Abstract
Inflammation plays a key role in the pathogenesis of atherosclerosis. The more we discover about the molecular pathways involved in atherosclerosis, the more we perceive the importance of monocytes in this process. Circulating monocytes are components of innate immunity, and many pro-inflammatory cytokines and adhesion molecules facilitate their adhesion and migration to the vascular endothelial wall. In addition to the accumulation of lipids and formation of atherogenic ‘foam’ cells, monocytes may promote atherosclerotic plaque growth by production of inflammatory cytokines, matrix metalloproteinases, and reactive oxidative species. However, the contribution of monocytes to atherogenesis is not only limited to tissue destruction. Monocyte subsets are also involved in intraplaque angiogenesis and tissue reparative processes.
The aim of this overview is to discuss the mechanisms of monocyte activation, the pivotal role and importance of activated monocytes in atherosclerotic coronary artery disease, their implication in the development of acute coronary events, and their potential in cardiovascular reparative processes such angiogenesis.
Key words::
| Abbreviations | ||
| ACS | = | acute coronary syndromes |
| GM-CSF | = | granulocyte–macrophage colony-stimulating factor |
| HSP | = | heat shock protein |
| IκB | = | inhibitor of κB |
| ICAM-1 | = | intercellular adhesion molecule-1 |
| IL | = | interleukin |
| LDL | = | low density lipoprotein |
| LOX-1 | = | lectin-like oxidized LDL receptor-1 |
| MCP-1 | = | monocyte chemoattractant protein-1 |
| M-CSF | = | macrophage colony-stimulating factor |
| NFκB | = | nuclear factor κB |
| ROS | = | reactive oxygen species |
| SR | = | scavenger receptor |
| TLR | = | Toll-like receptor |
| TNFα | = | tumour necrosis factor α |
| VCAM-1 | = | vascular cell adhesion molecule-1 |
| VEGF | = | vascular endothelial growth factor |
Key messages
Progression of atherosclerotic plaques ultimately depends on the persistent process of excessive lipid accumulation by monocyte-derived ‘foam’ cells leading to their death and repeated lipid accumulation.
Monocyte Toll-like receptors interact with a variety of ligands from bacteria, fungi, and viruses, but the host itself may be the origin of the ligands that bind the Toll-like receptors.
Introduction
In recent decades, much research has been directed towards understanding the pathogenesis of atherosclerosis and inflammation. Fatty streaks, which may be seen even in intrauterine human life and represent the earliest stage of an atherosclerotic lesion, are characterized by dense accumulation of monocyte-derived macrophages in the vascular wall, a phenomenon which is seen in every stage of atherosclerosis (Citation1–4).
Systemic inflammation, as reflected by high levels of pro-inflammatory cytokines and adhesion molecules, actively promotes the mobilization of monocytes to the sites of atherosclerotic lesions (Citation3). Moreover, monocytes themselves may be considered pro-inflammatory cells, producing a wide range of cytokines, chemokines, and reactive oxidative species. However, the role of monocytes in atherogenesis goes far beyond providing a source of lipid-loaded ‘foam’ cells and includes their participation in plaque progression, destabilization, rupture, and thrombus formation (Citation4,Citation5). Contribution of monocytes to the atherosclerotic process also comprises angiogenesis and tissue repair, and, indeed, certain monocyte subsets possess distinct angiogenic properties and constitute a substantial proportion of so-called endothelial progenitor cells (Citation5).
In this overview, we aim to analyse the mechanisms implicated in monocyte-related processes of the atherosclerotic plaque formation (i.e. atherogenesis). The role of monocytes in plaque destabilization and the development of acute coronary syndromes has been discussed in a recent review (Citation6).
Monocyte activation and inflammation
The primary function of circulating monocytes is non-specific host protection against foreign pathogens via their prompt elimination (innate immunity). This system is not antigen-specific and depends on pattern recognition receptors such as Toll-like receptors (TLR), CD14, scavenger receptors, and other receptor systems (Citation7). Monocytes/macrophages can phagocytize pathogens but they also promote pathogen neutralization and elimination by the production of numerous cytokines (Citation8). The ‘dark side’ of the process is the inevitable damage of the host's own tissues. In this respect the vascular wall is especially vulnerable, being located in direct proximity to the circulation.
Monocytes possess receptors that can detect highly conserved components (e.g. membrane constituents such as lipopolysaccharides) of various pathogens. These receptors include the prototypic pattern recognition receptor CD14 and a number of receptor families implicated in innate immunity including the TLRs, nucleotide oligomerization domain-like receptors, retinoic acid-inducible gene I-like receptors, C-type lectins, DNA-dependent activator of interferon-regulatory factors, and the recently identified HIN200 receptor AIM-2 (absent in melanoma-2). Each receptor family possesses unique roles following ligation with specific ligand(s), though there are still common pathways mediating receptor-stimulating effects and a synergy between receptors from different families (Citation9).
The TLRs are the most widely studied receptor system responsible for monocyte inflammatory responses. Currently, nine TLRs (TLR1–TLR9) have been characterized in humans. Most of these receptors (e.g. TLR4) are located on the surface of monocytes (and other cells) to detect bacterial components in the circulation, whilst TLR3 and TLR5 are intracellular receptors able to respond to virus DNA/RNA (Citation10). TLRs are highly conserved from Drosophila to humans and even share structural and functional similarities. TLR-mediated responses may be modulated by interactions so that different groups of pattern recognition receptors may augment each other's functions (Citation11–14). For example, TLR4, a receptor closely related with the atherosclerotic process, is functionally linked to lipopolysaccharide receptor CD14, and mutations in TLR4 gene have been associated with differences in responsiveness to lipopolysaccharides (Citation15).
TLRs have been repeatedly implicated in pathogenesis of atherosclerosis. For example, TLR4 is involved in both early and late phases of the atherosclerotic process, and a prominent reduction of atherosclerosis has been observed in TLR4-deficient mice (Citation16). TLR4 expression is increased on circulating monocytes and at sites of atherosclerotic lesions of patients with acute coronary syndromes (ACS) compared to stable patients’ angina (Citation17). Indeed, stimulation of TLR4 leads to the production of matrix metalloproteinases by monocytes/macrophages, resulting in degradation of the extracellular matrix and the promotion of plaque destabilization. An increased TLR2 expression on circulating monocytes has also been described in patients with infections and also in those with coronary artery disease (CAD) (Citation18–20). In fact, high TLR2 expression on monocytes is an independent risk factor for atherogenesis (Citation21).
How may TLRs be implicated in the chronic process of atherogenesis? Monocyte TLRs interact with a variety of ligands from bacteria, fungi, and viruses. However, the host itself may be the origin of the ligands that bind the TLRs.
Several of these receptors are not exclusively specific to bacterial pathogens and can be stimulated by molecules expressed during cardiovascular damage, thus causing activation of monocytes. TLR2 can be activated by heat shock protein (HSP) 70 and hyaluronan, TLR4 by HSP70, HSP60, oligosaccharides of hyaluronic acid, fibrinogen, heparan sulfate, and fibronectin, and TLR9 by chromatin-IgG complexes (Citation22–28). Irrespective of which ligand (i.e. exogenous or endogenous) causes activation of TLRs, monocytes respond by initiation of the same cascade of inflammatory reactions, resulting in massive cytokine production.
Of note, progression of atherosclerotic plaques ultimately depends on the persistent process of excessive lipid accumulation by monocyte-derived ‘foam’ cells, leading to their death and repeated lipid accumulation. It has been demonstrated that monocyte/macrophage pattern recognition receptors are capable of recognizing dying cells and promoting their elimination (Citation29,Citation30). Although the biological role of these reactions is by nature protective, atherogenesis is self-perpetuated by the inflammatory reactions triggered.
There are several transcriptional mechanisms that mediate the stimulation of TLRs and the production of inflammatory factors. Nuclear factor κB (NFκB) and mitogen-activated protein kinase pathways appear to be major participants is such processes (Citation31). NFκB is a transcription factor that plays a critical role in diverse cellular processes associated with proliferation, as well as cellular development, apoptosis, and the production of innate and adaptive immune responses. However, NFκB is also a direct regulator of the expression of pro-inflammatory genes.
In resting conditions, NFκB is located in the cell cytoplasm, being inhibited by the inhibitor of κB (IκB). Stimulation of TLRs triggers NFκB signalling pathway by activation of IκB kinase that phosphorylates IκB (Citation32). This is followed by ubiquitination and degradation of IκB. Once released, NFκB moves to the cell nucleus to initiate transcription of numerous cytokines, chemokines, adhesion molecules, and matrix metalloproteinases, which is accompanied by the production of reactive oxygen species (ROS) (Citation33). Persistent NFκB-mediated signal transduction in monocytes and other cells is associated with plaque progression, destabilization, and rupture (Citation34). Animal studies have also established that the NFκB pathway regulates expression of lipoxygenase, cyclo-oxygenase, and monocyte chemoattractant protein-1 (MCP-1), which are all involved in the modification of low density lipoprotein (LDL) and monocyte chemotaxis (Citation35–37). The expression of several adhesion molecules associated with the pathogenesis of atherosclerosis (P-selectin, E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1)) is also regulated by the NFκB pathway (Citation38,Citation39).
Additionally, NFκB accelerates differentiation of monocytes into macrophages by macrophage colony-stimulating factor (M-CSF) at the site of atherosclerotic lesion (Citation40). Furthermore, transcription of the extracellular matrix-degrading enzymes matrix metalloproteinase 9 and myeloperoxidase is also regulated by the NFκB pathway and is closely linked to the processes of plaque destabilization and atherothrombosis (Citation41,Citation42). Ultimately, most of the inflammatory cytokines up-regulated in atherosclerosis (e.g. tumour necrosis factor (TNFα), interleukin (IL)-1, IL-6, IL-10, IL-12) are produced by monocytes via a NFκB-dependent pathway. Finally, activation of NFκB is associated with smooth muscle cell proliferation and fibrous cap formation (Citation43). These data indicate that NFκB signalling is the principal transcriptional pathway of monocyte inflammatory response and may be involved at every stage of atherosclerotic disease ().
Figure 1. Implication of monocytes in atherogenesis. COX = cyclo-oxygenase; Ecs = endothelial cells; GM-CSF = granulocyte–macrophage colony-stimulating factor; HIF-1 = hypoxia-inducible factor; HSP = heat shock protein; ICAM-1 = intercellular adhesion molecule 1; IL = interleukin; LOX = lipoxygenase; LPS = lipopolysaccharide; MAPK = mitogen-activated protein kinase; MCP-1 = monocyte chemotactic protein-1; M-CSF = macrophage colony-stimulating factor; MMPs = matrix metalloproteinases; MPO = myeloperoxidase; NFκB = nuclear factor κB; SRs = scavenger receptors; TIR = Toll/interleukin-1 receptor; TLR4 = Toll-like receptor 4; TNF = tumour necrosis factor; VCAM-1 = vascular cell adhesion molecule 1; VEGF = vascular endothelial growth factor.
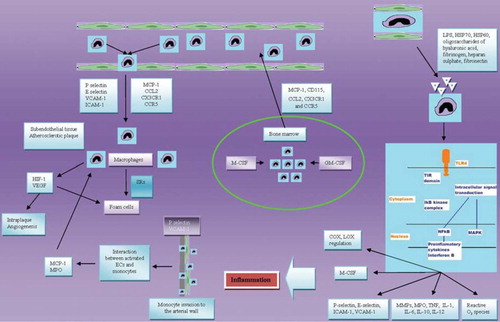
Cardiovascular effects of monocyte activation
Inflammatory cytokines are uniformly increased in patients with atherosclerosis, especially in its complicated forms, such as myocardial infarction (Citation44–46). Inflammatory activation of monocytes is accompanied by the release of various cytokines and an oxidative state that promotes oxidation of LDL and activation of endothelial cells. The latter leads to the increased expression of P-selectin, VCAM-1, and chemokines essential for the recruitment of monocytes into the arterial wall (Citation47). Activated endothelial cells in turn interact with monocytes by production of large amounts of MCP-1, resulting in a vicious cycle of self-activation, smooth muscle cell mitogenesis, and inhibition of antithrombotic properties of the vascular wall (e.g. modulation of synthesis of plasminogen activator inhibitor 1) (Citation48,Citation49). Myeloperoxidase secreted by activated monocytes plays a role in the formation of atherogenic dysfunctional lipoproteins by their carbamylation (Citation50), a process further enhanced by monocyte-derived ROS in subjects with hyperlipidemia (Citation51).
Additionally, matrix metalloproteinases, produced by monocytes, facilitate plaque destabilizaton and the development of atherothrombotic complications (Citation52). In addition to secretion of matrix metalloproteinases by monocytes themselves, a high expression of TNFα and ROS by these cells triggers matrix metalloproteinase production by other cell lines inside the vascular wall (Citation53). Not surprisingly, monocytes have been closely implicated in the development of ACS, and their high count at admission is strongly associated with an unfavourable prognosis ( and ).
Table I. Relationship between the monocyte counts/subtypes and left ventricular dysfunction/remodelling after myocardial infarction.
Table II. Monocytes and congestive heart failure.
Monocyte mobilization and homing in atherosclerosis
Monocytes exert their pro-atherogenic effects both in the circulation and inside the vascular wall where they are involved in various processes related to atherogenesis (e.g. ‘foam’ cell formation). Monocyte mobilization from the bone-marrow and their migration to tissues are of great interest. Circulating monocytes derive from hematopoietic precursor cells in the bone-marrow and are mobilized into the circulation and thence to the vascular wall by biological stimulants abundantly produced by damaged tissues.
The continuing entry, survival, and replication of mononuclear cells in atherosclerotic lesions depend partly on M-CSF, granulocyte–macrophage colony-stimulating factor (GM-CSF), and MCP-1 (Citation54). In addition to the mobilization of monocytes into circulation, chemoattractants (e.g. M-CSF, MCP-1) also support their proliferation and differentiation into macrophages. In keeping with its role in monocyte mobilization and recruitment, M-CSF contributes to the atherosclerotic plaque progression (Citation55). It has been recently demonstrated that a significant reduction of atherosclerosis in M-CSF-deficient mice results from a marked decrease in macrophage accumulation within the lesions (Citation56).
Monocyte mobilization from bone-marrow is also largely mediated by chemokines, such as MCP1 (CCL2), fractalkine (CX3CL1), and CCL5 (RANTES) and their monocyte receptors (CCR2, CX3CR1, and CCR5, respectively) that differentially alter the mobilization of subsets of circulating monocytes to the blood and promote macrophage accumulation within atherosclerotic lesions (Citation57). In addition, MCP-1 causes diapedesis of monocytes from the lumen to the subendothelial tissue, where they differentiate into ‘foam’ cells, and promotes the expression of endothelial cell adhesion molecules on the arterial intima (Citation58). The role of MCP-1 in monocyte-related processes seen in ACS has been recently reviewed (Citation52).
The recruitment of circulating monocytes to the vascular subendothelial tissue occurs via a tightly regulated multistep process mediated by a series of cell surface adhesion molecules. Endothelial cell activation causes up-regulation of endothelial cell adhesion molecules, including both P- and E- selectins, VCAM-1, and ICAM-1, a process mediated by pro-inflammatory cytokines (Citation59).
As the first step, activated endothelial cells at the target region of atherogenesis express increased levels of P- and E-selectins that mediate the tethering and rolling of circulating monocytes upon the endothelium (Citation60,Citation61). Following monocyte adhesion to the endothelium, the selectins stimulate monocyte expression of integrins (e.g. ICAM-1, VCAM-1), which are essential to their migration to tissues (Citation60). An ex vivo model of isolated carotid arteries shows that P-selectin blockade prevents monocyte attachment to the carotid endothelium (Citation62). ICAM and VCAM are over-expressed at sites of atherosclerotic lesions and support stronger adhesion of monocytes to the endothelium (Citation63). The complex process of monocyte–endothelium interactions in atherosclerosis also involves various chemokines, including CXCL1, CCL5, CXCL4, CXCL12 and their monocyte receptors CXCR2, CCR5, CXCR4 (Citation64,Citation65). Interestingly, the migration of different monocyte subsets through endothelium depends on different receptor-ligand interactions. Inflammatory circulating monocytes (i.e. mouse Ly6Chigh subset) are shown to differentiate into vascular wall macrophages and form ‘foam’ cells and express high levels of CCR2, but also to require CX3CR1 and CCR5 for effective recruitment (Citation66,Citation67). Ly6Clow monocytes predominantly enter the atherosclerotic wall in a CCR5-dependent manner but do not require CX3CR1 (highly expressed on this subset) or CCR2 (Citation67). Of interest, a gene polymorphism of CX3CR1 (fractalkine receptor) modulates the affinity of fractalkine to its receptor on monocytes and has been found to affect the risk of CAD (Citation68).
Accumulation of lipids in atherosclerotic plaque
Accumulation of lipids by monocyte-derived macrophages leads to the formation of lipid-loaded ‘foam’ cells in the vascular wall and represents a critical point of atherogenesis. The biological role of this process may be to remove excessive amounts of toxic oxidized LDL from the vascular wall. However, uncontrolled lipid accumulation followed by macrophage death results in a self-perpetuating process of atherosclerotic core enlargement, deposition of collagen, and the migration of smooth muscle cells into the intima. This ultimately leads to the (irreversible) formation of atherosclerotic plaques. Accumulation of modified LDL by macrophages is mediated by scavenger receptors (SRs), a large family of proteins consisting of at least eight classes. For example, SRs of Class A include SR-AI (CD204), SR-AII, SR-AIII, macrophage receptor with collagenous structure (MARCO), and SR with C-type lectin, whilst Class B includes SR-BI, SR-BII (lysosomal integral membrane protein-II), and SR-BIII. SR Class C includes Drosophila Class C SR, whilst Class D is CD68 (macrosialin), and Class E is lectin-like oxidized LDL receptor-1 (LOX-1) (Citation69,Citation70).
The impact of these SRs on atherogenesis seems to be complex. These receptors are involved in modified lipoprotein uptake. Furthermore they, like many other members of the family of the pattern recognition receptors, are linked with apoptotic cell clearance and initiation of signal transduction and serve as pattern recognition molecules for pathogens (Citation71). SRs also have potential to trigger both pro-inflammatory and anti-inflammatory responses (Citation72,Citation73).
SR-AI and SR-BI are responsible for the scavenging of atherogenic modified proteins and are directly involved in ‘foam’ cell formation and atherogenesis (Citation74). Circulating monocytes express high levels of CD36 and SR-AI. CD36 is a transmembrane glycoprotein with a high affinity to oxidized LDL, whilst SR-AI accumulates acetylated LDL (Citation75). Furthermore, the exposition of macrophages to oxidized LDL increases their CD36 expression resulting in uncontrolled lipid accumulation (Citation76). The lack of CD36 expression is associated with delayed progression of atherosclerosis (Citation77). The involvement of CD36 in atherosclerosis is further supported by findings of abundant receptor substrate (oxidized lipids) in the plaque region but not in the plaque-free part of a vessel (Citation77,Citation78). Recent experimental studies also suggest not only pro-atherogenic but a prothrombotic role of CD36 in the cardiovascular system (Citation77). In contrast, SR-BII is involved in cholesterol transfer from cells to high density lipoprotein (HDL) and liver and may exert anti-atherosclerotic properties (Citation79,Citation80). The role of other SR classes in atherosclerosis is less established.
Monocytes and intraplaque angiogenesis
Intimal and plaque neovascularization is a common finding in patients with advanced atherosclerosis which is often associated with plaque progression and destabilization (Citation81). Intraplaque vessels most frequently arise from vessels in the adventitia, adjacent to a plaque, rather than from the main artery lumen (Citation82). This process is largely driven by hypoxia in the plaque tissue due to impaired diffusion of oxygen though thickened intima.
Progressive hypoxia in advanced human atherosclerosis stimulates local expression of hypoxia- inducible factor-1 and vascular endothelial growth factor (VEGF) by monocytes/macrophages. Consequently, increased plaque vascularization promotes further accumulation of inflammatory cells and atheroma progression, through a vicious circle. Indeed, the pathway of hypoxia-inducible factor-1 is linked to both lesion progression (‘foam’ cell formation) and angiogenesis, and in a vulnerable atherosclerotic plaque an enlarged necrotic core contains an increased number of vasa vasorum and apoptotic macrophages (Citation83–86). It has recently been shown that an anti-angiogenic molecule, angiostatin, reduces accumulation of intraplaque macrophages and inhibits intraplaque angiogenesis with the secondary reduction of macrophage density and may thus promote plaque stability (Citation87).
Monocytes may be directly implicated in the process of angiogenesis, being both protective (to improve tissue perfusion) and detrimental (when inside atheroma) (Citation88). Pro-angiogenic factors are expressed by monocytes and stimulate the proliferation of endothelial cells as well as mobilization and homing of bone-marrow-derived endothelial progenitors. (Citation86). Activated macrophages may influence each phase of angiogenesis by producing pro-angiogenic factors (e.g. VEGF, fibroblast growth factor-2) (Citation89). However, more studies are needed to shed further light on this issue.
Monocyte–platelet interactions
Circulating monocytes may form aggregates with platelets, creating the so-called monocyte-platelet aggregates (MPAs). This process is believed to reflect mainly the removal of activated platelets from the circulation (Citation90). However, the nature of the interactions between monocytes and platelets appears to be much more complex and represents an additional pathway implicated in the regulation of monocyte activity. Conjugation with platelets activates expression of receptors to adhesion molecules (e.g. MAC-1) on monocytes (Citation91). Up-regulation of monocyte receptors to adhesive molecules, known to be closely associated with formation of atherosclerotic plaques, may facilitate monocyte migration to the sites of inflammation and atherogenesis. Indeed, the aggregation of leukocytes with platelets is promoted by different conditions associated with inflammation and endothelial dysfunction (Citation92,Citation93). Exposure of human blood C-reactive protein doubles the number of MPAs, whilst infusion of lipopolysaccharide (LPS) to mice increased the number of MPAs 4-fold (Citation94). Of interest, increased formation of platelet conjugates with monocytes, but not with lymphocytes or neutrophils, is characteristic for patients with diabetes, a recognized risk factor for atherosclerosis (Citation93). Furthermore, the number of MPAs appears to parallel the severity of diabetes and is higher in those patients with signs of proliferative retinopathy and nephropathy (Citation93). Not surprisingly, MPA formation is increased in patients with atherosclerosis. For example, patients with stable coronary artery disease have more than twice the counts of MPAs compared with healthy subjects (Citation95). Destabilization of atherosclerotic plaques and atherothrombosis has been associated with more dramatic (3-fold) increases in total MPA levels (Citation96). Nevertheless, a substantial gap in the understanding of mutual effects of monocytes and platelets drives extensive on-going research.
Conclusion
Monocytes are important cells of the innate immune system and play important roles in host defence. They are also strongly implicated in cardiovascular pathology and many key steps of atherosclerosis. Numerous pattern recognition molecules, integrins, cytokines, and chemokine receptors are implicated in monocyte-derived inflammatory responses, which include the promotion of plaque growth, rupture, and thrombosis. Specifically, NFκB-dependent signal transduction appears to be the dominant mediator in these processes. Numerous mechanisms are responsible for the maintenance of equilibrium between inflammatory and anti-inflammatory forces of the innate immune system, but these have been poorly characterized to date.
Although most contemporary research is focused on the detrimental roles of monocytes in the pathogenesis of atherosclerotic cardiovascular diseases, monocytes may have reparative actions in both tissue repair and angiogenesis. A better understanding of the monocyte involvement in such processes may provide clinicians with a better understanding of the pathophysiology of cardiovascular disorders, perhaps allowing the development of new therapeutic interventions in the future.
Acknowledgements
BP and ES are funded by research fellowships from the European Society of Cardiology. We acknowledge the support of Heart Research UK and the Peel Medical Research Trust, and the research and development programme of Sandwell and West Birmingham Hospitals NHS Trust.
Declaration of interest: The authors declare no conflicts of interest.
References
- Napoli C, D'Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, . Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia: intimal accumulation of low density lipoprotein and its oxdation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100: 2680–90.
- Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr, Rosenfeld ME, . A definition of initial, fatty streak, and intermediate lesions of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Atherosclerosis, American Heart Association. Circulation. 1994;89: 2462–78.
- Ross R. The pathogenesis of atherosclerosis—an update. N Engl J Med. 1986;314:488–500.
- Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulation of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–8.
- Elsheikh E, Uzunel M, He Z, Holgersson J, Nowak G, Sumitran-Holgersson S. Only specific subtype of human peripheral blood monocytes has endothelial-like functional capacity. Blood. 2005;106:2347–55.
- Shantsila E, Lip GY. Monocytes in acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2009;29:1433–8.
- Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
- Kantari C, Pederzoli-Ribeil M, Witko-Sarsat V. The role of neutrophils and monocytes in innate immunity. Contrib Microbiol. 2008;15:118–46.
- Monie TP, Bryant CE, Gay NJ. Activating immunity: lessons from the TLRs and NLRs. Trends Biochem Sci. 2009;34: 553–61.
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–45.
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, . Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82.
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, . Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–17.
- Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, . Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–81.
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5.
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, . Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8.
- Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, . Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–84.
- Methe H, Kim JO, Kofler S, Weis M, Nabauer M, Koglin J. Expansion of circulating Toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation. 2005;111:2654–61.
- Orihara K, Nagata K, Hamasaki S, Oba R, Hirai H, Ishida S, . Time-course of Toll-like receptor 2 expression, as a predictor of recurrence in patients with bacterial infectious diseases. Clin Exp Immunol. 2007;148:260–70.
- Kajiya T, Orihara K, Hamasaki S, Oba R, Hirai H, Nagata K, . Toll-like receptor 2 expression level on monocytes in patients with viral infections: monitoring infection severity. J Infect. 2008;57:249–59.
- Mizoguchi E, Orihara K, Hamasaki S, Ishida S, Kataoka T, Ogawa M, . Association between Toll-like receptors and the extent and severity of coronary artery disease in patients with stable angina. Coron Artery Dis. 2007;18:31–8.
- Kuwahata S, Fujita S, Orihara K, Hamasaki S, Oba R, Hirai H, . High expression level of Toll-like receptor 2 on monocytes is an important risk factor for arteriosclerotic disease. Atherosclerosis. 2010;209:248–54.
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, . Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34.
- Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177: 1272–81.
- Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61.
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, . Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195: 99–111.
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–94.
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor- mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–9.
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, . The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33.
- Devitt A, Moffatt OD, Raykundalia C, Capra JD, Simmons DL, Gregory CD. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. 1998;392:505–9.
- Devitt A, Parker KG, Ogden CA, Oldreive C, Clay MF, Melville LA, . Persistence of apoptotic cells without autoimmune disease or inflammation in CD14-/- mice. J Cell Biol. 2004;67:1161–70.
- Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111.
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:S81–96.
- van den Berg R, Haenen GR, van den Berg H, Bast A. Transcription factor NF-kappaB as a potential biomarker for oxidative stress. Br J Nutr. 2001;86 Suppl 1:S121–7.
- Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, . Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–22.
- Zhao L, Funk CD. Lipoxygenase pathways in atherogenesis. Trends Cardiovasc Med. 2004;14:191–5.
- Burleigh ME, Babaev VR, Oates JA, Harris RC, Gautam S, Riendeau D, . Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor deficient mice. Circulation. 2002;105:1816–23.
- Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, . Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–25.
- Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, . A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62.
- Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-selectin or intercellular adhesion molecule (ICAM)- 1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191:189–94.
- Brach MA, Henschler R, Mertelsmann RH, Herrmann F. Regulation of M-CSF expression by M-CSF: role of protein kinase C and transcription factor NF kappa B. Pathobiology. 1991;59:284–8.
- Tavora FR, Ripple M, Li L, Burke AP. Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc Disord. 2009;9:27.
- Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–91.
- Hoshi S, Goto M, Koyama N, Nomoto K, Tanaka H. Regulation of vascular smooth muscle cell proliferation by nuclear factor-kappaB and its inhibitor, I-kappaB. J Biol Chem. 2000;275:883–9.
- Kanda T, Takahashi T. Interleukin-6 and cardiovascular diseases. Jpn Heart J. 2004;45:183–93.
- Ikeda U, Ito T, Shimada K. Interleukin-6 and acute coronary syndrome. Clin Cardiol. 2001;24:701–4.
- Yao L, Huang K, Huang D, Wang J, Guo H, Liao Y. Acute myocardial infarction induced increases in plasma tumor necrosis factor-alpha and interleukin-10 are associated with the activation of poly(ADP-ribose) polymerase of circulating mononuclear cell. Int J Cardiol. 2008;123:366–8.
- Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–32.
- Lukacs NW, Strieter RM, Elner V, Evanoff HL, Burdick MD, Kunkel SL. Production of chemokines, interleukin-8 and monocyte chemoattractant protein-1, during monocyte: endothelial cell interactions. Blood. 1995;86:2767–73.
- Lundgren CH, Sawa H, Sobel BE, Fujii S. Modulation of expression of monocyte/macrophage plasminogen activator activity and its implications for attenuation of vasculopathy. Circulation. 1994;90:1927–34.
- Sirpal S. Myeloperoxidase-mediated lipoprotein carbamylation as a mechanistic pathway for atherosclerotic vascular disease. Clin Sci (Lond). 2009;116:681–95.
- Vasconcelos EM, Degasperi GR, de Oliveira HC, Vercesi AE, de Faria EC, Castilho LN. Reactive oxygen species generation in peripheral blood monocytes and oxidized LDL are increased in hyperlipidemic patients. Clin Biochem. 2009;42: 1222–7.
- Speidl WS, Toller WG, Kaun C, Weiss TW, Pfaffenberger S, Kastl SP, . Catecholamines potentiate LPS-induced expression of MMP-1 and MMP-9 in human monocytes and in the human monocytic cell line U937: possible implications for peri-operative plaque instability. FASEB J. 2004; 18:603–5.
- Poitevin S, Garnotel R, Antonicelli F, Gillery P, Nguyen P. Type I collagen induces tissue factor expression and matrix metalloproteinase 9 production in human primary monocytes through a redox-sensitive pathway. J Thromb Haemost. 2008;6:1586–94.
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–26.
- Irvine KM, Andrews MR, Fernandez-Rojo MA, Schroder K, Burns CJ, Su S, . Colony-stimulating factor-1 (CSF-1) delivers a proatherogenic signal to human macrophages. J Leukoc Biol. 2009;85:278–88.
- Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A. 1995;92:8264–8.
- Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, . Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–57.
- Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91.
- Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47:C7–12.
- Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J Clin Invest. 1995;95:2297–303.
- van der Wal AC, Das PK, Tigges AJ, Becker AE. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am J Pathol. 1992;141: 1427–33.
- Ramos CL, Huo Y, Jung U, Ghosh S, Manka DR, Sarembock IJ, . Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84:1237–44.
- Gerszten RE, Lim YC, Ding HT, Snapp K, Kansas G, Dichek DA, . Adhesion of monocytes to vascular cell adhesion molecule-1-transduced human endothelial cells: implications for atherogenesis. Circ Res. 1998;82:871–8.
- Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, . The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108: 1307–14.
- von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, . Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105:924–30.
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, . Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205.
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, . Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94.
- Apostolakis S, Lip GY, Shantsila E. Monocytes in heart failure: Relationship to a deteriorating immune overreaction or a desperate attempt for tissue repair? Cardiovasc Res. 2010; 85:649–60.
- Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A. 1979;76: 333–7.
- Krieger M. The other side of scavenger receptors: pattern recognition for host defense. Curr Opin Lipidol. 1997;8: 275–80.
- Devitt A, Gregory CD. Innate immune mechanisms in the resolution of inflammation. In: Rossi AG, Sawatzky DA. Progress in inflammation: the resolution of inflammation. Basel: Birkhauser Verlag; 2008. p. 39–56.
- de Winther MP, Gijbels MJ, van Dijk KW, van Gorp PJ, Suzuki H, Kodama T, . Scavenger receptor deficiency leads to more complex atherosclerotic lesions in APOE3Leiden transgenic mice. Atherosclerosis. 1999;144:315–21.
- Dansky HM, Charlton SA, Sikes JL, Heath SC, Simantov R, Levin LF, . Genetic background determines the extent of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1960–8.
- Teupser D, Stein O, Burkhardt R, Nebendahl K, Stein Y, Thiery J. Scavenger receptor activity is increased in macrophages from rabbits with low atherosclerotic response: studies in normocholesterolemic high and low atherosclerotic response rabbits. Arterioscler Thromb Vasc Biol. 1999;19: 1299–305.
- Silverstein RL, Febbraio M. CD36 and atherosclerosis. Curr Opin Lipidol. 2000;11:483–91.
- Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, . Targeted disruption of the Class B scavenger receptor, CD36, protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–56.
- Silverstein RL. Inflammation, atherosclerosis, and arterial thrombosis: Role of the scavenger receptor CD36. Cleve Clin J Medicine. 2009;76:S27–30.
- Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004; 24:2333–8.
- Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–5.
- Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, . Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90:270–6.
- Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, . Atherosclerotic plaque progression and vulnerability to rupture angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25: 2054–61.
- Moulton KS. Plaque angiogenesis: its functions and regulation. Cold Spring Harbor Symp Quant Biol. 2002;67: 471–82.
- Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, . Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–65.
- Gessi S, Fogli E, Sacchetto V, Merighi S, Varani K, Preti D, . Adenosine modulates HIF-1{alpha}, VEGF, IL-8, and foam cell formation in a human model of hypoxic foam cells. Arterioscler Thromb Vasc Biol. 2010;30:90–7.
- Fong GH. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis. 2008;11:121–40.
- Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130: 147–58.
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, . Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003; 100:4736–41.
- Ribatti D, Levi-Schaffer F, Kovanen PT. Inflammatory angiogenesis in atherogenesis—a double-edged sword. Ann Med. 2008;40:606–21.
- Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–11.
- Shantsila E, Lip GY. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb Haemost. 2009;102:916–24.
- Simon DI, Ezratty AM, Francis SA, Rennke H, Loscalzo J. Fibrin(ogen) is internalized and degraded by activated human monocytoid cells via Mac-1 (CD11b/CD18): a nonplasmin fibrinolytic pathway. Blood. 1993;82:2414–22.
- Arber N, Berliner S, Pras E, Arber L, Fishelson Z, Kahn Y, . Heterotypic leukocyte aggregation in the peripheral blood of patients with leukemia, inflammation and stress. Nouv Rev Fr Hematol. 1991;33:251–5.
- Kaplar M, Kappelmayer J, Veszpremi A, Szabo K, Udvardy M. The possible association of in vivo leukocyte-platelet heterophilic aggregate formation and the development of diabetic angiopathy. Platelets. 2001;12:419–22.
- Danenberg HD, Kantak N, Grad E, Swaminathan RV, Lotan C, Edelman ER. C-reactive protein promotes monocyte-platelet aggregation: an additional link to the inflammatory-thrombotic intricacy. Eur J Haematol. 2007; 78:246–52.
- Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, . Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998;31:352–8.
- Brambilla M, Camera M, Colnago D, Marenzi G, De Metrio M, Giesen PL, . Tissue factor in patients with acute coronary syndromes: expression in platelets, leukocytes, and platelet-leukocyte aggregates. Arterioscler Thromb Vasc Biol. 2008;28:947–53.
- Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, . Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002;39:241–6.
- Hojo Y, Ikeda U, Zhu Y, Okada M, Ueno S, Arakawa H, . Expression of vascular endothelial growth factor in patients with acute myocardial infarction. J Am Coll Cardiol. 2000;35:968–73.
- Iwama H, Uemura S, Naya N, Imagawa K, Takemoto Y, Asai O, . Cardiac expression of placental growth factor predicts the improvement of chronic phase left ventricular function in patients with acute myocardial infarction. J Am Coll Cardiol. 2006;47:1559–67.
- Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, . Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–8.
- Bruno S, Bussolati B, Scacciatella P, Marra S, Sanavio F, Tarella C, . Combined administration of G-CSF and GM-CSF stimulates monocyte-derived pro-angiogenic cells in patients with acute myocardial infarction. Cytokine. 2006;34:56–65.
- Dresske B, El Mokhtari NE, Ungefroren H, Ruhnke M, Plate V, Janssen D, . Multipotent cells of monocytic origin improve damaged heart function. Am J Transplant. 2006;6:947–58.
- Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y, Nakamura M. Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur J Heart Fail. 2006;8:810–5.
- de Lemos JA, Morrow DA, Blazing MA, Jarolim P, Wiviott SD, Sabatine MS, . Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results from the A to Z trial. J Am Coll Cardiol. 2007;50: 2117–24.
- Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y, . Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol. 2006;109:226–34.
- Sheu JJ, Chang LT, Chiang CH, Youssef AA, Wu CJ, Lee FY, . Prognostic value of activated toll-like receptor-4 in monocytes following acute myocardial infarction. Int Heart J. 2008;49:1–11.
- Dahl CP, Gullestad L, Fevang B, Holm AM, Landrø L, Vinge LE, . Increased expression of LIGHT/TNFSF14 and its receptors in experimental and clinical heart failure. Eur J Heart Fail. 2008;10:352–9.
- Satoh M, Iwasaka J, Nakamura M, Akatsu T, Shimoda Y, Hiramori K. Increased expression of tumor necrosis factor-alpha converting enzyme and tumor necrosis factor-alpha in peripheral blood mononuclear cells in patients with advanced congestive heart failure. Eur J Heart Fail. 2004;6: 869–75.