Abstract
Muscarinic acetylcholine receptors MAChRs from Bovine Tracheal Smooth Muscle (BTSM) plasma membranes are responsible for the cGMP rise and signal-amplitude peaks associated with smooth muscle contraction present in bronchial asthma. These MAChRs bind [3H]QNB and exhibit the classic G Protein Coupled-Receptor (GPCR) behavior towards muscarinic agonist and antagonists that is sensitive to sensitive to GTP analogs. Interestingly, the [3H]QNB binding activity was stimulated by cGMP and ATP, and was enhanced by IBMX and Zaprinast, inhibitors of cGMP-PDE. Cyclic GMP plus ATP affected the agonist-antagonist muscarinic binding activities. Thus, the high affinity agonist (Carbamylcholine) binding sites disappeared, whereas, 4-DAMP, a M3 selective antagonist displayed an additional high affinity-binding site. In contrast, non-selective (atropine) and M2-selective (methoctramine and gallamine) antagonists revealed one low binding site. Moreover, the 4-DAMP-mustard alkylation of the MAChRs blocked the cGMP effect indicating that the M3AChR is the main receptor target of cGMP. Interestingly, these cGMP effects were potentiated by an activator (Sp-8-pCPT-cGMPS), and diminished by an inhibitor (Rp-8-pCPT-CGMPS), of cGMP-dependent protein kinase (PKG-II), which was detected by Western blotting using specific PKG II antibodies. Finally, plasma membrane M3AChRs were phosphorylated in a cGMP-dependent manner and this novel post-translational reversible modification at M3AChRs may act as a feedback mechanism to terminate the cGMP dependent muscarinic signal transduction cascades at the sarcolema of BTSM.
Keywords::
Introduction
Muscarinic agonists activate Airway smooth muscle (ASM) through muscarinic receptors subtypes (MAChRs) (Lucchesi et al. Citation1990, Mak and Barnes Citation1990, Haddad et al. Citation1991). In ASM, there are two types of MAChRs, M2 and M3AChRs, which are members of a family of cell surface receptors that activate intracellular responses by coupling G proteins (GPCR) to specific effectors (Kostenis et al. Citation1999). High activity of MAChRs have been demonstrated in the plasma membrane fraction from bovine tracheal smooth muscle (BTSM) using [3H]QNB binding studies (Misle et al. Citation1994). Functional studies have shown that M2/M3AChRs subtypes are also present in BTSM plasma membranes (Alfonzo et al. Citation1998a, Citation2012).
Classically, activation of MAChRs increase intracellular cGMP levels (Schultz et al. Citation1973) and contractility of ASM from guinea pigs (Murad and Kimura Citation1974) and BTSM (Katsuki and Murad Citation1977). Likewise, muscarinic agonists in isolated strips of BTSM induce the generation of two cGMP peak amplitude signals at 20 s and 60 s (Guerra de González et al. Citation1999). The first (20 s) cGMP peak-signal is a product of the NO-sensitive soluble guanylyl cyclase (NO-sGC) (Uray et al. Citation2010) and the second one (60 s) is produced by the action of a Natriuretic peptide Receptor-Guanylyl cylase B (NPR-GC-B) (Borges et al. Citation2001, Bruges et al. Citation2007). In BTSM plasma membrane fraction, NPR-GC-B is regulated by two MAChRs subtypes in an opposite way, whereby the M3AChR stimulates the production of cGMP and the M2AChR subtype seems to inhibit NPR-GC-B activation (Alfonzo et al. Citation1998a, Citation1998b, Citation2012).
After a muscarinic agonist binds to MAchR, some molecular events occur to guarantee the activation/termination of this transduction cascade. Plasma membranes possess molecular mechanisms for the generation of second messengers as above-mentioned. By contrast, the capability to end this muscarinic activation, via a MAchR desensitization has been also described (Hosey et al. Citation1995, Krupnick and Benovic, Citation1998, Luo et al. Citation2008). For example, GPCR phosphorylation-dephosphorylation processes regulate the functional coupling between membrane receptors and G-proteins. Two classes of muscarinic phosphorylation have been reported: G-protein coupled receptor kinases (GRKs) and the G-protein independent kinases. The changes in the affinity of this MAChRs for agonists and antagonists are associated with protein kinases activities. For example, muscarinic receptor phosphorylation by GRKs (DebBurman et al. Citation1995, Hosey et al. Citation1995, Willets et al. Citation2001), induced receptor desensitization by the phosphorylation of serine/threonine residues, which promoted uncoupling the receptor from G proteins and thereby terminating signaling (Hosey et al. Citation1995, Krupnick and Benovic Citation1998, Luo et al. Citation2008).
Given the capability of MAChRs to induce cGMP signals at plasma membranes, we decided to explore the possible existence of cGMP regulatory feedback mechanisms on the MAChR activity in these plasma membranes fractions from BTSM. This last subcellular fraction is a simpler and reliable system to evaluate the direct effect of cGMP on M3AChR anchored to the plasma membranes instead of intact airway smooth muscle cells, which is a more complicated system due the plethora of regulatory mechanisms associated with cGMP, as an intracellular second messenger, in a number of physiological functions.
In this paper, we demonstrate for the first time that cGMP can affect the plasma membrane [3H]QNB binding activity. This novel cGMP biological effect is mediated through the activation of a cGMP-dependent protein kinase (PKG-II). Moreover, we provide evidence that the molecular signal transducing functional machinery comprised of M3AChRs, NPR-GC-B, PKG-II and cGMP-PDE, is located at the plasma membrane of BTSM.
Methods
The following compounds were purchased from Sigma Chemical Co. (USA): Trizma base, sucrose, DTT, ATP, cGMP, carbamylcholine, atropine sulfate, oxotremorine, gallamine, metacholine, methoctramine, AFDX-DS 116, 4-DAMP mustard, Digitonin, cholate, protein A/G-agarose beads and Sephadex G-50 (20–80 μm). Bovine serum albumin standard was obtained from Pierce (USA). Rabbit anti-m3, anti-m2AChR and anti-PKG-II antibodies from Santa Cruz Biotechnology (USA). Amersham ECL Western Blotting Reagent Pack and Hyperfilm® and L-[3H]QNB (45.5 Ci/mmol) from the Radiochemical Centre, Amersham (Bucks, UK) GE/Healthcare, Life Sciences. Precision Plus protein™ Western C™ Pack was obtained from BioRAD Labs. Protein Phosphatase Inhibitor set (containing Cypermethrin, Dephostatin, Okadaic acid, and NIPP-1 Bovine Thymus recombinant), Rp-8-pCPT-cGMPS and Sp-8-pCPT-cGMPS were obtained from Calbiochem (USA). [32P] γ ATP (3000 Ci/mmol) was purchased from Perkin-Elmer (USA). Other chemical reagents were obtained from E. Merck (Germany) and Fisher (USA).
Plasma membrane preparation
The plasma membrane fraction (P1) was prepared from bovine tracheal smooth muscle as previously described (González de Alfonzo et al. Citation1996). This plasma membrane fraction is highly enriched in active muscarinic receptors (Misle et al. Citation1994, Alfonzo et al. Citation1998a, Citation1998b). The plasma membrane fraction was diluted with 80 volumes of 20 mM Tris-HCl (pH 7.2) containing 0.5 mM DTT and centrifuged at 150,000 g for 30 min, washed and suspended in small volume of incubation buffer (50 mM Tris-HCl, pH 7.6) prior to use.
Muscarinic receptor activity measurement studies
The [3H]QNB binding studies were performed as described previously (Misle et al. Citation1995). Briefly, [3H]QNB binding assay was started by adding membrane protein (2–5 μg) in incubation buffer [50 mM Tris-HCl pH 7.8, and L-[3H]QNB (1,250 nM) ] to a final volume of 120 μl. Different drugs or compounds to be tested were added in the incubation media. After 1 h of incubation at 37°C, the incubation mixture was placed onto a pre-centrifuged Sephadex G-50 column (3 ml) equilibrated with 0.3 M sucrose-50 mM Tris-HCl (pH 7.6) and immediately centrifuged at 700 g for 1.5 min to remove free [3H]QNB. The column effluent containing 97–98% of the bound [3H]QNB was transferred to vials containing Aquasol®. Radioactivity was measured in a RackBeta liquid scintillation counter LKB, Wallac 1214/1219 and all the samples counted with approximately the same (30%) efficiency. Specific binding was calculated by subtracting non-specific binding (less than 1% of total binding measured with 1 μM atropine), from the total binding (Fields et al. Citation1978). In all binding experiments, no more than 5% of the fixed radioligand concentration was allowed to bind to the membranes to avoid ligand depletion and similar amounts of active receptors were employed in these binding assays.
Selective 4-DAMP mustard M3-receptor alkylation
Alkylation of plasma membranes with 4-DAMP mustard was performed in excess methoctramine, to protect the M2AchR subtype as described elsewhere (Michel et al. Citation1989, Reddy et al. Citation1995) with some modifications. Briefly, the aziridinium ion was produced from incubation of the M3-selective irreversible receptor antagonist, 4-DAMP mustard at 37°C for 30 min in incubation Buffer. Then, plasma membrane fractions were exposed to 4-DAMP mustard aziridinium ion (4 nM) as prepared for 60 min, in the presence of methoctramine (1 μM). Alkylation of the membranes was stopped by the addition of 1 mM DTT and the reaction proceeded for 10 min. Finally, the alkylated-membranes were recovered after centrifugation at 150,000 g for 30 min, washed with 50 mM Tris-HCl buffer (pH 7.6) and 1 mM DTT buffer and centrifuged 4× at 150,000 g for 30 min to remove methoctramine excess and suspended in 50 mM Tris-HCl (pH 7.6) buffer before assayed. Control plasma membranes were subjected in parallel to the similar procedure without 4-DAMP mustard.
Immunodetection of PKG II by Western blot
Plasma membranes proteins were separated on SDS-PAGE using a 10% gel and transferred to PDVF membranes (Immunoblots-PDVF) following the procedure described in the ECL Advance Western Blotting Detection Kit brochure of Amersham, Life Sciences®. The remaining steps to detect the PKG II by autoradiography were Protein Blots were blocked for 2–4 h at 4°C in phosphate buffered saline (PBS) containing 2% bovine albumin (BSA) and 0.05% (v/v) Tween 20. After thoroughly washing twice with PBS-Tween, the PDVF membrane was incubated for 4–6 h at 4°C with (1/1000) rabbit polyclonal IgG specific for PKG II from Santa Cruz Biotechnology, Inc. Then, the membrane was thoroughly washed with PBS-Tween with one change overnight and incubated for 4–6 h at 4°C with (1/1000) horseradish labeled anti-rabbit IgG. To identify the immunoreactivity proteins (positive bands), a Precision Plus protein™ Western C™ Pack from BioRAD and the enhanced chemiluminescence method as described by the manufacturer (Amersham ECL Western Blotting Reagent Pack from GE/Healthcare, Life Sciences) were employed.
Muscarinic AChR [32P]phosphorylation and selective immunoprecipitation assays
Native BTSM plasma membranes (P1 fraction) were thawed and diluted 80 times with 20 mM Tris-HCl pH 7.2 buffer containing 5 mM DTT and 1 mM PMSF, to remove cytosolic PKG-I and other soluble proteins. Washed plasma membranes (0.5–1.0 mg/ml of protein) was subjected to [32P]phosphorylation reactions at 37°C with 0.1 mM [γ32P]ATP (Specific activity: 3000 μCi/mole), 1 mM MgCl2, 20 mM potassium phosphate buffer (pH 7.2), 1 mM DTT, 10 μM IBMX, 100 μM protein phosphatase inhibitor kit containing (Cypermethrin, Dephostatin, Okadaic acid, and NIPP-1 Bovine Thymus recombinant) and other compound as indicated in Results. [32P]-labeled plasma membranes were collected by centrifugation at 12,000 g for 15 min at 0°C in an Eppendorf® centrifuge. [32P]labeled membranes were washed twice with a mixture solution containing, 20 mM EDTA, 1 mM PMSF and 20 mM potassium phosphate buffer (pH 7.2) and 100 μM protein phosphatase inhibitor kit. Later, [32P]-labeled membranes were solubilized by incubation at 4°C for 10 min (5 mg/ml) in a mixture containing 5 mM MgCl2, 20 mM potassium phosphate buffer, pH 7.2, 1 mM DTT, 1 mM EDTA, 0.1 mM PMSF (buffer I) and one ninth volume of a detergent mixture containing 0.1% Digitonin-0.02% sodium cholate, which specifically solubilized MAChRs as described elsewhere (Peterson and Schimerlik Citation1984). Detergent-solubilized proteins were recovered after centrifugation at 150,000 g for 30 min. The sediment was again re-extracted using the same procedure and both detergent-solubilized supernatants were pooled. Then, the digitonin/cholate-solubilized material was incubated with protein A/G-agarose beads for 1 h at 4°C. Later, the pre-clarified supernatant was overnight incubated with specific anti m3 or anti m2AChR antibodies at 4°C. Immunoprecipitates were collected by the addition of protein A/G-agarose beads for 6 h at 4°C. These beads were then collected by low speed centrifugation and washed three times using detergent-free buffer I. These immunoprecipitates were incubated with cracking SDS solution and [32P]-labeled polypeptides were separated on 10% SDS-PAGE as described (Laemmli Citation1970). Gels were dried and exposed for autoradiogram during 7–12 days at −80°C under X-film (Hyperfilm®).
Protein measurements.
The amount of protein was quantified using bovine serum albumin (BSA) as standard following the procedure described elsewhere (Bensadoun and Weinstein Citation1976).
Data analysis.
A computer-assisted non-linear regression program (InPlot, Graph Pad® software, U.S.A.) was used to analyze binding and competition experiments results, as previously reported (Misle et al. Citation1994, Citation1995). Also, the method used to calculate the association rate constants (kon) as described (Bennett and Yamamura Citation1985).
Results
In order to demonstrate that active M2 and M3AChRs are present in these plasma membranes, some initial experiments using the [3H]QNB binding studies were performed to evaluate the presence of agonist and antagonists binding sites.
Muscarinic drugs competitive binding studies on native plasma membranes
Muscarinic (agonists and antagonists) displacement experiments were performed in the absence or presence of GTPγS, due the fact that MAChRs are GPCRs, which are affected by GTP analogs. shows that in the absence of GTPγS, displacement curves for muscarinic agonists such as carbamylcholine, metacholine and oxotremorine rendered pseudo-Hill coefficients (np-H) lower than 1.0, suggesting the presence of agonist receptor heterogeneity. Nonetheless, GTPγS induced dramatic changes in the np-H, which reached values of 1.0 indicating the disappearance of the agonist dependent receptor heterogeneity ().
Table I. Agonist competition binding parameters of muscarinic receptors in tracheal smooth muscle plasma membranes.
Moreover, in the presence of GTPγS, the IC50 values for agonists became similar indicating the existence of ‘one receptor' agonist population. This agonist-dependent muscarinic receptor heterogeneity was analyzed by applying the ‘two sites' model as the best fitting for all agonists assayed. shows that two agonist-binding sites such as the high affinity (RH) and low affinity (RL) states were detected. These two agonist states are in a ratio 1:1 in the presence of 5 mM MgCl2. Interestingly, upon addition of 100 μM GTPγS, significant changes in the carbamylcholine, metacholine and oxotremorine binding parameters was observed with a complete disappearance of the RH states together with an increment in the relative amount of receptors in the RL state to 100% as shown in .
Muscarinic antagonist competitive binding studies on native membranes
Antagonist binding properties were also studied by using competition assays and in a similar experimental approach used for the muscarinic agonists. The compounds evaluated were 4-DAMP (selective M3 antagonist), AF-DX 116 and methoctramine (selective M2AchR subtype antagonists) in the presence or absence of GTPγS. These results are shown in . We have previously shown that in the basal conditions (without MgCl2) (Misle et al. Citation1994), all muscarinic antagonists displacement curves fit the ‘one site' model with np-H values close to 1.0 indicating that these drugs seem to bind to a homogeneous receptor-binding site. In the present work, methoctramine, and AF-DX 116 did not change their binding isotherms, either in the presence of Mg2+ or GTPγS showing np-H values close to 1.0 (). Surprisingly, the 4-DAMP (selective M3AChR subtype antagonist) showed competitive displacement curves with pseudo-Hill coefficients (np-H) lower than 1.0, suggesting the presence of antagonist receptor heterogeneity in these MAchRs. Further analysis of the latter antagonist competition curves obtained in the presence of MgCl2 by applying the ‘two sites' model unveiled two binding sites in the MAchRs. These new sites were designated as high antagonist affinity sites (Rh) and low antagonist affinity (Rl) sites. The Rh sites were unmasked in the presence of Mg2+ showing a pIC50 of 8.1 ± 0.2 for 4-DAMP, these novel Rh sites disappeared upon addition of GTPγS as shown in . Competitive displacement curves of 4-DAMP also showed changes induced by GTPγS. These latter displacement curves exhibit np-H values of 1.0 indicating the disappearance of the antagonist dependent receptor heterogeneity. The last behavior is similar to the agonist curves described above.
Table II. Antagonist competition binding parameters of muscarinic receptor in tracheal smooth muscle plasma membranes.
Muscarinic agonist and antagonist competitive binding studies in 4-DAMP alkylated membranes
Plasma membrane fractions from BTSM contain a mixture of M2 and M3AChR subtypes (Misle et al. Citation1994). In order to explore the possibility of obliterating one subtype of MAChR, the native plasma membrane fractions were treated with a selective M3-alkylating agent (4-DAMP mustard), while protecting the M2 subtype with methoctramine as described in Methods. As expected, the alkylated membranes showed IC50 values (), which belong to a M2 pharmacological profile (4-DAMP ≥ methoctramine > AF-DX 116). Interestingly, all antagonist-binding isotherms in the alkylated membranes showed np-H values of 1.0 and fitted for ‘one' homogeneous binding site, which was unaffected in the presence of GTPγS (). In order to assure the presence of active agonists binding sites in these remaining M2AChRs, some competition experiments were performed in the presence of the agonist carbamylcholine, which showed the classic high (RH) and low sites (RL) for this drug (). Moreover, the competition experiments with muscarinic agonists using alkylated membranes were affected by GTPγS, in such a way, that the high agonist sites (RH) completely disappeared in the presence of this guanine nucleotide analog displaying only the low agonist sites (RL) ().
Table III. Drugs competition binding parameters of muscarinic receptors in 4-DAMP alkylated plasma membranes from tracheal smooth muscle.
Effect of cGMP and PDE inhibitors on antagonist binding studies at plasma membranes
The effect of cGMP on the MAChRs binding properties was evaluated and shows the binding of [3H]QNB to MAChR in plasma membranes from BTSM, in the presence of 5 mM MgCl2 and increasing concentrations of cGMP. Moreover, also shows that this muscarinic binding activity was dependent upon the concentration of cGMP reaching the maximal increment (40%) between 3–10 nM cGMP, and decreasing to basal values after 100 nM of this cyclic nucleotide. The bimodal behavior above described in may be due to the existence of cyclic nucleotide phosphodiesterases (PDE) in these plasma membranes fractions. Thus, the effect of cGMP on the [3H]QNB binding was evaluated in the presence of two PDE inhibitors such as a non-selective PDE inhibitor, isobutyl methyl xanthine (IBMX), and a specific inhibitor of the cGMP-specific phosphodiesterase PDE V(Zaprinast). shows the effect of variable cGMP concentrations on the muscarinic binding activity in the presence of 10 μM IBMX or 100 nM Zaprinast. Thus, in the presence of IBMX or Zaprinast, there is a marked increment in the [3H]QNB binding at low cGMP concentrations (<10 nM), and remain constant at high cGMP concentrations. These PDE inhibitors abolished the bimodal response on the [3H]QNB binding activity exhibited in . These results suggest that the cGMP effect on the mAChR activity may be regulated by a Zaprinast-sensitive PDE V located in these plasma membranes fractions from BTSM.
Figure 1. Effect of cGMP in the presence or absence of ATP on the [3H]QNB binding from of plasma membranes from BTSM. Binding experiments were carried out in the presence of cGMP (○) and cGMP plus 5 mM ATP (•) and 1,500 pM [3H]QNB, 5 mM MgCl2 and 2–4 µg of membrane proteins were assayed at 37°C as described in Methods. Specific [3H]QNB binding expressed as percentage of binding in the absence of nucleotides. The binding activity in the absence of cGMP was 1,100 ± 130 fmoles/mg protein and 1,450 ± 170 fmoles/mg protein in the presence of cGMP plus ATP. Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.
![Figure 1. Effect of cGMP in the presence or absence of ATP on the [3H]QNB binding from of plasma membranes from BTSM. Binding experiments were carried out in the presence of cGMP (○) and cGMP plus 5 mM ATP (•) and 1,500 pM [3H]QNB, 5 mM MgCl2 and 2–4 µg of membrane proteins were assayed at 37°C as described in Methods. Specific [3H]QNB binding expressed as percentage of binding in the absence of nucleotides. The binding activity in the absence of cGMP was 1,100 ± 130 fmoles/mg protein and 1,450 ± 170 fmoles/mg protein in the presence of cGMP plus ATP. Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.](/cms/asset/6e37e1f3-9b6a-4356-960b-007afc9da4d1/imbc_a_851419_f0001_b.jpg)
Figure 2. Effect of PDE inhibitors on [3H]QNB binding in of plasma membranes from BTSM. Binding experiments were carried out at 37°C as described in Methods in the presence of 2–4 µg of membrane proteins, 1,500 pM [3H]QNB, 5 mM MgCl2, 5 mM ATP and (•) increasing concentrations of cGMP or (▪) the specific inhibitor of PDE V, zaprinast (100 nM) or (○) the non-specific inhibitor of PDEs, IBMX (10 μM). Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.
![Figure 2. Effect of PDE inhibitors on [3H]QNB binding in of plasma membranes from BTSM. Binding experiments were carried out at 37°C as described in Methods in the presence of 2–4 µg of membrane proteins, 1,500 pM [3H]QNB, 5 mM MgCl2, 5 mM ATP and (•) increasing concentrations of cGMP or (▪) the specific inhibitor of PDE V, zaprinast (100 nM) or (○) the non-specific inhibitor of PDEs, IBMX (10 μM). Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.](/cms/asset/abcdd747-de97-440f-81cc-5e2f0de0a72d/imbc_a_851419_f0002_b.jpg)
Effect of cGMP on the kinetic and saturation parameters of [3H]QNB binding
To characterize the effect of the cGMP on the muscarinic activity, saturation and kinetic parameters were estimated from [3H]QNB binding curves performed in the presence or absence of ATP at 10 nM cGMP. shows a typical [3H]QNB saturation experiment, whose kinetic parameters obtained from these assays are shown in . There is an increment, which is statistically significant (p < 0.01), in more than 42% in the Bmax observed in the presence of cGMP plus ATP in comparison with cGMP or ATP alone. However, the affinity (KD) of the MAchRs did not change in the presence of cGMP and ATP or in the presence of cGMP or ATP alone suggesting that cGMP only affects the total amount of muscarinic receptors (Bmax), but not the affinities of these receptors under scrutiny. Thus, kinetic curves of the muscarinic activity in the presence of 10 nM cGMP, 5 mM ATP, and 10 nM cGMP plus 5 mM ATP are shown in insert of .
Figure 3. Kinetic curves of the binding of [3H]QNB in of plasma membranes from BTSM. Saturation concentration of [3H]QNB was 1,500 pM, 5 mM MgCl2 and 2–4 µg of membrane proteins were assayed in the presence of IBMX (10 μM) at 37°C as described in Methods. Binding experiments were carried out in the presence of 10 nM cGMP (○), 5 mM ATP (▪) and 10 nM cGMP plus 5 mM ATP (•). Specific [3H]QNB binding expressed as fmoles/mg protein. Each point represents the mean ± SE of four different membrane preparations assayed by triplicate. *Statistical significant (p < 0.05). Insert shows the plot of the best line of fit through the first half-life of each curve from Figure 3, which is a method used to calculate the association rate constants (kon) as described (Bennett and Yamamura Citation1985).
![Figure 3. Kinetic curves of the binding of [3H]QNB in of plasma membranes from BTSM. Saturation concentration of [3H]QNB was 1,500 pM, 5 mM MgCl2 and 2–4 µg of membrane proteins were assayed in the presence of IBMX (10 μM) at 37°C as described in Methods. Binding experiments were carried out in the presence of 10 nM cGMP (○), 5 mM ATP (▪) and 10 nM cGMP plus 5 mM ATP (•). Specific [3H]QNB binding expressed as fmoles/mg protein. Each point represents the mean ± SE of four different membrane preparations assayed by triplicate. *Statistical significant (p < 0.05). Insert shows the plot of the best line of fit through the first half-life of each curve from Figure 3, which is a method used to calculate the association rate constants (kon) as described (Bennett and Yamamura Citation1985).](/cms/asset/c5722bd0-bb93-4438-bd04-fff6d6788505/imbc_a_851419_f0003_b.jpg)
Table IV. Effects of nucleotides on the [3H]QNB binding parameters in tracheal smooth muscle plasma membranes.
The kinetic parameter kon was estimated from the insert data as described elsewhere (Bennett and Yamamura Citation1985). Thus, the kinetic curves in the presence of 10 nM cGMP displayed kon = 6.52 ± 0.02 msec-1, whereas in the presence of 5 mM ATP, the kon = 9.49 ± 0.02 msec-1 and in the presence of 10 nM cGMP plus 5 mM ATP, the curves exhibited a kon = 11.63 ± 0.02 msec-1. These data suggest that saturation of the mAchR by the radioligand is 1.78 times faster in the presence of the cGMP plus ATP.
Effect of cGMP on the muscarinic agonist and antagonist binding parameters at plasma membranes from tracheal smooth muscle
To evaluate the ability of cGMP to affect muscarinic agonists and antagonists MAChRs binding parameters, competition experiments were carried out with ATP in the presence or absence of 10 nM cGMP. The competition binding parameters results are shown in . It can be seen that muscarinic agonists and antagonists seem to behave differently in the presence of the cGMP and ATP. Thus, a classic muscarinic agonist (Carbamylcholine) displays competition curves in the presence of ATP with high and low affinities agonist binding sites on the mAChR. Nevertheless, cGMP plus ATP causes the disappearance of the high affinities sites for Carbamylcholine (). Competition curves of muscarinic antagonists such as atropine, 4-DAMP, gallamine and methoctramine evidenced that these antagonists bind to one affinity state of the mAChR in the presence of ATP. Surprisingly, the inclusion of cGMP induces high and low affinities antagonist binding sites on the MAChRs, which were only detected by 4-DAMP (M3AChR selective antagonist), as shown in . However, these high binding sites were not detected by the M2-selective antagonists, such as methoctramine and gallamine or the non-selective, atropine.
Table V. Effect of cGMP in the presence of ATP on the agonist and antagonist competition binding parameters of muscarinic receptors in tracheal smooth muscle plasma membranes.
Antagonist-binding studies in 4-DAMP alkylated plasma membranes from BTSM
The [3H]QNB binding activity in the plasma membrane fractions from BTSM is the expression of a mixture of M2 and M3AchR subtypes as previously described (Misle et al. Citation1994, Alfonzo et al. 1998b). From previous experiments, there were some experimental evidences that suggest the M3AChR is the main target of cGMP. To validate that the muscarinic subtype involved in this cGMP effect was indeed the M3AChR, native plasma membrane fractions were 4-DAMP alkylated with excess of methoctramine (1 μM) to protect the M2 subtype and incubated in the presence of a selective-M3AChR subtype-alkylating agent (4-DAMP mustard) as described (Michel et al. Citation1989). shows the binding of the radioligand in the 4-DAMP-alkylated membranes in the presence of increasing concentrations of cGMP and 10 μM IBMX. Interestingly, after alkylating the M3 AChR subtype, the cGMP dependent [3H]QNB binding activity was abolished. The last evidence supports the rationale that M3AchR subtype is the endogenous target for the cGMP effect.
Figure 4. Effect of cGMP on the [3H]QNB binding in Control plasma membranes and 4-DAMP mustard-alkylated plasma membranes. The [3H]QNB binding curves were perrformed as described in Methods for both native membranes (•) and 4-DAMP mustard-alkylated membranes (○) in the presence of 5 mM ATP, 5 mM MgCl2 and increasing cGMP concentrations are indicated. Specific [3H]QNB binding is expressed as percentage of binding in the absence of nucleotides. Binding experiments were carried out at 1,500 pM [3H]QNB and 2–4 µg of membrane proteins were assayed at 37°C. The total binding activity was 1,570 ± 170 fmoles/mg protein in Control membranes and 549 ± 20 fmoles/mg protein in 4-DAMP mustard alkylated-membranes. Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.
![Figure 4. Effect of cGMP on the [3H]QNB binding in Control plasma membranes and 4-DAMP mustard-alkylated plasma membranes. The [3H]QNB binding curves were perrformed as described in Methods for both native membranes (•) and 4-DAMP mustard-alkylated membranes (○) in the presence of 5 mM ATP, 5 mM MgCl2 and increasing cGMP concentrations are indicated. Specific [3H]QNB binding is expressed as percentage of binding in the absence of nucleotides. Binding experiments were carried out at 1,500 pM [3H]QNB and 2–4 µg of membrane proteins were assayed at 37°C. The total binding activity was 1,570 ± 170 fmoles/mg protein in Control membranes and 549 ± 20 fmoles/mg protein in 4-DAMP mustard alkylated-membranes. Each point represents the mean ± SE of four different membrane preparations assayed in triplicate.](/cms/asset/bb817038-9193-4603-9c67-b33ad78d9313/imbc_a_851419_f0004_b.jpg)
Effect of inhibitors and activators of PKG on the muscarinic activity of plasma membranes
The behavior of the MAChR activity in the presence of ATP and cGMP suggests that may be an association between the [3H]QNB binding activity and the ability of cGMP to activate the cyclic nucleotide-dependent protein kinase (PKG-II) associated with these plasma membranes. Thus, experiments were performed using a specific activator of the PKG such as Sp-8-pCPT-cGMPS are shown in . It can be seen that there is an increment of the [3H]QNB binding on the membrane with increasing concentrations of this cGMP analog. The insert in shows that the activation of the [3H]QNB binding activity depends on the cGMP analog concentration. On the other hand, shows that increasing concentrations of specific inhibitor of the PKG (Rp-8-pCPT-cGMPS), abolished the cGMP effect on [3H]-QNB binding to the plasma membranes. An insert in shows a typical inhibition curve, which is obtained when binding data is plotted against inhibitor concentration. These experimental results suggest that the cGMP effects on the muscarinic activity are mediated through a PKG-II located at the plasma membrane of BTSM.
Figure 5. (A) Effect of cyclic nucleotide analogs (activator of PKG on the muscarinic activity of plasma membranes from BTSM. [3H]QNB binding in plasma membranes from BTSM was performed using 1,500 pM [3H]QNB and 2–3 µg of membrane proteins were assayed at 37°C as described in Methods. A titration of cGMP was performed in the presence of 5 mM ATP and 5 mM MgCl2 under increasing concentration of Sp-8-pCPT-cGMPS were 0 (▪), 0.25 (•), 0.5 (□), 1.0 (○) and 5.0 μM (◊), respectively. (Insert: Activation curve obtained by plotting binding data obtained at 1 nM cGMP). (B) Effect of cyclic nucleotide analogs (inhibitor) of PKG on the muscarinic activity of plasma membranes from BTSM. [3H]QNB binding in plasma membranes from BTSM was performed using 1,500 pM [3H]QNB and 2–3 µg of membrane proteins were assayed at 37°C as described in Methods. A titration of cGMP was performed in the presence of 5 mM ATP and 5 mM MgCl2 under increasing concentration of Rp-8-pCPT-cGMPS (Insert: Inhibition curve obtained by plotting binding data obtained at 10 nM cGMP). Sp-8-pCPT-cGMPS and Rp-8-pCPT-cGMPS concentrations were 0 (▪), 0.25 (•), 0.5 (□), 1.0 (○) and 5.0 μM (◊), respectively. Each point represents the mean ± SE of four different plasma membrane preparations assayed in triplicate.
![Figure 5. (A) Effect of cyclic nucleotide analogs (activator of PKG on the muscarinic activity of plasma membranes from BTSM. [3H]QNB binding in plasma membranes from BTSM was performed using 1,500 pM [3H]QNB and 2–3 µg of membrane proteins were assayed at 37°C as described in Methods. A titration of cGMP was performed in the presence of 5 mM ATP and 5 mM MgCl2 under increasing concentration of Sp-8-pCPT-cGMPS were 0 (▪), 0.25 (•), 0.5 (□), 1.0 (○) and 5.0 μM (◊), respectively. (Insert: Activation curve obtained by plotting binding data obtained at 1 nM cGMP). (B) Effect of cyclic nucleotide analogs (inhibitor) of PKG on the muscarinic activity of plasma membranes from BTSM. [3H]QNB binding in plasma membranes from BTSM was performed using 1,500 pM [3H]QNB and 2–3 µg of membrane proteins were assayed at 37°C as described in Methods. A titration of cGMP was performed in the presence of 5 mM ATP and 5 mM MgCl2 under increasing concentration of Rp-8-pCPT-cGMPS (Insert: Inhibition curve obtained by plotting binding data obtained at 10 nM cGMP). Sp-8-pCPT-cGMPS and Rp-8-pCPT-cGMPS concentrations were 0 (▪), 0.25 (•), 0.5 (□), 1.0 (○) and 5.0 μM (◊), respectively. Each point represents the mean ± SE of four different plasma membrane preparations assayed in triplicate.](/cms/asset/085e947b-c387-4f5f-9146-a3911cf99089/imbc_a_851419_f0005_b.jpg)
Identification of cGMP-dependent protein kinase (PKG-II) by immunoblotting in plasma membranes
To confirm that the above results are due to the presence of cGMP-dependent protein kinases on the plasma membranes, we assayed for the presence of a PKG-II using Western blot analysis. BTSM plasma membrane proteins were solubilized with SDS and resolved on SDS-PAGE and transferred to PDVF membranes by Western blotting as described in Methods. Immunoreactive PKG-II bands were detected using specific antibodies against PKG-II. A chemiluminescence autoradiography results is shown in . A polypeptide Mw(r) of 86 KDa, which correlates with the presence of PKG-II was found. In addition, an immunoreactive polypeptide band with Mw(r) approximately 32 KDa was detected. This latter band appeared to be a proteolytic fragment of 86 KDa as was previously observed in other systems (Draijer et al. Citation1995). This is the first report that PKG-II is located in the plasma membrane from BTSM.
Figure 6. Western blotting for PKG-II in P1 plasma membranes fractions from BTSM. Plasma membrane fraction (P1) (20 µg) was subjected to 10% PAGE-SDS. Proteins were transfer to PDVF membranes and Western blot analysis with rabbit antisera directed against the PKG-II was performed. In addition, to identify the immunoreactivity proteins (positive bands), a Precision Plus protein™ Western C™ Pack from BioRAD and the enhanced chemiluminescence method as described by the manufacturer (Amersham ECL Western Blotting Reagent Pack from GE/Healthcare, Life Sciences) were used to identify the positive bands.

Muscarinic AChR [32P]phosphorylation and selective immunoprecipitation assays
To demonstrate that, indeed, M3AChRs is phosphorylated in the presence of cGMP, native BTSM plasma membranes was subjected to [32P]phosphorylation using [γ32P]ATP, collected by centrifugation as described in Methods. Later, these [32P] labeled membranes were detergent-solubilized with 0.1% Digitonin-0.02% sodium cholate, which selectively solubilized MAchR. The detergent-solubilized material was incubated with specific m2 and m3AChR antibodies. Later, immunoprecipitates were collected with protein A/G-agarose beads and the [32P]-labeled polypeptides were separated on 10% SDS-PAGE as described. Gels were dried and exposed during 7–12 days at −80°C under X-film (Hyperfilm®). The [32P]-autoradiograms are shown in . Thus, lanes A and B show the m3AChR immunoprecipates, which was more phosphorylated, in the presence of cGMP (10 nM) (lane B), while in m2AChR immunoprecipates (Lanes C and D), the 32P incorporation was not changed on the presence of cGMP. These findings indicate that m3AChR is more phosphorylated in the presence of cGMP possibly by the PKG-II anchored to these plasma membrane fractions from BTSM.
Figure 7. Autoradiograms of m2 and m3-immunoprecipitated 32P-labeled polypeptides. Plasma membranes fraction was subjected to [32P]phosphorylation using [γ32P]ATP as described in Methods. Later, [32P]-labeled membranes were solubilized using a detergent mixture containing 0.1% Digitonin-0.02% sodium cholate and incubated with specific anti m3 or anti m2AChR antibodies as described in Methods. Immunoprecipitates were collected with protein A/G-agarose beads by low speed centrifugation an incubated with cracking SDS solution and subjected to 10% SDS-PAGE. Gels were dried and exposed during 7–12 days at −80°C under X-film (Hyperfilm®). 32P-labeled m3-AChR immunoprecipitated polypeptides in the absence (lane A) or presence (lane B) of 10 nM cGMP and 32P-labeled m2-AChR immunoprecipitated polypeptides in the absence (lane C) or presence (lane D) of 10 nM cGMP. Arrows indicates the molecular masses m3AChR of 67 KDa and 52 KDa for m2AChR. All 32P-labeled phosphoproteins were adjusted to the same level for comparison.
![Figure 7. Autoradiograms of m2 and m3-immunoprecipitated 32P-labeled polypeptides. Plasma membranes fraction was subjected to [32P]phosphorylation using [γ32P]ATP as described in Methods. Later, [32P]-labeled membranes were solubilized using a detergent mixture containing 0.1% Digitonin-0.02% sodium cholate and incubated with specific anti m3 or anti m2AChR antibodies as described in Methods. Immunoprecipitates were collected with protein A/G-agarose beads by low speed centrifugation an incubated with cracking SDS solution and subjected to 10% SDS-PAGE. Gels were dried and exposed during 7–12 days at −80°C under X-film (Hyperfilm®). 32P-labeled m3-AChR immunoprecipitated polypeptides in the absence (lane A) or presence (lane B) of 10 nM cGMP and 32P-labeled m2-AChR immunoprecipitated polypeptides in the absence (lane C) or presence (lane D) of 10 nM cGMP. Arrows indicates the molecular masses m3AChR of 67 KDa and 52 KDa for m2AChR. All 32P-labeled phosphoproteins were adjusted to the same level for comparison.](/cms/asset/9cf33a58-1f37-4774-b923-299c69788ca8/imbc_a_851419_f0007_b.jpg)
Discussion
In the present paper, we report a novel function of cGMP on airway smooth muscle (ASM), via the activation of a plasma membrane-associated cGMP dependent protein kinase (PKG II), and cGMP-dependent phosphorylation of M3AChRs and regulation of the receptor activity at plasma membranes from BTSM. Muscarinic agonist stimulate MAChRs and increase cGMP levels in ASM (Murad and Kimura Citation1974, Katsuki and Murad Citation1977) and generate two cGMP signal peaks (20-s and 60-s) in BTSM (Guerra de González et al. Citation1999). In this study, we investigated whether a cGMP negative feedback mechanism may act to attenuate muscarinic receptor activation. Thus, in a broken-cell system, such as plasma membranes isolated from BTSM, the MAChRs function was evaluated, using the [3H]QNB binding activity. Initially, cGMP in the presence of ATP induced a ‘bimodal response' in the [3H]QNB binding activity, which was abolished by a selective-PDE V (Zaprinast) and unselective (IBMX) PDEs inhibitors. These last findings support the existence of a cGMP-specific PDEs at the plasma membranes capable of cGMP hydrolyzes. Membrane-bound PDEs have been reported in smooth muscle (Burns et al. Citation1992, Corbin and Francis Citation1999). Hence, the specific effect of Zaprinast confirms the presence of PDE V located at the plasma membranes from BTSM, which may play an important physiological role in the regulation of cGMP intracellular concentration (Kulkarni and Patil Citation2004, Bender and Beavo Citation2006).
The [3H]QNB binding activities displayed by these plasma membrane fractions from BTSM is consistent with the expression of a mixture of M2 and M3AChR subtypes (Misle et al. Citation1994, Alfonzo et al. Citation1998b). Thus, the alkylation of the M3AChR subtype with 4-DAMP mustard, in the presence of excess of methoctramine to protect the M2 subtype (Michel et al. Citation1989), abolished the cGMP activation effect on the [3H]QNB binding in these plasma membranes. Moreover, a ratio M3/M2AChR (3:1) was obtained, after 4-DAMP alkylation of these membranes. These results indicate that the M3AchR subtype is the main endogenous target for the cGMP effect.
It is possible that cGMP via PKG-II phosphorylation induced the following biological actions on the M3AChR binding activity: (1) The increment of the Bmax for [3H]-QNB binding activity (>40%) indicating that ‘new [3H]-QNB binding sites' are displayed. (2) A faster [3H]-QNB binding for these M3AChRs as indicated by the rise (almost twice) of Kon parameter from saturation assays. (3) A significant change in Hill coefficients (nH) from saturation kinetics, which increased from 1.0 close to 2.0 suggesting a positive cooperativity or a possible homodimer formation of M3AChRs. (4) These ‘new [3H]-QNB binding sites' exhibited similar affinity constants (KD) for this labeled antagonist. To explain the origin of these ‘new [3H]-QNB binding sites', it is important to point out, that there are not new synthesized or exposed of ‘recycled or hidden' M3AChRs from endosomes because our experimental findings were obtained with isolated plasma membranes fractions, where these intracellular processes are absent.
M3AChR belongs to the class A GPCR, which are regulated by three principal mechanisms: Desensitization, internalization, and down-regulation. The last two processes such as internalization and down-regulation are ruled out in our experimental conditions using isolated plasma membranes fractions from BTSM leaving the receptor desensitization as unique mechanism to explain these cGMP effects.
Receptor desensitization mechanism is related to phosphorylation. Classically, GPCR phosphorylation-dephosphorylation processes regulate the functional coupling between membrane receptors and G-proteins. Two classes of muscarinic phosphorylation have been reported: The G-protein coupled receptor kinases (GRKs) phosphorylation and the G-protein independent kinases. The changes in the affinity for agonists and antagonists of MAChRs are associated with increased protein kinase activity following receptor activation. Thus, receptor desensitization, is induced by phosphorylation by GRKs (DebBurman et al. Citation1995, Hosey et al. Citation1995, Willets et al. Citation2001), which is initiated by the phosphorylation of serine/threonine residues, that promotes uncoupling the receptor from G protein and terminating signaling (Hosey et al. Citation1995, Krupnick and Benovic Citation1998, Luo et al. Citation2008).
It has been proposed that GPCR are in equilibrium between ground receptors (R) and activated receptors (R*), with the latter coupled to heterotrimeric G-proteins leading to a binary complex as G-protein coupled receptor (G–R*) (Leff Citation1995). Classically, uncoupled receptors (R) show a higher affinity for antagonists while the coupled receptors (G–R*) exhibit lower affinity for them, being vice versa for agonists (Burgisser et al. Citation1982).
The MAChR population present in these plasma membranes from BTSM is coupled to G-proteins and functionally active as previously demonstrated (Alfonzo et al. Citation1998b, Citation2012). This statement is supported further by this work, where G-protein dependent desensitization of these MAChRs was observed in the presence of GTPγS for all tested muscarinic agonists, in competition curves, showing ‘high and low affinities' agonist binding sites, being shifted to ‘low affinity sites' by GTPγS. Our results on muscarinic agonists and antagonists binding activities seem to be similar in the presence of the [cGMP plus ATP] in comparison with the behavior displayed with irreversible G-proteins activators. In this present paper, the phosphorylation of the M3AChR by PKG-II may correlate to changes in the affinity of the receptor for agonists or antagonists. Thus, a muscarinic agonist (carbamylcholine) displays competition curves in the presence of ATP with ‘high and low affinities' agonist binding sites. Nevertheless, [cGMP plus ATP] causes the disappearance of the ‘high affinity' sites, which were shifted to ‘low affinity sites' for muscarinic agonist (carbamylcholine) indicating a receptor desensitization mechanism. It has been reported in cardiac cells, agonist-dependent phosphorylation of the MAChRs correlates with a decreased affinity of the receptors for muscarinic agonists (Hosey Citation1992). gallamine (Tränkle et al. Citation2001) and methoctramine (Michel and Whiting Citation1988) demonstrated that these antagonists bind to one ‘low affinity' receptor state, in the presence of MgCl2, GTPγS or ATP alone. Unexpectedly, the inclusion [cGMP plus ATP] induced ‘high and low' affinities antagonist binding sites, which were only detected by 4-DAMP (M3AChR selective antagonist). However, these ‘high affinity' antagonist binding sites were not disclosed by the M2-selective antagonists, such as methoctramine and gallamine or the non-selective, atropine, which exhibited only ‘one low binding site' in competition curves. Thus, the specific M3AChR phosphorylatio.
In relation to muscarinic antagonists, competition curves of atropine, n by the PKG II affects the antagonists binding activity, especially, the higher affinity shown by 4-DAMP, (M3AChR selective antagonist), inducing low affinity states towards the muscarinic agonists. These evidences support the rationale that M3AChR is involved in this cGMP-dependent process.
The molecular mechanisms of antagonist binding to muscarinic cholinergic receptors is complex (Caulfield and Birdsall Citation1998), which was classically interpreted according to a two-step isomerization model induced by the antagonist molecule (Henis and Sokolovsky Citation1983, Eller and Jarv Citation1988). Molecular biology studies using point mutations and irreversible affinity labeling of the M1AChR led to the proposal of a tandem two-site model (Jakubik et al. Citation2000), and the possibility for the receptor to bind two ligand molecules. It may be proposed that PKG-II phosphorylation of M3AChR induced a similar molecular mechanism of homodimer/oligomer formation and the appearance of ‘new antagonists binding sites' as described. This rationale is supported by the fact that the M3 AChR displays a greater propensity to form homodimer/oligomer structure at higher density receptor population (Zeng and Wess Citation1999, Angers et al. Citation2002, Milligan Citation2007, Alvarez-Curto et al. Citation2010, Palczewski Citation2010, McMillin et al. Citation2011), which may be our case, considering the alkylation M3/M2AChR ratio of 3:1 obtained in these plasma membranes from BTSM. Thus, GPCR complexes may affect the efficiency of agonist-induced G protein activation (White et al. Citation2007), the extent of receptor phosphorylation (Song et al. Citation2007), or signaling selectivity (Rovira et al. Citation2010) and ‘communication' between protomers through an allosteric mechanism (Han et al. Citation2009). It can be proposed that this homodimer formation, induced by PKG-II phosphorylation, stabilizes or ‘freezes' the M3AChR population, in a refractory state to agonist activation, and prone to antagonist binding, which helps to understand the molecular mechanisms of muscarinic antagonist drug action as shown in . Additional research is needed to establish that indeed PKG-II phosphorylation induced the homodimer/oligomer formation as above postulated.
Figure 8. Proposed model of molecular regulation of M3AChR induced by PKG-II phosphorylation at plasma membrane from BTSM. All molecular compnents are membrane bound entities. (A) Muscarinic agonist (Acetylcholine) binds to M3AChR and activates the heterotrimeric Gq16, which stimulates the NPR-GC and increases the cGMP production. (B) Cyclic GMP activates the PKG-II and produces M3AChR phosphorylation inducing a conformation changes to produce M3AChR dimer, which stabilizes or ‘freezes' the M3AChR population, in a refractory state to agonist activation, and prone to antagonist binding, which helps to understand the molecular mechanisms of muscarinic antagonist drug action.
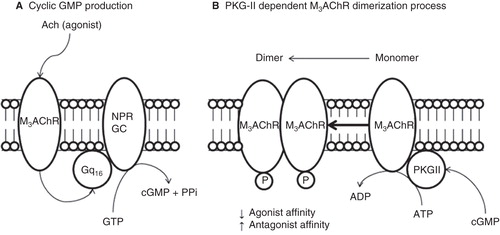
The cGMP analogs such as, a PKG-specific activator named Sp-8-pCPT-cGMPS (Butt et al. Citation1992) augmented the [3H]QNB binding, whereas Rp-8-pCPT-cGMPS, a PKG-specific inhibitor (Butt et al. Citation1994) suppressed the cGMP effect on [3H]QNB binding. Consequently, these experimental results support the involvement of PKG-II in this cGMP-dependent M3AchR desensitization. Further confirmation of the existence of a PKG-II in these plasma membranes from BTSM was obtained probing immunoblots with a specific PKG-II antibody. Thus, two polypeptides bands with apparent molecular weight of 86 KDa and 32 KDa were observed. The 86 KDa band having the apparent molecular weight of the PKG-II as described (Hofmann et al. Citation1992) and the second ones may be a PKG-II proteolytic product. Cyclic GMP-dependent protein phosphokinases (PKG) exist as two isoenzymes termed PKG-I and PKG-II (Lohmann et al. Citation1997, Francis et al. Citation2010). PKG-II enzymes have been located in particulate fractions from intestinal mucosa, (Vaandrager and De Jonge Citation1994, Vaandrager et al. Citation1997), brain (El-Husseini et al. Citation1995), and bone (Pfeifer et al. Citation1996). Therefore, the PKG-II detected in the present work is associated to the BTSM plasma membranes, may be a similar isoenzyme as detected in plasma membranes fractions from enterocytes (Vaandrager and De Jonge Citation1994).
To strengthen the role of PKG-II in M3AChR phosphorylation, we performed experiments using the same plasma membranes (P1) from BTSM, which were exposed to [γ32P]ATP and it was found that M3AChR immunoprecipates showed a higher 32P incorporation in the presence of cGMP. By contrast, the 32P incorporation into the M2AChR did not show significant changes with this cyclic nucleotide. These results support the rationale that cGMP activates a PKG-II anchored to BTSM sarcolema, which can phosphorylated the M3AChR. The putative PKG-II phosphorylation site(s) on M3AChR is under intense investigation in our laboratory.
Interestingly, the M3AChR is a prototypic class A GPCR that preferentially couples to G proteins family, is involved in numerous important physiological functions (Wess Citation1996, Caulfield and Birdsall Citation1998, Wess et al. Citation2007). Recently, M3AChR on airway smooth muscle is involved in the cholinergic tone contributes to airflow obstruction and chronic airway inflammation in asthma and COPD, where anticholinergics are effective bronchodilators by blocking the muscarinic receptor subtype (Kistemaker et al. Citation2012). Thus, understanding the molecular mechanism of the M3AChR has considerable relevance for designing novel classes of drugs that can modulate M3AChR function for therapeutic purposes in some of the pathological conditions above mentioned.
In summary, this is the first report on the ability of cGMP, possibly via a PKG-II, to increase the [3H]QNB antagonist binding activity of M3AChR subtypes at plasma membranes isolated from BTSM. Moreover, the involvement of PKG-II being responsible of a cGMP-dependent phosphorylation of the M3AChRs is described in this work. This is a novel mechanism presents in ASM cells to guarantee a feedback control of cGMP on the M3AChRs.
Conclusions
The MAchRs activities from bovine tracheal smooth muscle (BTSM) plasma membranes are modified in the presence of cGMP and ATP. These effects are potentiated by Sp-8-pCPT-cGMPS (PKG-II activator) and diminished by Rp-8-pCPT-cGMPS (PKG-II inhibitor) suggesting the existence of cGMP-dependent protein kinase (PKG II), which was detected by Western blotting using specific PKG II antibodies in the BTSM plasma membranes. Under phosphorylating (plus ATP) conditions, cGMP affected the agonist and antagonist binding activities. These original findings indicated that M3AChRs are regulated by cGMP via a PKG II anchored at the plasma membrane from BTSM. This novel post-translational reversible modification (phosphorylation) at M3AChRs dependent on a cGMP-PKG-II, which may act as a feedback mechanism to finish the cGMP dependent muscarinic signal transduction cascades at the sarcolema of BTSM.
Moreover, a molecular signal transducing functional machinery integrated by M3AChRs, PKG-II, cGMP-PDE, is located at the plasma membrane from BTSM supporting the rationale that the sarcolema contains and it is able to activate and shut down, these muscarinic receptors-linked signal cascades. The relevance of this cGMP effect, via PKG-II on M3AChRs might have a function in the airway smooth muscle contraction presents in bronchial asthma, which is an interesting biomedical research project to explore in the future.
Acknowledgements
This work was supported by grants from CDCH-UCV # PG -09-7401-2008/2 (RGA) and CDCH-UCV # PI-09-7726.2009/2 (ILB) and CDCH-UCV # PG 09-7772-2009/1(MJA). Marcelo A. Alfonzo-Gonzalez is a Graduate Student of the PhD program in Physiological Sciences/Graduate School-Faculty of Medicine at Universidad Central de Venezuela (UCV). The authors wish to recognize Dr Alfredo J. Misle for his technological help on the performance of experimental assays.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alfonzo MJ, Guerra de González L, Villaroel SS, Francis J, Misle AJ, Napoleón V, et al. 1998a. Signal transduction pathways through mammalian guanylyl cyclases. New Adv Cardiovasc Physiol Pharmacol 1184:147–172.
- Alfonzo MJ, Becemberg IL, Villaroel SS, Herrera VN, Misle AJ, Alfonzo RG. 1998b. Two opposite signal transducing mechanisms regulate a G-protein-coupled guanylyl cyclase. Arch Biochem Biophys 350:19–25.
- Alfonzo MJ, Placeres-Uray F, Hassan-Soto W, Borges A, Gonzalez de Alfonzo R, Lippo de Becemberg I. 2012. Two guanylylcyclases regulated the muscarinic activation of Airway Smooth Muscle. In: Sugi H, editor. Current basic and pathological approaches to the function of muscle cells and tissues – from molecules to humans. Croatia. InTech. 113–132.
- Alvarez-Curto E, Ward RJ, Pediani JD, Milligan G. 2010. Ligand regulation of the quaternary organization of cell surface M3 muscarinic acetylcholine receptors analyzed by fluorescence resonance energy transfer (FRET) imaging and homogeneous time-resolved FRET. J Biol Chem 285:23318–23330.
- Angers S, Salahpour A, Bouvier M. 2002. Dimerization: An emerging concept for G protein-coupled receptor ontogeny and function. Ann Rev Pharmacol Toxicol 42:409–435.
- Bender AT, Beavo JA. 2006. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol Rev 58:488–520.
- Bennett JP Jr, Yamamura HI. 1985. Neurotransmitter, hormone, or drug receptor binding methods. In: Yamamura HI, Enna SJ, Kuhar MJ, editors. Neurotransmitter receptor binding. New Jersey: Raven. pp 61–89.
- Bensadoun A, Weinstein D. 1976. Assay of proteins in the presence of interfering materials. Anal Biochem 70:241–250.
- Borges A, Villarroel SS, Winand NJ, Becemberg IL, Alfonzo MJ, Alfonzo RG. 2001. Molecular and biochemical characterization of a CNP-sensitive guanylyl cyclase in bovine tracheal smooth muscle. Am J Respir Cell Mol Biol 25:98–103.
- Bruges G, Borges A, Villarroel SS, Becemberg IL, Francis G, Pláceres F, et al. 2007. Coupling of M3 acetylcholine receptor to Gq16 activates a natriuretic peptide receptor guanylyl cyclase. J Recept Signal Transduct Res 27:189–216.
- Burgisser E, De Lean A, Lefkowitz RJ. 1982. Reciprocal modulation of agonist and antagonist binding to muscarinic cholinergic receptor by guanine nucleotide. Proc Natl Acad Sci USA 79:1732–1736.
- Burns F, Rodger IW, Pyne NJ. 1992. The catalytic subunit of protein kinase A triggers activation of the type V cyclic GMP-specific phosphodiesterase from guinea-pig lung. Biochem J 283:487–491.
- Butt E, Nolte C, Schulz S, Beltman J, Beavo JA, Jastorff B, et al. 1992. Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochem Pharmacol 43:2591–2600.
- Butt E, Eigenthaler M, Geneiser HG. 1994. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur J Pharmacol 269:265–268.
- Caulfield MP, Birdsall NJ. 1998. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50:279–290.
- Corbin JD, Francis SH. 1999. Cyclic GMP phosphodiesterase-5: Target of sildenafil. J Biol Chem 274:13729–13732.
- DebBurman SK, Kunapuli P, Benovic JL, Hosey MM. 1995. Agonist-dependent phosphorylation of human muscarinic receptor in Spodoptera frugiperda insect cell membranes by G protein-coupled receptor kinases. Mol Pharmacol 47:224–233.
- Draijer R, Vaandrager AB, Nolte C, de Jonge HR, Walter U, van Hinsbergh VW. 1995. Expression of cGMP-dependent protein kinase I and phosphorylation of its substrate, vasodilator-stimulated phosphoprotein, in human endothelial cells of different origin. Circ Res 77:897–905.
- El-Husseini AE, Bladen C, Vincent SR. 1995. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem 64:2814–2817.
- Eller M, Jarv J. 1988. Two-step isomerization of quinuclidinyl benzilate-muscarinic receptor complex. Neurochem Int 12:285–289.
- Francis SH, Busch JL, Corbin JD. 2010. cGMP-dependent protein kinases and cGMP. Phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62:525–563.
- Fields JZ, Roeske WR, Morkin E, Yamamura HI. 1978. Cardiac muscarinic cholinergic receptors. Biochemical identification and characterization. J Biol Chem 253:3251–3258.
- González de Alfonzo R, Bécemberg IL, Alfonzo MJ. 1996. A Ca2+/CaM dependent protein kinase associated with Ca2+ transport in sarco(endo) plasmic vesicles from tracheal smooth muscle. Life Sci 58:18–24.
- Guerra de González L, Misle A, Pacheco G, Napoleon de Herrera V, González de Alfonzo R, Lippo de Bécemberg I, et al. 1999. Effects of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) and N omega (ω)-nitro-L-arginine methyl ester (NAME) on cyclic GMP levels during muscarinic activation of tracheal smooth muscle. Biochem Pharmacol 58:563–569.
- Haddad EB, Landry Y, Gies JP. 1991. Muscarinic receptor subtypes in guinea pig airways. Am J Physiol 261:L327–L333.
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. 2009. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695.
- Henis YI, Sokolovsky M. 1983. Muscarinic antagonists induce different receptor conformations in rat adenohypophysis. Mol Pharmacol 24:357–365.
- Hofmann F, Dostmann W, Keilbach A, Landgraf W, Ruth P. 1992. Structure and physiological role of cGMP-dependent protein kinase. Biochim Biophys Acta 1135:51–60.
- Hosey MM. 1992. Diversity of structure, signaling and regulation within the family of muscarinic cholinergic receptors. FASEB J 6:845–852.
- Hosey MM, Benovic JL, DebBurman SK, Richardson RM. 1995. Multiple mechanisms involving protein phosphorylation are linked to desensitization of muscarinic receptors. Life Sci 56:951–955.
- Jakubik J, El-Fakahany EE, Tucek S. 2000. Evidence for a tandem two-site model of ligand binding to muscarinic acetylcholine receptors. J Biol Chem 275:18836–18844.
- Katsuki S, Murad F. 1977. Regulation of adenosine cyclic 3′,5′-monophosphate and guanosine cyclic 3′,5′-monophosphate levels and contractility in bovine tracheal smooth muscle. Mol Pharmacol 13:330–341.
- Kistemaker LE, Oenema TA, Meurs H, Gosens R. 2012. Regulation of airway inflammation and remodeling by muscarinic receptors: Perspectives on anticholinergic therapy in asthma and COPD. Life Sci 91:1126–1133.
- Kostenis E, Zeng FY, Wess J. 1999. Structure-function analysis of muscarinic receptors and their associated G proteins. Life Sci 64:355–362.
- Krupnick JG, Benovic JL. 1998. The role of receptor kinases and arrestins in G-protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38:289–319.
- Kulkarni SK, Patil CS. 2004. Phosphodiesterase 5 enzyme and its inhibitors: Update on pharmacological and therapeutical aspects. Methods Find Exp Clin Pharmacol 26:789–799.
- Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685.
- Leff P. 1995. The two-state model of receptor activation. Trends Pharmacol Sci 16:89–97.
- Lohmann SM, Vaandrager AB, Smolenski A, Walter U, De Jonge HR. 1997. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem Sci 22:307–312.
- Lucchesi PA, Scheid CR, Romano FD, Kargacin ME, Mullikin-Kilpatrick D, Yamaguchi H, et al. 1990. Ligand binding and G protein coupling of muscarinic receptors in airway smooth muscle. Am J Physiol 258:C730–C738.
- Luo J, Busillo JM, Benovic JL. 2008. M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol 74:338–347.
- Mak JC, Barnes PJ. 1990. Autoradiographic visualization of muscarinic receptor subtypes in human and guinea pig lung. Am Rev Respir Dis 141:1559–1568.
- Michel AD, Whiting RL. 1988. Methoctramine reveals heterogeneity of M2 muscarinic receptors in longitudinal ileal smooth muscle membranes. Eur J Pharmacol 145:305–311.
- Michel AD, Stefanich E, Whiting RL. 1989. Direct labeling of rat M3-muscarinic receptors by [3H]-4DAMP. Eur J Pharmacol 166:459–466.
- McMillin SM, Heusel M, Liu T, Costanzi S, Wess J. 2011. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J Biol Chem 286:28584–28598.
- Milligan G. 2007. G protein-coupled receptor dimerisation: Molecular basis and relevance to function. Biochim Biophys Acta 1768–1825–835.
- Misle AJ, Lippo de Becemberg I, Gonzalez de Alfonzo R, Alfonzo MJ. 1994. Methoctramine binding sites sensitive to alkylation on muscarinic receptors from tracheal smooth muscle. Biochem Pharmacol 48:191–195.
- Misle AJ, Lippo de Becemberg I, Gonzalez de Alfonzo R, Alfonzo MJ. 1995. Subcellular distribution and pharmacological characterization of muscarinic receptors in tracheal smooth muscle. Acta Cient Venez 46:166–173.
- Murad F, Kimura H. 1974. Cyclic nucleotide levels in incubations of guinea pig trachea. Biochim Biophys Acta 343:275–286.
- Palczewski K. 2010. Oligomeric forms of G-protein coupled receptors (GPCRs). Trends Biochem Sci 35:595–600.
- Peterson GL, Schimerlik MI. 1984. Large scale preparation and characterization of membrane-bound and detergent-solubilized muscarinic acetylcholine receptor from pig atria. Prep Biochem 14:33–74.
- Pfeifer A, Aszodi A, Seidler U, Ruth P, Hofmann F, Fassler R. 1996. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science 274:2082–2086.
- Reddy H, Watson N, Ford AP, Eglen RM. 1995. Characterization of the interaction between muscarinic M2 receptors and beta-adrenoceptor subtypes in guinea-pig isolated ileum. Br J Pharmacol 114:49–56.
- Rovira X, Pin JP, Giraldo J. 2010. The asymmetric/symmetric activation of GPCR dimers as a possible mechanistic rationale for multiple signalling pathways. Trends Pharmacol Sci 31:15–21.
- Schultz G, Hardman JG, Schultz K, Baird CE, Sutherland EW. 1973. The importance of calcium ions for the regulation of guanosine 3′:5′-cyclic monophosphate levels. Proc Natl Acad Sci USA 70:3889–3893.
- Song GJ, Jones BW, Hinkle PM. 2007. Dimerization of the thyrotropin-releasing hormone receptor potentiates hormone-dependent receptor phosphorylation. Proc Natl Acad Sci USA 104:18303–18308.
- Tränkle C, Kostenis E, Mohr K. 2001. Muscarinic allosteric modulation: M2/M3 subtype selectivity of gallamine is independent of G-protein coupling specificity. Naunyn Schmiedebergs Arch Pharmacol 364:172–178.
- Uray FP, de Alfonzo RG, de Becemberg IL, Alfonzo MJ. 2010. Muscarinic agonists acting through M2 acetylcholine receptors stimulate the migration of an NO-sensitive guanylyl cyclase to the plasma membrane of bovine tracheal smooth muscle. J Recept Signal Transduct Res 30:10–23.
- Vaandrager AB, De Jonge HR. 1994. Effect of cyclic GMP on intestinal transport. Adv Pharmacol 26:253–283.
- Vaandrager AB, Edixhoven M, Bot AG, Kroos MA, Jarchau T, Lohmann S, et al. 1997. Endogenous type II cGMP-dependent protein kinase exists as a dimer in membranes and can be functionally distinguished from the type I isoforms. J Biol Chem 272:11816–11823.
- Wess J. 1996. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol 10:69–99.
- Wess J, Eglen RM, Gautam D. 2007. Muscarinic acetylcholine receptors: Mutant mice provide new insights for drug development. Nat Rev Drug Discov 6:721–733.
- White JF, Grodnitzky J, Louis JM, Trinh LB, Shiloach J, Gutierrez J, et al. 2007. Dimerization of the class A G protein-coupled neurotensin receptor NTS1 alters G protein interaction. Proc Nat Acad Sci USA 104:12199–12204.
- Willets JM, Chaliss RA, Kelly E, Nahorski SR. 2001. G protein-coupled receptor kinases 3 and 6 use different pathways to desensitize the endogenous M3 muscarinic acetylcholine receptor in human SH-SY5Y cells. Mol Pharmacol 60:321–330.
- Zeng Y, Wess J. 1999. Identification and molecular characterization of m3 muscarinic receptor dimmers. J Biol Chem 274:19487–19497.