declaration of Helsinki, and the International Council for Harmonisation E6 Guideline for Good Clinical Practice (GCP; 1996), China GCP and applicable regulatory requirements.
Data sharing statement
The authors certify that this manuscript reports original clinical trial data: NCT03523390.
For all new products or new indications approved in both the European Union and the USA after 1 January 2014, Merck (CrossRef Funder ID: 10.13039/100009945) will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, upon researcher's request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company's data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck has a co-research, co-development, or co-marketing/co-promotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
Supplementary Data
Download MS Word (20.1 KB)Acknowledgments
The authors thank the patients and their families and the investigators, co-investigators and study teams at each of the participating centers.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/fon-2021-1342
Additional information
Funding
References
- D'Angelo SP , BhatiaS, BrohlASet al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: long-term data and biomarker analyses from the single-arm phase II JAVELIN Merkel 200 trial. J. Immunother. Cancer8(1), e000674 (2020).
- Powles T , ParkSH, VoogEet al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N. Engl. J. Med.383(13), 1218–1230 (2020).
- Apolo AB , EllertonJA, InfanteJRet al. Avelumab as second-line therapy for metastatic, platinum-treated urothelial carcinoma in the phase Ib JAVELIN solid tumor study: 2-year updated efficacy and safety analysis. J. Immunother. Cancer8(2), e001246 (2020).
- Barlesi F , VansteenkisteJ, SpigelDet al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): an open-label, randomised, phase III study. Lancet Oncol.19(11), 1468–1479 (2018).
- Keilholz U , MehnertJM, BauerSet al. Avelumab in patients with previously treated metastatic melanoma: phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer7(1), 12 (2019).
- Hassan R , ThomasA, NemunaitisJJet al. Efficacy and safety of avelumab treatment in patients with advanced unresectable mesothelioma: phase 1b results from the JAVELIN Solid Tumor trial. JAMA Oncol.5(3), 351–357 (2019).
- Kelly K , InfanteJR, TaylorMHet al. Safety profile of avelumab in patients with advanced solid tumors: a pooled analysis of data from the phase I JAVELIN Solid Tumor and phase II JAVELIN Merkel 200 clinical trials. Cancer124(9), 2010–2017 (2018).
- D'Angelo SP , RussellJ, LebbeCet al. Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol.4(9), e180077 (2018).
- Choueiri TK , MotzerRJ, RiniBIet al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann. Oncol.31(8), 1030–1039 (2020).
- Bavencio (avelumab) . Prescribing information. EMD Serono (2020). www.bavencio.com/content/dam/web/health-care/biopharma/web/Bavencio/us/redesign/hcp/bavencio-hcp-home/bavencio-pi.pdf
- Bavencio (avelumab) . Summary of product characteristics. Merck KGaA, Darmstadt, Germany (2020). www.ema.europa.eu/documents/product-information/bavencio-epar-product-information_en.pdf
- Rajman I , HiranoM, HonmaW, ZhaoS. New paradigm for expediting drug development in Asia. Drug Discov. Today25(3), 491–496 (2020).
- Peng L , QinBD, XiaoKet al. A meta-analysis comparing responses of Asian versus non-Asian cancer patients to PD-1 and PD-L1 inhibitor-based therapy. Oncoimmunology9(1), 1781333 (2020).
- Ma Y , LinQ, YangYet al. Clinical pharmacokinetics and drug exposure-toxicity correlation study of docetaxel based chemotherapy in Chinese head and neck cancer patients. Ann. Transl. Med.8(5), 236 (2020).
- Doi T , IwasaS, MuroKet al. Phase 1 trial of avelumab (anti-PD-L1) in Japanese patients with advanced solid tumors, including dose expansion in patients with gastric or gastroesophageal junction cancer: the JAVELIN Solid Tumor JPN trial. Gastric Cancer22(4), 817–827 (2019).
- Heery CR , O'Sullivan-CoyneG, MadanRAet al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose-escalation trial. Lancet Oncol.18(5), 587–598 (2017).
- Enrico D , PaciA, ChaputN, KaramouzaE, BesseB. Antidrug antibodies against immune checkpoint blockers: impairment of drug efficacy or indication of immune activation?Clin. Cancer Res.26(4), 787–792 (2020).