
Abstract
In recent years there has been enormous interest in researching oxidative stress. Reactive oxygen species (ROS) are derived from the metabolism of oxygen as by-products of cell respiration, and are continuously produced in all aerobic organisms. Oxidative stress occurs as a consequence of an imbalance between ROS production and the available antioxidant defence against them. Nowadays, a variety of diseases and degenerative processes such as cancer, Alzheimer’s and autoimmune diseases are mediated by oxidative stress. Heat stress was suggested to be an environmental factor responsible for stimulating ROS production because of similarities in responses observed following heat stress compared with that occurring following exposure to oxidative stress. This manuscript describes the main mitochondrial sources of ROS and the antioxidant defences involved to prevent oxidative damage in all the mitochondrial compartments. It also deals with discussions concerning the cytotoxic effect of heat stress, mitochondrial heat-induced alterations, as well as heat shock protein (HSP) expression as a defence mechanism.
Introduction: the chemistry of oxygen
Mitochondria are the main site of oxygen metabolism, accounting for approximately 85–90% of the oxygen consumed by the cell [Citation1,Citation2]. Reactive oxygen species (ROS) are by-products of oxygen metabolism, being chemical species with one unpaired electron derived from molecular oxygen. In the ground state, molecular oxygen is a bi-radical. It contains two unpaired electrons in the outer shell (also known as a triplet state). Since the two single electrons have the same spin, oxygen can only react with one electron at a time and therefore it is not very reactive with the two electrons in a chemical bond. If one of the two unpaired electrons is excited and changes its spin, the ground state of the oxygen changes. The species formed is known as singlet oxygen and is a powerful oxidant because the two electrons with opposing spins can quickly react with other pairs of electrons, especially double bonds [Citation3].
Although small concentrations of reactive oxygen species (ROS) are used in cellular signalling [Citation4], uncontrolled increases of ROS and reactive nitrogen species (RNS) concentrations lead to free radical mediated chain reactions which indiscriminately target proteins [Citation5], lipids [Citation6], polysaccharides [Citation7] and DNA [Citation8,Citation9], and trigger the intrinsic pathway of apoptosis. Various kinds of stress including heat and cold stress lead to an overproduction of ROS [Citation10].
Heat stress and ROS production
Superoxide anion
Heat stress increases mitochondrial superoxide anion () levels [Citation11,Citation12].
is the first by-product of the one-electron reduction of oxygen. This reaction occurs at specific sites of the electron transport chain (ETC) which is an important source of
during hyperthermia [Citation13,Citation14]. Generation of
has also been closely associated with the univalent reduction of oxygen by mitochondrial semiquinones [Citation1]. Semiquinone radicals have been directly observed in the portal blood of hyperthermic rats by electron paramagnetic resonance spectroscopy [Citation15]. In Escherichia coli, flavin adenine dinucleotide autoxidation from nicotinamide adenine dinucleotide (NADH) has been reported to be a primary source of
formation, which also increases in ubiquinone mutants in a temperature-dependent manner [Citation16]. Perhaps thermal denaturation of flavin-containing redox proteins might enhance the rate of reaction of reduced flavoproteins with oxygen to form
. This latter species increases when a higher ratio of reduced redox centres exists [Citation17].
Heat stress was also shown to decrease superoxide dismutase 1 (SOD-1) mRNA levels, cytoplasmic SOD protein and enzyme activity, leading to the increase of ROS generation [Citation18]. Furthermore, heat stress leads to an overproduction of transition metal ions (TMI), which can make electron donations to oxygen, forming superoxide anions [Citation19,Citation20]. In that way, it was demonstrated that heat stress increases the rate of iron release from ferritin, and there is a positive relationship between the magnitude of iron release and the amount of xanthine oxidase (XO) in the type O form, and hence the rate of production [Citation21].
is the precursor of most ROS and a mediator in oxidative chain reactions [Citation3]. It is highly reactive and does not diffuse easily throughout the cell membrane. Superoxide anions can dismute to produce hydrogen peroxide (H2O2) either spontaneously or through a reaction catalysed by SOD enzyme. In addition,
may react with other radicals including NO. The product, peroxynitrite (ONOO−), is also a very powerful oxidant [Citation22,Citation23].
Hydrogen peroxide and hydroxyl radical
Hydrogen peroxide (H2O2) is the second key player in mitochondrial ROS. It is the product of superoxide detoxification by MnSOD. Heat stress was shown to increase H2O2 [Citation24,Citation25] and hydroxyl radical formation [Citation26]. The generation of OH· corresponds to an increased generation of [Citation26].
Since hyperthermia enhances the production of TMI, electron capture by oxygen leads to the formation of H2O2 [Citation19–21]. H2O2 is further reduced to the extremely reactive hydroxyl radical via the Fenton reaction, producing the extremely reactive hydroxyl radical (OH·) [Citation27].
Due to its small size and relatively benign reactivity, compared to the rest of ROS, H2O2 can diffuse freely across cell membranes. Hence, it is able to mediate toxic effects far from the site of ROS production.
OH· has a very short half-life and reacts with any molecule in close proximity [Citation28]. H2O2 is able to induce reversible covalent modifications of cysteine (Cys) thiol residues located in active and allosteric sites of specific proteins, resulting in alterations of their activity and function [Citation29]. Any protein containing a deprotonated Cys residue is susceptible to oxidation by H2O2 [Citation30]. Protein thiols can undergo further two-electron oxidations by H2O2 to form sulphinic (R–SO2H) or sulphonic acid (R–SO3H). Different proteins can be modified by H2O2, including phosphatases, transcription factors, ion channels, antioxidant and metabolic enzymes, structural proteins and protein kinases, among others [Citation31].
Nitric oxide and peroxynitrite
Nitric oxide is a vasodilator resulting from the breakdown of arginine to citrulline, in a reaction catalysed by a family of nicotinamide adenine dinucleotide phosphate (NADPH)-dependent enzymes called nitric oxide synthase (NOS) [Citation32].
Severe hyperthermia increases NOS levels and stimulates the release of NO [Citation15,Citation33,Citation34]. The formation of NO· in mitochondria may have important consequences because this compound binds to haem groups from cytochromes (in particular cytochrome oxidase) and inhibits respiration. This may, in turn, stimulate formation [Citation35].
ONOO− is a highly damaging agent with a vast repertoire of targets and detrimental cellular effects. ONOO− modifies proteins by nitrating tyrosine residues, forming dityrosine and oxidising tryptophan and Cys [Citation36]. The main mitochondrial targets of ONOO− are complexes I, II, IV and V of the ETC, aconitase, creatine kinase, superoxide dismutase, mitochondrial membranes and mitochondrial DNA (mtDNA).
NADPH oxidase-mediated ROS
In addition to mitochondria, ROS are generated by NADPH oxidase while converting NADPH to NADP+ [Citation37]. Heat treatment (HT) was shown to increase the NADP+/NADPH ratio without affecting NADPH subunits, as well as the NADPH oxidase 1 (NOX1) mRNA expression [Citation38]. These data suggest that HT activates NADPH oxidase through NOX1 up-regulation. The NOX1 knockdown significantly inhibited ROS production [Citation38]. These findings suggest that NOX1 is required for heat-induced ROS production. NOX1-derived ROS production is mediated by the ERK signalling pathway in response to HT [Citation39]. NOX-mediated ROS production leads to the activation of heat inducible factor-1 (HIF-1) [Citation38].
Detection of intracellular ROS production
Numerous methods were used to estimate intracellular ROS production in response to heat exposure, including chemiluminescence, cytochrome c reduction, nitro blue tetrazolium (NBT) reduction, electron spin resonance and spin trapping, aromatic traps, as well as flow cytometry. All these methods confirmed the heat-induced increase in ROS [Citation17,Citation26,Citation40,Citation41].
Heat stress and the mitochondrial antioxidant defence system
Mitochondrial antioxidant defence system
In order to maintain the steady-state concentrations of ROS, mitochondria have their own antioxidant system allowing the scavenging and neutralisation of the radicals generated. As summarised in , the mitochondrial antioxidant system includes two major groups: enzymatic and non-enzymatic low molecular weight antioxidants such as vitamin E.
Table 1. The mitochondrial antioxidant system.
Mitochondrial antioxidant system and thermal sensitivity
The modulation of antioxidant enzymes affects the thermal sensitivity of the cell. MnSOD plays an important role in thermotolerance. Lowering SOD enzyme levels results in a significant reduction in thermal resistance [Citation49], whereas overexpression of MnSOD by stable transfection provides cellular resistance against the cytotoxic effects of hyperthermia [Citation18,Citation50,Citation51]. These results suggest that superoxide free radicals, or their reaction products, are responsible for much of the cytotoxicity of hyperthermia. Indeed, some authors demonstrated that treatment of cells with catalase conferred partial protection against heat-induced apoptosis, whereas treatment with SOD had no effect [Citation40]. In light of these results, H2O2 appears to be of greater importance.
Heat stress was also shown to decrease or inhibit SOD expression and activities [Citation52–54]. This inhibition can be attributed to thermal/oxidative inactivation of the enzyme [Citation55].
Furthermore, several studies demonstrated that heat stress results in a dramatic decrease in glutathione (GSH) levels. GSH depletion is coupled with an increased ROS production [Citation25,Citation56–59] and increased sensitivity to hyperthermia [Citation60]. It is considered as an early event of heat-induced apoptosis [Citation61].
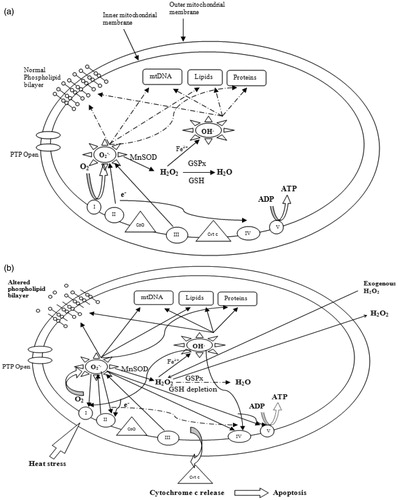
It is also important to note that heat stress disturbs the function and the structure of complex I of the ETC. In addition, it has been shown that the impairment of this complex induces MnSOD and/or increases ROS production [Citation62–64]. As a result, a complex I defect may not be accompanied by increased ROS production, if MnSOD activity is elevated. However, MnSOD as the sole scavenger might not be sufficient to prevent oxidative damage. Its action needs to be coupled with glutathione peroxidase (GSPx)/GSH for a complete detoxification of H2O2 in order to avoid the formation the highly reactive OH· [Citation63]. Since heat stress results in both drastic decrease in SOD and GSH levels, and because mitochondria do not contain catalase, ROS have a pivotal role as intracellular mediators of heat-induced oxidative stress, and heat-induced cytotoxicity [Citation40]. Consequently, free radicals are propagated through non-discriminative chain reactions and target lipids, proteins, carbohydrates and nucleic acids, and result in mitochondrial dysfunction through Bax induction and cytochrome c release [Citation65], as illustrated in .
Figure 1. Generation and targets of reactive oxygen species in mitochondria. (a) A schematic diagram of ROS generation and their targets under thermal neutral conditions. Solid arrows indicate generation and diffusion of molecules. Dashed arrows indicate attenuated/blocked reactions. (b) An illustration of ROS generation and their targets under heat stress conditions. GSH depletion leads to increasing and OH√ production, and therefore mitochondrial damage.
Heat stress and the respiratory chain
The energy released as electron flow through the respiratory chain is converted into a H+ gradient through the inner mitochondrial membrane [Citation66]. This gradient, in turn, dissipates through the adenosine triphosphate (ATP) synthase complex (complex V) and is responsible for the ATP synthesis [Citation67]. The amount of cellular ATP is regulated by mitochondrial and glycolytic ATP synthesis.
The energetic activity of mitochondria changes under the heat shock effect [Citation68] because of the thermosensitivity of the respiratory chain to elevated temperatures [Citation68,Citation69].
Heat stress inhibits mitochondrial ATP synthesis and results in the dysfunction of the ETC [Citation25,Citation70,Citation71]. It has been shown that ROS oxidises protein thiols of complexes I, II, IV and V (F0F1 – ATPase) [Citation72,Citation73] and carbonylates several glycolytic enzymes [Citation74]. In addition, ROS-activated poly(ADP-ribose) polymerase reduces both NAD+ and ATP [Citation75]. Also, it has been shown that heat stress inactivates the complex I of the respiratory chain and causes its dissociation into smaller components without affecting the other complexes [Citation71], resulting in the slowdown of the electron flow through the ETC [Citation76,Citation77]. As a result, the respiratory chain becomes more reduced, the oxygen uptake reduces and the physiological steady-state concentration of formation increases [Citation78]. The decrease of electron flow along the respiratory chain results in a decrease in respiration, and therefore the depletion of ATP synthesis [Citation25,Citation76].
Heat stress and lipid peroxidation
Enhanced ROS production in mitochondria during heat stress leads to non-specific modification of lipids, proteins and nucleic acids, resulting in bioenergetic dysfunction. Mitochondrial membrane constituents are particularly susceptible to oxidative damage by ROS. The major phospholipid components of the mitochondrial membranes are rich in unsaturated fatty acids that are potentially susceptible to oxygen radical attack, because of the presence of double bonds, which undergo peroxidation through a chain of oxidative reactions.
Malondialdehyde (MDA) is the principal product of polyunsaturated fatty acid (PUFA) peroxidation. Exposure to heat stress results in higher mitochondrial and plasmatic MDA levels [Citation11]. Lipoperoxidation of mitochondrial membrane is another source of free radicals production, due to their self-propagating nature.
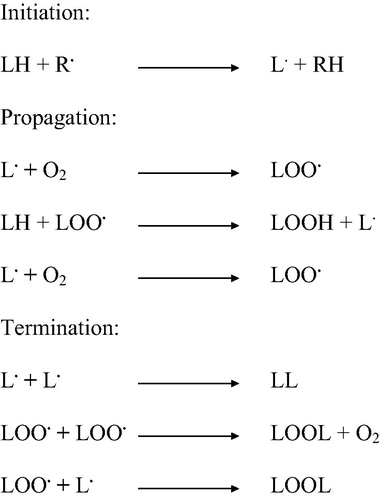
Peroxidation of polyunsaturated fatty acids of the mitochondrial membrane occurs in three distinct steps: initiation, propagation and termination [Citation20,Citation79]. This reaction is initiated by the abstraction of a hydrogen atom by ROS, especially by hydroxyl radicals [Citation80]. PUFA are attacked by a radical at an internal position or near the end of the conjugated system, generating a peroxyl radical (LOO·) [Citation81]. When attacked at an internal position, the peroxyl radical undergoes either a cyclisation or metal catalysed reaction leading to produce reactive alkoxyl radicals. After cyclisation, the fatty acid may form a hydroperoxide or undergo another cyclisation, which produces aldehydes, including MDA and 4-hydroxy-2-nonenal (HNE) [Citation82]. While MDA can react with DNA bases, resulting in gene mutations, HNE reacts mostly with proteins, leading to functional alterations [Citation83].
During the propagation step, peroxyl radicals react again with fatty acid to produce lipid free radicals, and this reaction is propagated. During termination, the two radicals react with each other, and the process comes to an end [Citation20]. All of these steps are summarised in .
Figure 2. A schematic diagram of the different steps of the peroxidation of PUFA. This reaction is initiated when ROS abstract a hydrogen atom from the PUFA, leading to the generation of a peroxyl radical and the production of lipid free radicals.
Heat stress and mitochondrial protein oxidation
Heat stress was also shown to cause mitochondrial protein denaturation. This damage is triggered by the overproduced ROS. Oxidatively damaged mitochondrial proteins include subunits of the pyruvate decarboxylase complex, subunits of the ATP synthase, and enzymes of the tricarboxylic acid (TCA) cycle [Citation80]. During heat exposure, a large number of mitochondrial proteins are oxidised. A total of 82 oxidised proteins have been identified with molecular weights ranging from 24 to 108 kDa [Citation11].
Heat-induced ROS can damage and/or inhibit proteins in several ways, including:
Direct oxidation of amino acids by ROS. In this case, the oxidation of Cys residues forms disulphide bonds, oxidation of Met residues forms Met sulphoxide, and oxidation of arginine, lysine, proline, histidine, serine and threonine residues creates carbonyl groups in side chains [Citation20,Citation23,Citation84].
Reactions with lipid peroxidation products. For instance, HNE can form adducts with Cys, histine and lysine residues, resulting in an altered enzyme function [Citation85]. It was also shown that inhibition of mtETC results in elevated MDA levels [Citation86,Citation87].
Reactions with reactive nitrogen species that are formed by reaction of NO with ROS [Citation88].
Oxidation causing the break of peptide backbone [Citation89].
Direct ROS interaction with metal co-factors such as the iron-sulphur enzyme aconitase of the TCA cycle [Citation90,Citation91]. The oxidative degradation of protein is enhanced in the presence of metal co-factors that are capable of redox recycling, such as Fe.
There are many forms of heat-induced oxidative damage of proteins, including oxidation of amino acid residues side chains, formation of protein–protein cross linkage, and oxidation of the protein backbone resulting in protein fragmentation [Citation92]. Although all amino acids are susceptible to oxidation by ROS/RNS, certain side chains such as lysine, arginine, histidine, proline and threonine are particularly susceptible to oxidation because of their high sensitivity to metal catalysed oxidation [Citation81]. Because protein oxidation leads to a formation of carbonyl groups which are easily detectable, the level of protein carbonyls has been used as a quantitative marker of protein oxidation and oxidative stress [Citation89].
Protein oxidation can be reversible or irreversible. Reversible oxidation of the sulphydryl group includes intramolecular or intermolecular protein cross-linkages and glutathionylation [Citation93]. Irreversible protein oxidation includes nitrosylation of Cys sulphydryl groups, tyrosine, methionine, and tryptophan by ONOO−. Nitration of tyrosine residues may inhibit its phosphorylation or adenylation, important for protein function [Citation94]. Oxidative stress can induce disulphide bond-mediated protein cross-linkage or secondary oxidative modifications such as adduct formation between oxidised proteins and lipid peroxides or glycation products. These products can generate aggregation of bulky protein complexes, which may inactivate both 26S and 20S proteosome, leading to accumulation of damaged proteins and cell death [Citation95].
Heat stress and mitochondrial DNA damage
MtDNA encodes several proteins which are involved in the ETC, the main source of ROS in cells. It has been demonstrated that following oxidative stress, mtDNA damage is not only more extensive, but also persists longer than the damage in the nuclear DNA (nDNA) [Citation96,Citation97]. Several studies have demonstrated that mtDNA mutations lead to ETC complex dysfunction, increased production of ROS, and oxidative damage. An inherited or random primary mtDNA mutation initially induces a respiratory defect, leading to the increase of the leakage of ROS from the ETC. Subsequently, ROS may trigger the accumulation of secondary mtDNA mutations exacerbating mitochondrial respiratory defects and consequently increasing ROS production and lipid peroxidation from mitochondria [Citation28]. Other studies are necessary to make a direct correlation between oxidatively induced mtDNA damage and heat exposure.
At the cellular level, the DNA repair is directly impaired by hyperthermia. The hyperthermia-induced DNA repair deficiency is enhanced by inhibitors of the cellular heat-shock response [Citation98].
Heat stress and uncoupling proteins
Mammalian uncoupling proteins (UCPs) belong to a transporter family of proteins. As the name suggests, UCP uncouple respiration from ATP synthesis by dissipating the mitochondrial proton gradient [Citation99]. UCPs 1, 2, 3, 4, and 5 are members of the anion carrier protein family located in the inner mitochondrial membrane. UCP1 is present exclusively in brown adipose tissue, which is the major site of thermogenesis in small rodents [Citation100]. It is now accepted that UCP1 is a key molecule in thermogenesis, in particular in cold- and diet-induced heat production [Citation101,Citation102]. UCP2 is ubiquitously expressed [Citation103], whereas UCP3 expression is seen in skeletal muscle, adipose tissue, and heart [Citation104,Citation105]. UCP4 is mainly expressed in brain and other neural tissues. UCPs are implicated in the mediation of thermogenesis, in the regulation of lipids as fuel substrates, in the control of insulin secretion, in the control of ROS production, and in protection against their deleterious impacts [Citation106–109]. In fact, mild mitochondrial uncoupling via the action of adenine nucleotide translocator (ANT) as well as UCP decreases ROS production [Citation110,Citation111].
Heat stress down-regulates UCPs at the transcriptional level. It was shown that both UCP protein levels and their corresponding mRNA decreased dramatically in response to heat stress exposure. This decrease is associated with an important increase in ROS production [Citation12]. It is also important to note that ANT mRNA levels are not affected by exposure to heat stress [Citation41].
Heat stress and apoptosis
Mitochondria play a key role in the commitment to cell death through diverse signalling pathways activated by cell death stimuli [Citation112].
There is evidence that hyperthermia induces apoptotic cell death by a mitochondria-induced pathway [Citation25,Citation113,Citation114]. Two major mechanisms are activated during the outer mitochondrial membrane (OMM) permeabilisation to cytochrome c during heat stress [Citation112].
The first relies on the OMM pores such as Bax/Bak oligomeric pores [Citation115,Citation116] and Bax (Bak)/voltage-dependent anion channel (VDAC) hybrids [Citation117], which are regulated by the anti and pro-apoptotic Bcl2 family proteins which control directly the OMM permeabilisation to cytochrome c. Hyperthermia induces apoptosis through mitochondrial Bax translocation and cytochrome c release into the cytosol [Citation25].
The second is the non-specific OMM rupture by the persisting opening of the permeability transition pore (PTP) [Citation112,Citation118,Citation119], due to mitochondrial Ca2+ overload. Ca2+ is mainly taken up into the mitochondrial matrix by mitochondrial calcium uniporter in the inner mitochondrial membrane in the presence of Δψ. The ER and mitochondria are interconnected with respect to Ca2+ transfer via microdomains at their close apposition [Citation120–122]. Hyperthermia causes mitochondrial Ca2+ and ROS overload which are the key inducers of PTP opening [Citation25], resulting in the matrix swelling and OMM rupture [Citation122–124]. The resultant OMM permeabilisation releases apoptotic factors such as cytochrome c and Smac/DIABLO (a mitochondrial protein that potentiates some forms of apoptosis [Citation125]) from the mitochondria into the cytosol [Citation126,Citation127].
Once released, cytochrome c binds to apoptotic protease activating factor 1 (Apaf-1) and forms the Apaf-1/procaspase-9 apoptosome, thereby activating the initiator caspase-9, which in turn activates the downstream effectors, caspases-3 and 7 [Citation50]. However, it seems that ROS or mitochondrial Ca2+ overload may assist the apoptotic cytochrome c release via the two step mechanisms by which cytochrome c is first mobilised from peroxidised cardiolipin in the cristae or from ROS/Ca2+ remodelled cristae to the releasable intermembrane compartment, and subsequently released through the critical Bax-mediated OMM permeabilisation [Citation25,Citation128].
Moreover, it has been shown that heat stress induces the necrosis pathway [Citation25,Citation114]. In fact, apoptosis and necrosis have been shown to be more tightly linked than once believed. Necrosis serves as a substitute for caspase-mediated apoptosis during development [Citation129]. FAS receptor ligation, which normally induced the classical caspase-mediated apoptotic pathway, induced necrosis in caspase-8-deficient Jurkat cells [Citation130]. Moreover, PTP is involved in the necrosis process. The anti-apoptotic proteins, Bcl-2 and Bcl-XL, have been shown to confer protection against both apoptosis and necrosis [Citation131,Citation132].
Heat stress and heat shock proteins (HSPs)
Although synthesis of most cellular proteins is impaired under heat stress, this cannot apply to heat shock proteins. HSPs represent a heterogeneous group of molecular chaperones with different molecular weight and biological functions. HSPs are usually divided into small HSPs (molecular weight <40 kDa), and the Hsp60, Hsp70, Hsp90 and Hsp100 protein families. All these families share their chaperone function, which facilitates the folding, unfolding and refolding of nascent or stress denatured proteins, thus preventing their improper folding or further damage and degradation [Citation133]. HSPs unselectively bind to hydrophobic protein sequences liberated by denaturation, so that they prevent their irreversible interaction with neighbour proteins, thus preventing the loss of the protein function. A variety of non-toxic chaperone inducers in chemical compounds can be isolated from medicinal plants [Citation134].
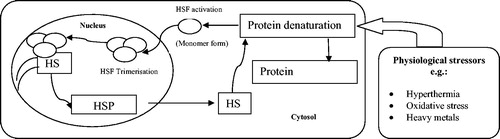
HSP synthesis is triggered by the activation of so-called ‘heat shock transcription factors’ (HSFs) by a signalling cascade. HSFs are present in the cytosol in an inactive state characterised by a monomeric and non-DNA binding form [Citation135]. Upon activation, they are hyperphosphorylated by protein kinases and translocate into the nucleus where they are trimerised. HSF trimer complexes bind to specific sites on HSP gene promoter regions called ‘heat shock elements’ (HSEs) [Citation136]. HSP mRNA is then transcribed and leaves the nucleus to the cytosol where the new HSP is synthesised ().
Figure 3. A schematic diagram of the mechanism of HSP synthesis. Denatured proteins accumulate in response to physiological stressors, and activate HSFs. HSFs are hyperphosphorylated in the nucleus and bind to the HSEs. HSP mRNA is then transcribed. HSP are synthesized and ensure the renaturation of denatured proteins.
Whereas HSPs expression is not restricted to heat stress conditions, it is generally accepted that HSPs prevent cells from lethal thermal damage. Today, at least Hsp27 and Hsp70 families are associated with heat exposure [Citation137–139]. Indeed, it was demonstrated that HSF1 is able to respond to signalling by stress factors, including elevated temperature [Citation140]. Deckers et al. showed that temperature-induced Hsp70 promoter activation was modulated by both temperature as well as duration of hyperthermia [Citation141]. Other studies suggest that hydrogen peroxide increases nuclear translocation and DNA binding activity site of HSF1, which makes a redox mechanism in heat-induced signal transduction pathways during apoptosis very likely [Citation40].
HSPs may be induced as a mechanism of cellular defence, which would support the hypothesis of an inverse relationship between heat-induced cytotoxicity and HSP induction. It was demonstrated that the lack of Hsp27 and Hsp70 synthesis during heat exposure allows cells to undergo apoptosis [Citation40,Citation59]. Moreover, they abolish heat-induced ROS production [Citation59]. It was also shown that Hsp27 and Hsp70 suppress the apoptogenic PTP opening [Citation142]. In this model, heat stress induces misfolding of mitochondrial membrane proteins, which expose their hydrophilic residues to the bilayer phase and then cluster to form aqueous pores that are permeable to ions and molecules up to a molecular mass of about 1500 Da. Chaperone proteins bind to the pore complex to shut off conductance. If the number of misfolded protein clusters exceeds the number of chaperones available to block the PTP conductance, unregulated pores accumulate [Citation124].
Both Hsp27 and Hsp70 inhibit cytochrome c release and thereby the intrinsic pathway of apoptotic cell death [Citation143–145]. At the post-mitochondrial level, Hsp70 binds to Apaf-1, thereby preventing the recruitment of procaspase-9 to apoptosome [Citation146,Citation147]. Indeed, Hsp27 binds to released cytochrome c, and blocks the interaction of Apaf-1/procaspase-9 [Citation148]. Hence, both Hsp70 and Hsp27 protect cells from heat-induced mitochondrial apoptosis.
Protection against heat stress
There is an abundance of oxidisable substrates in the cell, including proteins, lipids, carbohydrates, and DNA. Thus, antioxidants may function to prevent the formation of or to detoxify free radicals, to scavenge ROS or their precursors. As defined by Halliwell [Citation149], ‘an antioxidant is any substance that, when present at low concentrations compared to those of an oxidisable substrate, significantly delays or prevents oxidation of that substrate.’
Several studies have shown that extracellular administration of antioxidants such as GSH and taurine conferred partial thermoprotection [Citation150–153]. However, Sreedhar et al. [Citation59] reported that GSH, NAC or vitamin E are ineffective in inhibiting or delaying heat-induced apoptosis in BC-8 cells (a rat histiocytoma). Katschinski [Citation40] reported that treatment with catalase protects cells from hyperthermia-induced apoptosis.
Another approach to protecting cells from oxidative damage is targeting antioxidant compounds, including small molecule antioxidants and antioxidant enzymes, specifically to mitochondria. It was recently reported that targeting antioxidants to mitochondria protects from heat-induced oxidative damage. Mito-TEMPO was shown to block the generation of both intracellular and mitochondrial ROS, whereas inhibitors of reduced NADPH oxidase, nitric oxide synthase, cyclooxygenase and lipoxygenase did not. Furthermore, Mito-TEMPO inhibited hyperthermia-induced MDA production, cardiolipin peroxidation and heat-induced apoptosis [Citation65].
Whereas sulphydryl compounds have been widely investigated because of their potential radioprotectant properties, they fail to prevent hyperthermia-induced cell killing. Studies with synchronised Chinese hamster cells heated at 43 °C for 1 h in the presence of 16 mM cysteamine demonstrated that the potentiation of heat killing occurred in all phases of the cell cycle. Similarly, enhancement of hyperthermia-induced cell killing was seen for asynchronous cells exposed to 2-aminoethylisothiourium bromide, Cys and 5-thio-D-glucose, but the magnitude of the effect differed for the various sulphydryl compounds [Citation154,Citation155].
In addition to antioxidants, glucose appears to protect cells against H2O2 and heat shock by providing NADPH through its metabolism via the pentose phosphate cycle [Citation156]. Furthermore, glycerol was shown to protect cells from heat-induced oxidative stress [Citation157], probably through increasing energy availability for heat-treated cells.
Hyperthermia in cancer therapy
Drug resistance represents the major cause of treatment failure in human malignancies, and can be induced by different mechanisms, of which the pleiotrope ‘multidrug resistance’, mediated by the transmembrane glycoprotein p170 efflux pump, has gained particular interest. Pre-clinical data suggest that hyperthermia is a good candidate to overcome various modes of drug resistance [Citation158,Citation159], and this has been demonstrated in particular for the platinum-derived cisplatin [Citation160–162]. Excellent reviews on the interaction of hyperthermia with a wide range of anticancer agents in models employing cells in tissue culture or rodent tumours have been presented by Hahn [Citation163], Dahl [Citation164] and Urano [Citation165].
Local and regional hyperthermia improves treatment results when conjugated with radiotherapy and/or chemotherapy [Citation166,Citation167]. In the past, the most often performed treatment modality involving hyperthermia was its local or regional application with radiation. Now substantial clinical data exist, demonstrating for the treatment of locally advanced superficial tumours of different tissues the improved efficacy and relative lack of normal tissue toxicity of combined radiation and hyperthermia treatments as compared to radiation therapy alone [Citation166–169].
In cancer patients, many changes were found during and after the application of various hyperthermia modalities. These changes concern blood, nutrient and oxygen supply of the tumour, metabolic changes, signal transduction, immunology, as well as pharmacological effects [Citation170,Citation171]. In superficial tumours, an improved tumor control of the combined treatment was shown for recurrent or metastatic melanoma [Citation168] and recurrent breast cancer [Citation172].
It is well known that heat develops an independent cytotoxic effect on various cell types. In addition, hyperthermia acts in a synergic way when combined with radiation or cytostatic drugs at lower temperatures (40.5–44 °C) in vitro and in vivo [Citation173,Citation174]. Interestingly, some authors demonstrated that heat exposure enhanced radiosensitivity by depressing DNA-PK kinase activity during double strand break repair [Citation174]. Moreover, heat stress activates hypoxia-inducible factor 1 (HIF-1) through ERK-NADPH oxidase-mediated ROS production, and this enhances tumour oxygenation by up-regulating HIF-1 target genes involved in tumour perfusion/vascularisation and metabolism. These findings suggest that HIF-1 may play a beneficial role in radiosensitising tumours after HT [Citation38].
Conclusion
Heat stress is cytotoxic in both normal and cancer cells. It induces oxidative damage, disturbs the mitochondrial homeostasis and the cellular function knockdowns. Heat-induced ROS target proteins, lipids, polysaccharides and DNA, and increase the rate of cell killing. The mitochondrion is the main site of ROS production, and their main target. If the mitochondria antioxidant system fails to maintain the steady-state level of free radicals, the heat-induced oxidative damage alters the ETC, the ATP synthesis, the uncoupling respiration, as well as the structural conformation of enzymes, lipids and proteins. Since the heat shock response fails to repair damaged proteins and stop the oxidative damage propagation, it is not surprising that heat stress is usually associated with apoptosis and necrosis. Combined with chemo- and/or radiotherapy, hyperthermia increases the sensitivity of tumour tissues, disturbs their metabolism, enhances their oxygenation and ameliorates the rate of cellular mortality. However, studies are necessary to clarify the available thermal doses and drug combinations.
Acknowledgements
The authors thank Gillian Murphy, Emeritus Professor in the Department of Oncology at the University of Cambridge, for her assistance and help with English editing of this paper.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979;59:527–604
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 1994;91:10771–8
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;555:335–44
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47–95
- Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci 2000;899:191–208
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, et al. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem 1994;269:26066–75
- Kaur H, Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation: Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett 1994;350:9–12
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA 1988;85:6465–7
- LeDoux SP, Driggers WJ, Hollensworth BS, Wilson GL. Repair of alkylation and oxidative damage in mitochondrial DNA. Mutat Res 1999;434:149–59
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 1973;134:707–16
- Mujahid A, Pumford NR, Bottje W, Nakagawa K, Miyazawa T, Akiba M, et al. Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. J Poult Sci 2007;44:439–45
- Mujahid A, Sato K, Akiba Y, Toyomizu M. Acute heat stress stimulates mitochondrial superoxide production in broiler skeletal muscle, possibly via downregulation of uncoupling protein content. Poult Sci 2006;85:1259–65
- Brooks GA, Hittelman KJ, Faulkner JA, Beyer RE. Temperature, skeletal muscle mitochondrial functions, and oxygen debt. Am J Physiol 1971;220:1053–9
- Salo D, Donovan C, Davies K. Hsp70 and other possible heat shock or oxidative stress proteins are induced in skeletal muscle, heart, and liver during exercise. Free Radic Biol Med 1991;11:239–46
- Hall DM, Buettner GR, Matthes RD, Gisolfi CV. Hyperthermia stimulates nitric oxide formation: Electron paramagnetic resonance detection of .NO-heme in blood. J Appl Physiol 1994;77:548–53
- Messner KR, Imlay JA. The identification of primary sites of superoxide and hydrogen peroxide formation in the aerobic respiratory chain and sulfite reductase complex of Escherichia coli. J Biol Chem 1999;274:10119–28
- Davidson JF, Schiestl RH. Mitochondrial respiratory electron carriers are involved in oxidative stress during heat stress in Saccharomyces cerevisiae. Mol Cel Biol 2001;21:8483–9
- El-Orabi NF, Rogers C, Edwards HG, Schwartz DD. Heat-induced inhibition of superoxide dismutase and accumulation of reactive oxygen species leads to HT-22 neuronal cell death. J Thermal Biol 2011;36:49–56
- Freeman ML, Spitz DR, Meredith MJ. Does heat shock enhance oxidative stress? Studies with ferrous and ferric iron. Radiat Res 1990;124:288–93
- Agarwal A, Prabhakaran SA. Mechanism, measurement and prevention of oxidative stress in male reproductive physiology. Indian J Exp Biol 2005;43:963–74
- Powers RH, Stadnicka A, Kalbfleish JH, et al. Involvement of xanthine oxidase in oxidative stress and iron release during hyperthermic rat liver perfusion. Cancer Res 1992;52:1699–703
- Beckman JS, Koppenol WH. Nitric oxide superoxide and peroxynitrite: The good the bad and the ugly. Am J Physiol 1996;271:C1424–37
- Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynetrite reactions and formation in mitochondria. Free Radic Biol Med 2002;33:1451–64
- Kikusato M, Toyomizu M. Crucial role of membrane potential in heat stress-induced overproduction of reactive oxygen species in avian skeletal muscle mitochondria. PLoS One 2013;8:e64412
- Zhao QL, Fujiwara Y, Kondo T. Mechanism of cell death induced by nitroxide and hyperthermia. Free Radic Biol Med 2006;40:1131–43
- Flanagan SW, Moseley PL, Buttner GR. Increased flux of free radicals in cells subjected to hyperthermia: Detection by electron paramagnetic resonance spin trapping. FEBS Lett 1998;431:285–6
- Liochev SI, Fridovich I. Superoxide and iron: Partners in crime. IUBMB Life 1999;48:157–61
- Kirkinezos IG, Moraes T. Reactive oxygen species and mitochondrial diseases. Cell Dev Biol 2001;12:449–57
- Breton-Romero R, Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol 2014;2:529–34
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell 2007;26:1–14
- Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal 2008;10:1713–65
- Tatoyan A, Giulivi C. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J Biol Chem 1998;237:11044–8
- Chen CF, Wang D, Leu FJ, Chen HI. The protective role of nitric oxide and nitric oxide synthases in whole-body hyperthermia-induced hepatic injury in rats. Int J Hyperthermia 2012;28:421–30
- Matsumoto H, Hayashi S, Hatashita M, Ohnishi K, Ohtsubo T, Kitai R, et al. Nitric oxide is an initiator of intercellular signal transduction for stress response after hyperthermia in mutant p53 cells of human glioblastoma. Cancer Res 1999;59:3239–44
- Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schöpfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 1996;328:85–92
- Ischiropoulos H, al Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett 1995;364:279–82
- Segal AW, Abo A. The biochemical basis of the NADPH oxidase of phagocytes. Trends Biochem Sci 1993;18:43–7
- Moon EJ, Sonveaux P, Porporato PE, Danhier P, Gallez B, Batinic-Haberle I, et al. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc Natl Acad Sci 2010;107:20477–82
- Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamataet T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene 2008;27:4921–32
- Katschinski D, Boos K, Schindler S, Fandrey J. Pivotal role of reactive oxygen species as intracellular mediators of hyperthermia-induced apoptosis. J Biol Chem 2000;28:21094–8
- Mujahid A, Yoshiki Y, Akiba Y, Toyomizu M. Superoxide radical production in chicken skeletal muscle induced by acute heat stress. Poult Sci 2005;84:307–14
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem 1995;64:97–112
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutase (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem 2001;276:38388–93
- Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, et al. Dual function of the selenoprotein PHGPx during sperm maturation. Science 1999;285:1393–6
- Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 2000;351:183–93
- Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 2009;11:2685–700
- Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem 1991;266:22028–34
- Ham AJ, Liebler DC. Vitamin E oxidation in rat liver mitochondria. Biochem 1995;34:5754–61
- Omar RA, Yano S, Kikkawa Y. Antioxidant enzymes and survival of normal and simian virus 40-transformed mouse embryo cells after hyperthermia. Cancer Res 1987;47:3473–6
- Li JJ, Oberley LW. Overexpression of manganese-containing superoxide dismutase confers resistance to the cytotoxicity of tumor necrosis factor alpha and/or hyperthermia. Cancer Res 1997;57:1991–8
- Kuninaka S, Ichinose Y, Koja K, Toh Y. Suppression of manganese superoxide dismutase augments sensitivity to radiation, hyperthermia and doxorubicin in colon cancer cell lines by inducing apoptosis. Br J Cancer 2000;83(7):928–934
- Moriyama-Gonda N, Igawa M, Shiina H, Urakami S, Shigeno K, Terashima M. Modulation of heat-induced cell death in PC-3 prostate cancer cells by the antioxidant inhibitor diethyldithiocarbamate. BJU Int 2002;90:317–25
- Yang CY, Lin MT. Oxidative stress in rats with heatstroke-induced cerebral ischemia. Stroke 2002;33:790–94
- Morrison JP, Coleman MC, Aunan ES, Walsh SA, Spitz DR, Kregel KC. Aging reduces responsiveness to BSO- and heat stress-induced perturbations of glutathione and antioxidant enzymes. Am J Physiol Regul Integr Comp Physiol 2005;289:R1035–41
- Lushchk VI, Bagnyukova TV. Temperature increase results in oxidative stress in goldfish tissues. 2. Antioxidant and associated enzymes. Comp Biochem Physiol C Toxicol Pharmacol 2006;143:36–41
- Mitchell JB, Russo A, Kinsellaa TJ, Glatstein E. Glutathione elevation during thermotolerance induction and thermosensitization by glutathione depletion. Cancer Res 1983;43:987–91
- Mitchell JB, Russo A. Thiols, thiol depletion, and thermosensitivity. Radiation Res 1983;95:471–85
- Russo A, Mitchell JB, McPherson S. The effects of glutathione depletion on thermotolerance and heat stress protein synthesis. Br J Cancer 1984;49:753–8
- Sreedhar AS, Pardhasaradhi BVV, Khar A, Srinivas UK. A cross talk between cellular signaling and cellular redox state during heat-induced apoptosis in a rat histiocytoma. Free Radic Biol Med 2002;32:221–7
- Shrieve DC, Li GC, Astromoff A, Harris JW. Cellular glutathione, thermal sensitivity, and thermotolerance in Chinese hamster fibroblasts and their heat-resistant variants. Cancer Res 1986;46:1684–7
- Macho A, Harsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, et al. Glutathione depletion is an early and calcium elevation is a late event thymocyte apoptosis. J Immunol 1997;158:4612–19
- Pitkanen S, Robinson BH. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest 1996;98:345–51
- Luo X, Pitkanen S, Kassovska-Bratinova S, Robinson BH, Lehotay DC. Excessive formation of hydroxyl radicals and aldehydic lipid peroxidation products in cultured skin fibroblasts from patients with complex I deficiency. J Clin Invest 1997;99:2877–82
- Robinson BH. Diagnosis of mitochondrial energy metabolism defects in tissue culture. Induction of MnSOD and Bcl-2 in mitochondria from patients with complex I (NADH-Coq reductase) deficiency. Biofactors 1998;7:229–30
- Wang Z, Cai F, Chen X, Luo M, Hu L, Lu Y. The role of mitochondria derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS One 2013;8:e75044
- Mitchell P. Vectoriel chemiosmotic processes. Annu Rev Biochem 1977;46:996–1005
- Noji H, Yoshida M. The rotary machine in the cell ATP synthase. J Biol Chem 2001;276:1665–8
- Voinikov VK, Rudikovsky AV, Pobezhimova TP, Varakina NN. The effect of heat shock proteins on maize mitochondria activity. Plant Physiol (Life Sci Adv) 1989;8:1–4
- Chou M, Chen Y-M, Lin C-Y. Thermotolerance of isolated mitochondria associated with heat shock proteins. Plant Physiol 1989;89:617–21
- Monti E, Supino R, Colleoni M, Costa B, Ravizza R, Gariboldi MB. Nitroxide TEMPOL impairs mitochondrial function and induces apoptosis in HL60 cells. J Cell Biochem 2001;82:271–6
- Downs CA, Heckathorn SA. The mitochondrial small heat shock protein protects NADH: Ubiquinone oxidoreductase of the electron transport chain during heat stress in plants. FEBS Lett 1998;430:246–50
- Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, et al. Specific modification of mitochondrial protein thiols in response to oxidative stress: A proteomics apporoach. J Biol Chem 2002;277:17048–56
- England K, O’Driscoll C, Cotter TG. Carbonylation of glycolytic proteins is a key response to drug-induced oxidative stress and apoptosis. Cell Death Differ 2004;11:252–60
- Colussi C, Albertini MC, Coppola S, Rovidati S, Galli F, Ghibelli L. H2O2-induced block of glycolysis as an active ADP-ribosylation reaction protecting cells from apoptosis. FASEB J 2000;14:2266–76
- Troyano A, Sancho P, Fernandez C, de Blas E, Bernardi P, Aller P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ 2003;10:889–98
- Pobezhimova T, Voinikov V, Varakina N. Inactivation of complex I of the respiratory chain of maize mitochondria incubated in vitro by elevated temperature. J Therm Biol 1996;5:283–8
- Rustin P, Lance C. Malate metabolism in leaf mitochondria from the crassulacean acid metabolism plant Kalanchoë blossfeldiana pollen. Plant Physiol 1986;81:1039–43
- Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J 1972;128:617–30
- Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res 1998;39:1529–42
- Rhoads DM, Umbach AL, Subbaiah CC, Siedow JN. Mitochondrial reactive oxygen species. Contribution to oxidative stress and interorganellar signaling. Plant Physiol 2006;141:357–66
- Valko M, Rhodes CJ, Moncol J, Izakivic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006;160:1–40
- Pinchuk I, Schnitzer E, Lichtenberg D. Kinetic analysis of copper-induced peroxidation of LDL. Biochem Biophys Acta 1998;1389:155–72
- Trachootham D, Lu W, Ogasawara MA, Valle NR, Huang P. Redox regulation of cell survival. Antioxid Redox Signal 2008;10:1343–74
- Yokoyama Y, Beckman JS, Beckman TK, Wheat JK, Cash TG, Freeman BA, et al. Circulating xanthine oxidase: Potential mediator of ischemic injury. Am J Physiol 1990;258:G564–70
- Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med 2003;24:149–59
- Taylor NL, Heazlewood JL, Day DA, Millar AH. Differential impact of environmental stresses on the pea mitochondrial proteome. Mol Cell Proteomics 2005;4:1122–33
- Winger AM, Millar AH, Day DA. Sensitivity of plant mitochondrial terminal oxidases to the lipid peroxidation product 4-hydroxy-2-nonenal (HNE). Biochem J 2005;387:865–70
- Sakamoto A, Tsukamoto S, Yamamoto H, Udea-Hashimoto M, Takahashi M, Suzuki H, et al. Functional complementation in yeast reveals a protective role of chloroplast 2-Cys peroxiredoxin against reactive nitrogen species. Plant J 2003;33:841–51
- Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J 1997;324:1–18
- Verniquet F, Gaillard J, Neuburger M, Douce R. Rapid inactivation of plant aconitase by hydrogen peroxide. Biochem J 1991;276:643–8
- Flint DH, Tuminello JF, Emptage MH. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem 1993;268:22369–76
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease and oxidative stress. J Biol Chem 1997;272:20313–16
- Stadtman ER. Role of oxidant species in aging. Curr Med Chem 2004;11:1105–12
- Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med 2001;30:463–88
- Poppek D, Grune T. Proteosomal defense of oxidative protein modifications. Antioxid Redox Signal 2006;8:173–84
- Melegh B, Bock I, Gàti I, Méhes K. Multiple mitochondrial DNA deletions and persistent hyperthermia in a patient with Brachmann–de Lange phenotype. Am J Med Genet 1996;65:82–8
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 1997;94:514–19
- Eppink B, Krawczyk PM, Stap J, Kannar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int J Hyperthermia 2012;28:509–17
- Palmieri F. Mitochondrial carrier proteins. FEBS Lett 1994;346:48–54
- Himms-Hagen J. Brown adipose tissue metabolism and thermogenesis. Annu Rev Nutr 1985;5:69–94
- Champigny O, Ricquier D. Effects of fasting and refeeding on the level of uncoupling proteins mRNA in rat brown adipose tissue, evidence for diet-induced and cold- induced responses. J Nutr 1990;120:1730–36
- Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 1997;387:90–94
- Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, et al. Uncoupling protein-2, a novel gene linked to obesity and hyperinsulinemia. Nat Genet 1997;15:269–72
- Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, Seydou J, et al. Uncoupling protein-3, a new member of the mitochondrial carrier family with tissue specific expression. FEBS Lett 1997;408:39–42
- Acin A, Rodriguez M, Rique H, Canet E, Boutin JA, Galizzi JP. Cloning and characterization of the 59 flanking region of the human uncoupling protein-3 (UCP3) gene. Biochem Biophys Res Commun 1999;258:278–83
- Adams SA. Uncoupling protein homologs: Emerging views of physiological function. J Nutr 2000;130:711–14
- Boss O, Hagen T, Lowell BB. Uncoupling proteins 2 and 3, potential regulators of mitochondrial energy metabolism. Diabetes 2000;49:143–56
- Dullo AG, Samec S. Uncoupling proteins, their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br J Nutr 2001;86:123–39
- Criscuolo F, Gonzales-Barroso MM, Maho YL, Ricquier D, Bouillaud F. Avian uncoupling protein expressed in yeast mitochondria prevents endogenous free radical damage. Proc Biol Sci 2005;272:803–10
- Casteilla L, Rigoulet M, Penicaud L. Mitochondrial ROS metabolism: Modulation by uncoupling proteins. IUBMB Life 2001;52:181–8
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otn M, et al. A signaling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J 2003;22:4103–10
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004;305:626–9
- Callahan TE, Marins J, Welch WJ, Horn JK. Heat shock attenuates oxidation and accelerates apoptosis in human neutrophils. J Surg Res 1998;85:317–22
- Du J, Di HS, Guo L, Li ZH, Wang GL. Hyperthermia causes bovine mammary epithelial cell death by a mitochondrial-induced pathway. J Therm Biol 2007;33:37–47
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 2000;345:271–8
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic Bax and Bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–30
- Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol 2001;152:237–50
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341:233–49
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 2001;276:12030–34
- Rizzuti R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998;280:1763–6
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. Bax and Bak regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003;300:135–9
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol Cell Biol 2003;4:552–65
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem 1997;272:3346–54
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: A new paradigm of pore structure and function. FEBS Lett 2002;512:1–7
- Adrain C, Creagh ME, Martin SJ. Apoptosis associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J 2001;20:6627–36
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–18
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000;6:513–19
- Ott M, Robertson JD, Gogvadeze V, Zhivotovsky B, Orrenus S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 2002;99:1259–63
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 2001;21:4752–60
- Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol 1998;143:1353–60
- Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, et al. Bcl-2 inhibition of neuronal death: Decreased generation of reactive oxygen species. Science 1993;262:1274–77
- Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, et al. Retardation of chemical hypoxia induced necrotic cell death by Bcl-2 and ICE inhibitors: Possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene 1996;12:2045–50
- Morimoto RI, Tissieres A, Georgopoulos C. Stress Proteins in Biology and Medicine. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990
- Ahmed K, Furusawa Y, Tabuchi Y, Emam HF, Piao JL, Hassan MA, et al. Chemical inducers of heat shock proteins derived from medicinal plants and cytoprotective genes response. Int J Hyperthermia 2012;28:1–8
- Gupta M, Kumar S, Dangi SS, Jangir BL. Physiological, biochemical and molecular responses to thermal stress in goats. Int J Livestock Res 2013;3:27–38
- Sreedhar AS, Pardhasaradhi BVV, Khar A, Srinivas UK. Heat induced expression of CD95 and its correlation with the activation of apoptosis upon heat shock in rat histiocytic tumor cells. FEBS Lett 2000;472:271–5
- Botzler C, Schmidt J, Luz A, Jennen L, Issels R, Multhoff G. Differential Hsp70 plasma-membrane expression on primary human tumors and metastases in mice with severe combined immunodeficiency. Int J Cancer 1998;77:942–8
- Roigas J, Wallwn ES, Loenings SA. Heat shock protein (Hsp72) surface expression enhances the lyses of a human renal cell carcinoma by IL-2 stimulated NK-cells. Adv Exp Med Biol 1998;451:225–9
- Nadin SB, Cuello-Carrión FD, Sottile ML, Ciocca DR, Vargas-Roig LM. Effects of hyperthermia on Hsp27 (HSPB1), Hsp72 (HSPA1A) and DNA repair proteins hMLH1 and hMSH2 in human colorectal cancer hMLH1-deficient and hMLH1-proficient cell lines. Int J Hyperthermia 2012;28:191–201
- Srinivas UK, Swamynathan SK. Role of heat shock transcription factors in stress response and during development. J Biosci 1996;21:103–21
- Deckers R, Debeissat C, Fortin PY, Moonen CTW, Couillaud F. Arrhenius analysis of the relationship between hyperthermia and Hsp70 promoter activation: A comparison between ex vivo and in vivo data. Int J Hyperthermia 2012;28:441–50
- He L, Lemasters JJ. Heat shock suppresses the permeability transition in rat liver mitochondria. J Biol Chem 2003;278:16755–60
- Samali A, Robertson JD, Peterson E, Manero F, Van Zeijil L, Paul C, et al. Hsp27 protects mitochondria of thermotolerant cells against apoptotic stimuli. Cell Stress Chaperones 2001;6:49–58
- Klein SD, Brune B. Heat shock protein 70 attenuates nitric oxide-induced apoptosis in RAW macrophages by preventing cytochrome c release. Biochem J 2002;362:635–41
- Sancho P, Troyano A, Fernandez C, De Blas E, Aller P. Differential effects of catalase on apoptosis induction in human promonocytic cells. Relationship with heat shock protein expression. Mol Pharmacol 2003;63:581–9
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000;2:469–75
- Saleh A, Srinivasula SM, Balkhir L, Robbins PD, Alnemri ES. Negative regulation of Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2000;2:476–83
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2000;2:645–52
- Halliwell B. Antioxidants: The basics – What they are and how to evaluate them. Adv Pharmacol 1997;38:3–20
- Kapiszewska M, Hopwood LE. Mechanisms of membrane damage for CHO cells heated in suspension. J Cancer Res Clin Oncol 1988;114:23–9
- Malayer JR, Pollard JW, Hansen PJ. Modulation of thermal killing of bovine lymphocytes and preimplantation of mouse embryos by alanine and taurine. Am J Vet Res 1992;53:689
- Ealy AD, Drost M, Barros CM, Hansen PJ. Thermoprotection of preimplantation bovine embryos from heat shock by glutathione and taurine. Cell Biol Int Rep 1992;16:125–31
- Kamwanja LA, Chase CC, Gutierrez JA, Guerriero V, Olson TA, Hammond AC, Hansen PJ. Responses of bovine lymphocytes to heat shock as modified by breed and antioxidant status. J Animal Sci 1994;72:438–44
- Kim JH, Kim SH, Hahn EW, Song CW. 5-Thio-o-glucose selectively potentiates hyperthermic killing of hypoxic tumor cells. Science 1978;200:206–7
- Kapp DS, Hahn GM. Thermosensitization by sulfhydryl compounds of exponentially growing Chinese hamster cells. Cancer Res 1979;39:4630–5
- Lord-Fontaine S, Averill-Bates DA. Heat shock inactivates cellular antioxidant defenses against hydrogen peroxide: Protection by glucose. Free Radical Biol Med 2002;32:752–65
- Henle KJ, Warters RL. Heat protection by glycerol in vitro. Cancer Res 1982;42:2171–6
- Yarema RR, Ohorchak A, Zubarev GP, Mylyan YP, Oliynyk YY, Zubarev MG, et al. Hyperthermic intraperitoneal chemoperfusion in combined treatment of locally advanced and disseminated gastric cancer: Results of a single-centre retrospective study. Int J Hyperthermia 2014;30:159–65
- Coss RA, Storck CW, Wells TC, Kulp KK, Wahl M, Leeper DB. Thermal sensitisation by lonidamine of human melanoma cells grown at low extracellular pH. Int J Hyperthermia 2014;30:75–8
- Hettinga JV, Konings AW, Kampinga HH. Reduction of cellular cisplatin resistance by hyperthermia – A review. Int J Hyperthermia 1997;13:439–57
- Hildebrandt B, Loeffel J, Deja M, Kerner T, Rick O, Bechstein W, et al. Whole body hyperthermia induces a renewed remission in a patient with refractory germ cell tumor after high-dose chemotherapy. Ann Hematol 1998;77:S222
- Petryk AA, Giustini AJ, Gottesman RE, Kaufman PA, Hoopes PJ. Magnetic nanoparticle hyperthermia enhancement of cisplatin chemotherapy cancer treatment. Int J Hyperthermia 2013;29:845–51
- Hahn GM. Hyperthermia and Cancer. New York: Plenum Press; 1982
- Dahl O. Mechanisms of thermal enhancement of chemotherapeutic cytotoxicity. In: Urano M, Douple E, editors. Hyperthermia and Oncology. Volume 4. Utrecht: VSP; 1994. pp 9–28
- Urano M, Douple E. Hyperthermia and Oncology. Volume 4. Utrecht: VSP; 1994
- Wust P, Hildebrant B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol 2002;3:487–97
- Viglianti BL, Stauffer P, Repasky E, Jones E, Vujaskovic Z, Dewhirst M. Hyperthermia. In: Hong WK, Bast RC, Hait WN, Kufe DW, Pollok RE, Weichselbaum RR, et al, editors. Cancer Medicine, 8th ed. Beijing, China: People’s Medical Publishing House; 2010. pp 528–40
- Overgaard J, Gonzalez Gonzalez D, Hulshof MCCM, Arcangeli G, Dahl O, Mella O, et al. Randomised trial of hyperthermia as adjuvant to radiotherapy for recurrent or metastatic malignant melanoma. Lancet 1995;345:540–3
- Franckena M. Review of radiotherapy and hyperthermia in primary cervical cancer. Int J Hyperthermia 2012;28:543–8
- Dayanc BE, Bansal S, Gure AO, Gollnick SO, Repasky EA. Enhanced sensitivity of colon tumor cells to natural killer cell cytotoxicity after mild thermal stress is regulated through HSF1-mediated expression of MICA. Int J Hyperthermia 2013;29:480–90
- Csoboz B, Balogh GE, Kusz E, Gombos I, Peter M, Crul T, et al. Membrane fluidity matters: Hyperthermia from the aspects of lipids and membranes. Int J Hyperthermia 2013;29:491–9
- Vernon CC, van der Zee J, Liu FF. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: Results from five randomized controlled trials. Int J Radiat Oncol Biol Phys 1996;35:731–44
- Schildkopf P, Ott OJ, Frey B, Wadepohl M, Sauer R, Fietkau R, et al. Biological rationales and clinical applications of temperature controlled hyperthermia – Implications for multimodal cancer treatments. Curr Med Chem 2010;17:3045–57
- Ihara M, Takeshita S, Okaichi K, Okumura Y, Ohnishi T. Heat exposure enhances radiosensitivity by depressing DNA-PK kinase activity during double strand break repair. Int J Hyperthermia 2014;30:102–9