
Abstract
We report a novel mutation at codon 24 of the α2-globin gene (HBA2: c.75T > A) found in a Sundanese family. This novel mutation was detected during prenatal diagnosis. The couple already had a 7-year-old boy who exhibited clinically severe α-thalassemia intermedia (α-TI), and he was found to be a compound heterozygote for the novel mutation at codon 24 and the previously described Hb Adana (HBA2: c.179G > A) at codon 59 of the α2-globin gene. The father was a carrier of the novel point mutation and showed normal hemoglobin (Hb) and a low mean corpuscular volume (MCV) and mean corpuscular Hb (MCH) value.
α-Thalassemia (α-thal) is an autosomal recessive disorder, characterized by microcytic hypochromic red blood cells with eventual mild anemia in the healthy carrier (Citation1). The defect is mostly caused by large α-globin gene deletions that abolish the expression of one or more α-globin genes, causing mild conditions such as α+ heterozygosity (–α/αα), homozygosity (–α/–α) or α0 heterozygosity (– –/αα). More severe conditions are Hb H disease (–α/– –) and Hb Bart’s hydrops fetalis (– –/– –), the latter being lethal during fetal life because no α-globin genes are expressed (Citation2).
Although nondeletional α-thal is less frequent compared to the large deletion types, it is commonly manifested as more severe phenotypes because the mutations are mostly associated with protein stability (Citation3). The severity is also determined by the localization of the mutation on the α-globin gene (α2 or α1). As previously reported by Liebhaber et al. (Citation4), the α2-globin gene is predominantly expressed, 2- to 3-fold higher than the α1-globin gene. Recently, a mild thalassemia syndrome due to compound heterozygosity for the codon 24 (HBA2: c.75T > G) of the α2-globin gene and a single α-globin gene deletion were described in a Surinamase patient (Citation5). Here we report a different mutation at the same codon 24 (HBA2: c.75T > A).
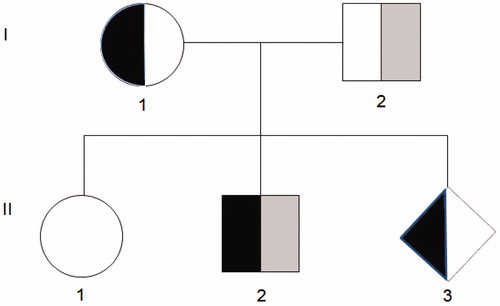
A Sundanese Indonesian couple was referred to our clinic (Yayasan GenNeka, Eijkman Institute for Molecular Biology, Jakarta, Indonesia) for prenatal diagnosis because their second child (II-2) has been affected with severe α-thal intermedia (α-TI) and has been receiving blood transfusions regularly every 4-8 weeks since he was 3 years old. The first child (II-1) has not been studied yet, but no clinical complaints have been reported. The pedigree of the family is depicted in .
Figure 1. Pedigree of the family. The codon 59 (HBA2: c.179G > A) mutation was found in the mother (I-1), the second child (II-2) and the fetus (II-3), whereas the novel codon 24 (HBA2: c.75T > A) mutation was carried by the father (I-2) and in compound heterozygosity by the second child (II-2).
Hematology profiles of the couples were consistent with α-thal carriers. DNA analysis for the parents was carried out using routine multiplex polymerase chain reaction (m-PCR) to detect the common deletions found in Indonesia: Southeast Asian (– –SEA/), Thailand (– –THAI/), Filipino (– –FIL/) (Citation6), the 3.7 kb (rightward) (–α3.7/) and 4.2 kb (leftward) (–α4.2/) (Citation7). None of the common deletions were detected; therefore, DNA analysis was performed using the PCR-RFLP (restriction fragment length polymorphism) method (Citation8) to detect the codon 59 (HBA2: c.179G > A) of the α2-globin gene and Hb Constant Spring (Hb CS or HBA2: c.427T > C) (Citation9), the two most common point mutations found in the Indonesian population. The mother was detected to be a carrier of codon 59, while the father was normal for both point mutations. DNA analysis of the father was continued using the multiplex ligation-dependent probe amplification (MLPA) technique (P140 HBA; MRC Holland, Amsterdam, The Netherlands) to detect deletion or duplication of the α-globin gene cluster [including the hypersensitive-40 (HS-40) region until the θ-globin gene], but no abnormalities were found. Then both α-globin genes were amplified by PCR and analyzed by direct sequencing (Citation10,Citation11) using the BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on the ABI PRISM™ 3130 Genetic Analyzer (Applied Biosystems). Sequencing results revealed that the father was heterozygous for the novel mutation at codon 24 of the α2-globin gene (). The clinical and molecular data are summarized in .
Figure 2. Sequencing result of the α2-globin gene in the father showing heterozygosity for codon 24 (HBA2: c.75T > A).
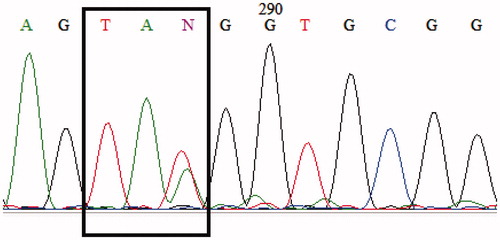
Table 1. Summary of the Hematological Profile and Molecular Findings of the Family.
Amniocentesis was performed for prenatal diagnosis and DNA was extracted from the amniotic fluid. A 2 mL amount out of 15 mL of the amniotic fluid was cultured using the amniomax C-100 GIBCO (Grand Island, NY, USA) medium for backup and confirmation. Apolipoprotein B (APO-B) and D1S80 variable number tandem repeat (VNTR) analyses were carried out to exclude maternal contamination as previously described (Citation8). Sequencing analysis of the fetus’ DNA detected the codon 59 mutation but not the codon 24 mutation. DNA analysis using DNA extracted from cultured cells showed the same result.
The affected child (II-2) has been suffering severe anemia since he was 3 years old (Hb 4.0 g/dL), and required regular blood transfusions to maintain the steady state Hb at 6.6-8.4 g/dL. However, his Hb level could decrease to as low as 4.5 g/dL during infections. During the first year, he received regular blood transfusions every 8 weeks (3 years old), after which the frequency of blood tranfusions was increased to every 4 weeks. Iron chelation was initiated after he had received five tranfusions when the serum ferritin level reached 1059.6 μg/L. A physical examination at 7 years old revealed splenomegaly (schuffner II) and liver enlargement (3 cm). Sequencing results using forward and reverse primers revealed that this affected child was a compound heterozygote for the codon 24 and codon 59 mutations, both on the α2-globin gene (results not shown).
We believe that the severe phenotype of this genetic compound heterozygosity can be explained by the increased hemolysis caused by the enhanced semi-hemizygous expression of the unstable Hb Adana due to the presence of the codon 24 defect in trans. Hb Adana is an unstable variant because the small and non polar glycine residue at α59 is internal and makes close spatial contact with the glycine residue at position α25 of the β helix replaced by the charged and larger aspartic acid residue. As previously described by Cürük et al. (Citation12), the Gly→Asp replacement affects the stability of tertiary structure of the α subunit. Some cases of compound heterozygotes for Hb Adana and a deletional form of thalassemia mutation have been reported. Coinheritance of Hb Adana on the α2-globin gene and a two-gene deletion results in Hb H-like hydrops fetalis (Citation13,Citation14), whereas a similar case with Hb Adana on the α1-globin gene was reported as less severe in clinical manifestation (Citation12,Citation15). Compound heterozygotes for Hb Adana on the α2-globin gene and a one-globin gene deletion manifested as varied phenotypes (Citation9,Citation16), while the cases with a combination of Hb Adana and a nondeletional α-globin gene mutation is quite rare. However, in our experience, cases with this combination manifested quite severely compared to those cases with Hb Adana and deletional mutations (Citation9). Hb Adana on the α2-globin gene is relatively frequent in Indonesia (Citation7) with a frequency of about 16.0% in α-thal patient groups (Citation8).
The phenotype of the compound heterozygote for this novel codon 24 mutation and Hb Adana is not easy to predict. This new mutation located in exon 1 of the α2-globin gene results in a premature termination, which generated short mRNA that should be eliminated via nonsense-mediated decay (NMD) (Citation17). The codon 24 mutation seems to be a mild α-thal defect that was confirmed by the hematological profile of the father similar to those observed in one α-globin gene deletion carriers (–α/αα). Therefore the phenotype should be similar to those in cases with compound heterozygous Hb Adana and one α-globin gene deletion (Citation9,Citation16). Although the phenotype of compound heterozygotes for Hb Adana on the α2-globin gene and one α-globin gene deletion varied, most are mild (Citation9). A severe phenotype present in the patient might be due to the activation of a cryptic splice site at the novel codon 24 mutation that may generate an abnormal α-globin chain. Further study is required to address this hypothesis.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper.
Acknowledgements
The authors thank the patients who were willing to be volunteers for this study, as well as Mewahyu Dewi (M. Biomed), Dr. Maria Dewi Indrawati and Dr. Widya Tirtha Kamandalu (The Eijkman Institute for Molecular Biology, Jakarta, Indonesia) for their helpful advice and encouragement during the preparation of this manuscript. The authors also sincerely thank Jan P. Schouten (MRC Holland) for providing the MLPA Kit and Al Harahap (Graduate Associate Teacher, Writing Program, University of Arizona, USA) for valuable comments, suggestions and proofreading the manuscript. Our thanks also to Dr. Baba Inusa [MRCP, FRCPCH, DCP (Haem), FMCPaed] (Evelina Children’s Hospital, Guy’s and St. Thomas’ National Health Service Foundation Trust, London, UK) for further suggestions and proofreading of the manuscript.
References
- Weatherall DJ, Clegg JB. The Thalassaemia Syndromes, 4th ed. Oxford: Blackwell Science, 2001
- Weatherall DJ. The molecular basis for phenotypic variability of the common thalasssemias. Mol Med Today. 1995;1(1):15–20
- Harteveld CL, Higgs DR. Review α-thalassaemia. Orphanet J Rare Dis. 2010;5:13
- Liebhaber SA, Cash FE, Ballas SK. Human α-globin gene expression: The dominant role of the α2-locus in mRNA and protein synthesis. J Biol Chem. 1986;261(32):15327–15333
- Giordano PC, Cnossen MH, Joosten AMS, et al. Codon 24 (TAT>TAG) and codon 32 (ATG>AGG) (Hb Rotterdam): two novel α2 gene mutations associated with mild α-thalassemia found in the same family after newborn screening. Hemoglobin. 2010;34(4):354–365
- Liu YT, Old JM, Miles K, et al. Rapid detection of α-thalassaemia deletions and α-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108(2):295–299
- Setianingsih I, Harahap A, Nainggolan IM. Thalassemia in Indonesia: phenotypes and molecular defects. Adv Exp Med Biol. 2003;531:47–56
- Nainggolan IM, Harahap A, Setianingsih I. Hydrops fetalis associated with homozygosity for Hb Adana [α59(E8)Gly→Asp (α2)]. Hemoglobin. 2010;34(4):394–401
- Nainggolan IM, Harahap A, Ambarwati DD, et al. Interaction of Hb Adana (HBA2: c.179G>A) with deletional and nondeletional α+-thalassemia mutations: diverse hematological and clinical features. Hemoglobin. 2013;37(3):297–305
- Dode C, Rochette J, Krishnamoorthy R. Locus assignment of human α globin mutations by selective amplification and direct sequencing. Br J Haematol. 1990;76(2):275–281
- Molchanova TP, Pobedimskaya DD, Postnikov YuV. A simplified procedure for sequencing amplified DNA containing the α2- or α1-globin gene. Hemoglobin. 1994;18(3):251–255
- Çürük MA, Dimovski AJ, Baysal E, et al. Hb Adana or α259(E8)Gly→Aspβ2, a severely unstable α1-globin variant, observed in combination with the –(α)20.5 kb α-thal-1 deletion in two Turkish patients. Am J Hematol. 1993;44(4):270–275
- Chan V, Chan VW, Tang M, et al. Molecular defects in Hb H hydrops fetalis. Br J Haematol. 1997;96(2):224–228
- Henderson S, Pitman M, McCarthy J, et al. Molecular prenatal diagnosis of Hb H hydrops fetalis caused by Haemoglobin Adana and the implications to antenatal screening for α-thalassaemia. Prenat Diagn. 2008;28(9):859–861
- Durmaz AA, Akin H, Ekmekci AY, et al. A severe α-thalassemia case compound heterozygous for Hb Adana in α1 gene and 20.5 kb double gene deletion. J Pediatr Hematol Oncol. 2009;31(8):592–594
- Douna V, Papassotiriou I, Garoufi A, et al. A rare thalassemic syndrome caused by interaction of Hb Adana [α59(E8)Gly→Asp] with an α+thalassemia deletion: clinical aspects in two cases. Hemoglobin. 2008;32(4):361–369
- Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14(10):1074–1081