Defective apoptosis is a fundamental hallmark feature of chronic lymphocytic leukemia (CLL) biology. Therefore, significant effort in cure of this disease is toward sensitizing malignant lymphocytes to programmed cell death. In parallel to mitochondrial oxidative stress, endoplasmic reticulum (ER) stress also triggers an essential pathway for B-CLL cell apoptosis, suggesting that genetic or pharmacologic manipulation of ER signaling could represent an important therapeutic strategy [Citation1,Citation2]. ER stress activated unfolded protein response (UPR) leads to up-regulation of downstream ER chaperone molecules such as PERK, eIF2α, glucose regulatory protein GRP78 and pro-apoptotic CHOP and direct activation of initiator caspase 4 [Citation3]. Once activated, caspase 4 in turn activates caspase 9 or 3, thereby leading to mitochondrial outer membrane permeabilization (MOMP)-independent cell death. Several anti-cancer agents including homoharringtonine, sorafenib, obatoclax and flavopiridol are identified to induce ER stress mediated, caspase- dependent apoptosis, as a caspase inhibitor in turn can reverse this process [Citation4,Citation5].
The regulatory role of the Bcl-2 family proteins in ER stress mediated signaling pathways has been well documented. Combining an ER stress inducing agent with a mitochondrial targeting agent demonstrated a synergistic effect, suggesting a link between these two pathways [Citation6]. Agents such as trichosanthin (TCS) and cardiotoxin III are shown to activate both mitochondrial and ER signaling pathways of apoptosis. While loss of the ATM gene was correlated with increased ER stress, activation of the MEK/ERK signaling pathway linked with high levels of oncogenes such as Mcl-1, Bcl-2 or Tcl-1 was inversely correlated with ER stress induced apoptosis. Inhibiting Mcl-1 with siRNA sensitized tumor cells to apoptosis initiated by ER stress inducers thapsigargin or tunicamysin [Citation7,Citation8].
In the past decade, extensive studies have established the connection between ER stress and autophagy in several tumor models (). While autophagy is activated by all classic stimuli of this process, only unfolded protein ER stress-mediated autophagy protects the cells from cell death. The connection between ER stress and autophagy was first established using the tyrosine kinase inhibitor imatinib, which was demonstrated to exhibit a cytoprotective effect by inducing autophagy and forming autophagosomes in chronic myeloid leukemia (CML) cells [Citation9]. This mechanism was indeed in association with the ER stress response, but not in reflection of cABL/Bcl-2 activities. Consistently, the combination of imatinib with autophagy inhibitors enhanced imatinib induced apoptosis [Citation10]. Mechanistically, the association between ER stress and autophagy was mediated through phosphorylation of eIFα, which impacts protein translation and mediates LC3 conversion and autophagosome formation, shifting the equilibrium toward a pro-survival mechanism [Citation11]. Importantly, abrogating autophagy restored cells vulnerable to ER stress, suggesting that autophagy plays an important role in cell survival after ER stress [Citation12].
Figure 1. Crosstalk between multiple pathways including mitochondrial stress, ER stress and autophagy. UPR, unfolded protein response; PERK, PKR-like ER-localized eIF2α kinase; EIF2α, translation initiation factor; CHOP, C/EBP-homologous protein.
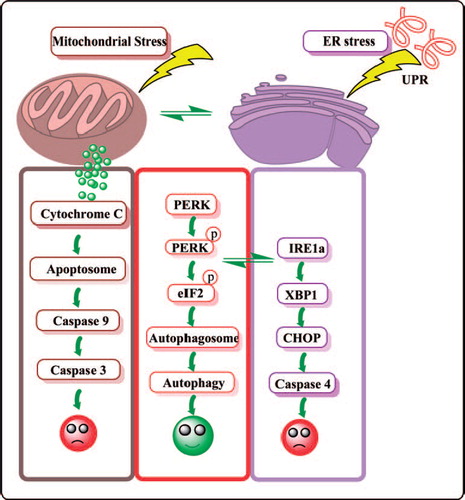
Mahoney et al. in this issue of Leukemia and Lymphoma demonstrate that nelfinavir, an ER stress inducing agent, exhibits modest apoptosis as a single agent in CLL [Citation13]. Detailed studies revealed that treatment with this agent also induced signature proteins of autophagic response, leading to pro-survival mechanisms [Citation13]. Nelfinavir causes two types of cell death programs, caspase dependent and caspase independent apoptosis characterized by induction of ER stress and autophagy. Autophagy was regarded as a pro-survival mechanism because an inhibitor of autophagy combined with nelfinavir increased nelfinavir-induced cell death in lung cancer cells [Citation14]. This proof-of-principle is now established in a CLL model system, proposing a new paradigm of treating CLL. These results engender several unanswered questions. While authors report up-regulation of IRE1, whether ATF6 and PERK mediated signal transduction axes [Citation15] become activated in CLL cells after nelfinavir treatment is unknown. What could be the effect of nelfinavir on mRNA translation, which appears to be a primary mode of action of ER stress inducing agents [Citation16]? As unmitigated ER stress can eventually lead to cell death [Citation15], does extended treatment with nelfinavir result in apoptosis? What could be the biological effect of nelfinavir in normal lymphocytes? Finally, the novel B cell receptor (BCR) targeting kinase inhibitors that are currently in clinical trial for CLL including GS-1101 and ibrutinib induce modest apoptosis in vitro. Do these agents induce ER stress and/or autophagy? Is lymphocytosis observed following treatment an indication of the mechanism of survival? Screening these compounds for ER stress response or autophagic response might provide additional information on the mechanism of action of these agents and combination strategies.
Supplementary Material
Download Zip (978.4 KB)Potential conflict of interest:
Disclosure forms provided by the authors are available with the full text of this article at http://www.informahealthcare.com/lal.
References
- Lust S, Vanhoecke B, Van Gele M, et al. Xanthohumol activates the proapoptotic arm of the unfolded protein response in chronic lymphocytic leukemia. Anticancer Res 2009;29:3797–3805.
- Rosati E, Sabatini R, Rampino G, et al. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 2010;116: 2713–2723.
- Carew JS, Nawrocki ST, Krupnik YV, et al. Targeting endoplasmic reticulum protein transport:a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood 2006;107:222–231.
- Jie H, Donghua H, Xingkui X, et al. Homoharringtonine-induced apoptosis of MDS cell line MUTZ-1 cells is mediated by the endoplasmic reticulum stress pathway. Leuk Lymphoma 2007;48:964–977.
- Rahmani M, Davis EM, Crabtree TR, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol 2007;27:5499–5513.
- Du Y, Wang K, Fang H, et al. Coordination of intrinsic, extrinsic, and endoplasmic reticulum-mediated apoptosis by imatinib mesylate combined with arsenic trioxide in chronic myeloid leukemia. Blood 2006;107:1582–1590.
- Jiang CC, Lucas K, Avery-Kiejda KA, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res 2008;68:6708–6717.
- Kriss CL, Pinilla-Ibarz JA, Mailloux AW, et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response:a novel mechanism of leukemic progression in mice. Blood 2012;120: 1027–1238.
- Ohtomo T, Miyazawa K, Naito M, et al. Cytoprotective effect of imatinib mesylate in non-BCR-ABL-expressing cells along with autophagosome formation. Biochem Biophys Res Commun 2010;391:310–315.
- Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009;119:1109–1123.
- Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007;14:230–239.
- Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006;26: 9220–9231.
- Mahoney E, Maddocks K, Flynn J, et al. Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: a new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk Lymphoma 2013;54: 2685–2692.
- Gills JJ, Lopiccolo J, Tsurutani J, et al. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res 2007;13:5183–5194.
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086.
- Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 2012;3:307–321.