Abstract
Advent of recombinant technology in protein synthesis has given birth to a new range of biopharmaceuticals. These therapeutic peptides and proteins are now emerging as an imperative part of various treatment protocols especially in the cancer therapeutics. Despite extensive research efforts, oral delivery of therapeutic peptide or protein is still a challenge for pharmaceutical industries and researchers. Number of factors including high proteolytic activity and low pH conditions of gastrointestinal tract act as major barriers in the successful delivery of intact protein/peptide to the targeted site. Low permeability of protein/peptide across the intestinal barrier is also a factor adding to the low bioavailability. Therefore, because of the short circulatory half-life exhibited by peptides in vivo, they need to be administered frequently resulting in increased cost of treatment and low patient compliance. Nano-carrier-based delivery presents an appropriate choice of drug carriers owing to their property to protect proteins from degradation by the low pH conditions in stomach or by the proteolytic enzymes in the gastrointestinal tract. This review focuses on recent aspects and patents on oral delivery of therapeutic proteins and peptides with special emphasis on nano-carrier-based approach.
Introduction
The successful use of proteins and peptides in treating some of the dreadful diseases including cancer has revolutionized the pharmaceutical as well as biotechnological world. This is because of the targeted nature of pharmacologically active proteins and peptides, i.e. treatment of disease instead of the mere symptoms unlike blockbuster chemical drugs. Till date, the delivery of these therapeutic protein/peptides is restricted majorly to parentral route due to short circulatory half-life and oral bioavailability (Rabanel et al., Citation2012). Chemical modifications like addition of novel functional group or protease inhibitors have been successfully used to increase the bioavailability of protein drugs in the laboratory although they still show some safety concerns when administered in vivo. Therefore, there have been several attempts to administer these proteins and peptides by a non-invasive route.
There is a need to design a delivery system that not only protects the protein/peptide from enzymatic degradation but also aid in enhancing its absorption without altering its biological activity. Nano-carriers are promising carrier systems for delivery of proteins/peptides. They exhibit high permeability properties because of their size range and high surface to volume ratio. The surface of these particles can be modified towards hydrophilic or lipophillic properties. These systems enhance the stability of proteins/peptides in vivo leading to increased circulation half lives in the body. illustrates the various stages involved in the absorption of nano-encapsulated protein/peptide when administered orally. By incorporating mucoadhesive properties, the residence time of these systems can be prolonged and the active can reach the site of action in effective concentrations. The following section discusses these systems in details. summarizes some of the patents related to delivery of therapeutic proteins through oral administration using nano-carriers.
Figure 1. Nano-carrier-based approach for the successful oral delivery of protein/peptide drugs.
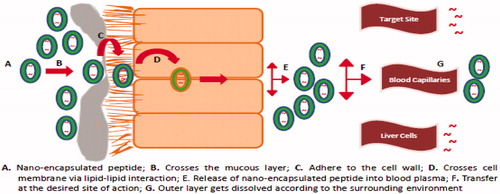
Table 1. Some of the patents related to the oral delivery of proteins and peptides.
Nano-carriers
Nanoparticles
Nanoparticles have emerged as potential drug delivery systems and are used for targeting of therapeutic peptides to particular organs/tissues via the peroral route of administration. Significant work is being done to modify the nanoparticles for better understanding of the factors affecting mucoadhesion and their subsequent uptake by the GI tract. Florence and group have reported the role of various biodegradable polymers in increasing the bioavailability of nanoparticles. In one of the studies performed by this group, approximately 10% of orally administered invasin-coated latex nanoparticles (500 nm in size) were found in the systemic circulation of rats (Hussain & Florence, Citation1998). Their work suggested the existence of optimum colloidal size for efficient entrapment of nanoparticles within the mucous and for epithelial cells to grip the particulates to trigger endocytic events (Florence & Hussain, Citation2001).
Polymeric nanoparticles
In order to improve the oral bioavailability of proteins and peptides, new range of biodegradable polymeric nanoparticles has been used which can enhance the stability, control the release and the pharmacokinetic parameters. Moreover, the surface of these polymers can be easily functionalized towards off opsonization (Chan et al., Citation2010). They are also known to show reduced toxicity in the peripheral healthy tissues. The protein to be delivered can be dissolved, entrapped or encapsulated and then delivered to the target area.
Kafka and group prepared polymeric nanoparticles and investigated the use of poly(ethylcyanoacrylate) as a polymer. These nanoparticles were loaded with chemical sterilant D-Lys6-Gonadotropin releasing hormone. These were studied under both in vivo and in vitro conditions. In vitro conditions tested were the artificial gastric juice, simulated intestinal fluids and brushtail possum plasma. The D-Lys6-Gonadotropin releasing hormone was intact in all the three conditions while the amount of released bioactive component varied. Less than 5% of the bioactive was released over 6 h in artificial gastric juice and simulated intestinal fluid in contrast to 60% that is released over 1 h in brushtail possum plasma. The nanoparticles were also administered in vivo into the caecum of the brushtail possum. Sufficient amount of the bioactive component was released into the systemic circulation. It was evident from the release of the luteinizing hormone that a significant amount also reached to the pituitary gland (Kafka et al., Citation2011).
Mucoadhesive polymeric nanoparticles
Clearance of some drugs delivered through alimentary tract is very fast for them to be properly absorbed. Mucoadhesion allows the drug to be attached to the mucous membrane and thus helps in increasing the residence time. The bioadhesive nature allows it to be protected from the mucociliary clearance system leading to an increase in bioavailability of the drug. The mucous membrane is relatively permeable and thus allows faster absorption of drug molecule (Carvalho et al., Citation2010).
Successful delivery of intact protein/peptide across the GI tract is directly related to the contact between the drug delivery system and the tissue surface, i.e. the residence time. Therefore, the use of adhesive as a part of drug delivery system may quantitatively increase its residence time, specifically for the oral cavity (Peppas & Robinson, Citation1995). A list of polymers and their specific features that are used to coat the nanoparticles is given in .
Table 2. Polymers used for the coating of nano-carriers encapsulating proteins and peptides.
Many prevailing mucoadhesive carriers have also been further modified to make them more effective. Graft co-polymers can be utilized for the oral delivery of peptides and therapeutics. Marks et al. formulated polymers from poly-ethylene glycol and aldehyde-modified poly ethylene glycol. Different percentages of aldehyde-modified poly ethylene glycol used were 0.06%, 0.6% and 3.3%. The swelling, release and adhesion studies were carried out for all the aldehyde formulations. Aldehyde modified with 3.3% of poly ethylene glycol formulation displayed a release of ∼80% after 3 h and adhesion test results showed an increase of 10–30 µJ over the other two formulations. The probable reason could be that aldehyde binds to the amines of the glycoproteins found in the mucous layer and makes poly-ethylene glycol more viable for effective delivery of proteins/peptides (Marks & Lowman, Citation2011).
Permeation enhancers for the mucoadhesive nanoparticles have been investigated by Makhlof and his associates. Polyelectrolyte complexes were prepared by the interaction of spermine and polyacrylic acid. The results for permeation enhancement were studied using fluoroscein isothiocyanate dextran for both in vitro (caco-2 cell line) and in vivo conditions. Confocal microscopy revealed a strong and prolonged penetration of fluoroscein isothiocyanate dextran when loaded with spermine-polyacrylic acid nanoparticles (5.56-folds) as compared to free fluoroscein isothiocyanate dextran. Cytotoxicity carried out on caco-2 cell lines revealed the safety of delivery system at the concentration used for permeation enhancement. Flouresceine-labeled polymer helped to study the strong association of the polyelectrolyte complex to caco-2 cells. The complex was also efficient in delivering calcitonin to rats when administered orally, as significant reduction in blood calcemia was observed. In comparison to spermine solution, Mg-polyacrylic acid nanoparticles and free drug, spermine-polyacrylic acid nanoparticles encapsulating calcitonin showed improved hypocalcemic effects by 2.7, 2.8 and 15.2 folds, respectively (Makhlof et al., Citation2011c).
Dunnhaupta et al. evaluated and compared the distribution of thiolated mucoadhesive poly acrylic acid and chitosan nanoparticles on intestinal mucosa. These nanoparticles were further modified by conjugation with cysteine and 2-iminothiolane. Nanoparticles were prepared by ionic gelation and labeled with fluorescent dye Alexa Fluor 488 and fluorescein diacetate. Unmodified and modified polymeric nanoparticles were examined in vitro for their diffusion behavior through natural porcine intestinal mucus and displayed 526 μmol/g for chitosan and 513 μmol/g poly acrylic acid of free thiol groups therefore, suggesting that mucoadhesion of thiolated nanoparticles is higher than the diffusion into the intestinal mucosa. Modified nanoparticles displayed ∼6-fold increase in mucoadhesion compared to unmodified ones. Chitosan–iminothiolane complex showed 2-fold higher mucoadhesive properties compared to poly acrylic acid–cysteine nanoparticles (Dunnhaupta et al., Citation2011).
In another study by Lichen Yina, the mucoadhesion and drug permeation of nanoparticles have been improved by using trimethyl chitosan–cysteine conjugate. The said polymeric conjugate of different molecular weights was combined with insulin by self assembly method to form the polyelectrolyte complex. The formulation showed better mucoadhesion (approximately 2–5 folds) and increased insulin transport (1.7–2.6 folds) as compared to trimethyl chitosan/insulin nanoparticles. The increase in mucoadhesion properties was attributed to the disulfide linkages formed between trimethyl chitosan and the mucin. In vitro studies showed a considerable increase in modified insulin internalization (1.7–3.0 folds) and uptake by the Peyer’s patches (1.7–5.0 folds). Biocompatibility assessment studies concluded that it could be used as a safe oral delivery system with enhanced mucoahesion and permeability characteristics (Yina et al., Citation2009).
Quaternization of nanoparticles can make them more favourable for use as a drug delivery system. Amphiphilic polyallylamine polymers were synthesized and subsequently quaternized with methyl iodide. The quaternized polymers provided better protection from the proteolytic enzymes with reasonably high complexation efficiency of around 78–93%. The quaternized polymers also showed low toxicity levels (15-fold improvement in IC50 value) as compared to polyallylamine (Thompson et al., Citation2009).
Chitosan-based polymeric nanoparticles
Chitosan is a cationic polymer that is derived from alkaline deacetylation of chitin. The positive charge over chitosan interacts with negatively charged sialic acid residues of mucosal surfaces leading to mucoadhesion. Chitosan-based nanoparticles have been primarily used for delivery of low molecular weight proteins and peptides (Liu et al., Citation2007).
Trapani et al. prepared chitosan-based nanoparticles for oral administration of glutathione. The formulation was composed of three type of nanoparticles – based on chitosan, chitosan/cyclodextrin and chitosan/sulphobutyl ether-β-cyclodextrin (SBE7m-β-CD). After studying the loading efficiency it was seen that glutathione was loaded more efficiently into the SBE7m-β-CD nanoparticles (25% higher) as glutathione forms a complex with cyclodextrin. However, studies carried out in frog intestine model confirmed that both chitosan and chitosan/cyclodextrin nanoparticles could induce 1.4 times better permeabilization of the intestinal epithilia while chitosan/(SBE7m-β-CD) nanoparticles displayed 2-fold higher absorption capability in all the segments of the duodenum as compared to the control (Trapani et al., Citation2010).
Chitosan-based nanoparticles despite having good bio-adhesion, biocompatibility and permeation enhancing capabilities; their use is limited because of instability in acidic environments of GI tract.
To overcome the problem of degradation of chiotsan in acidic environment, Rekha et al. used a chitosan derivative lauryl succinyl chitosan for oral administration of insulin using a cross-linking polymer sodium tripolyphosphate. Release kinetics showed miroparticles were stable in acidic pH. The stability was conferred to the presence of succinyl carboxyl groups. Hydrophobic moieties were responsible for controlling the release of insulin under the intestinal pH. Confocal microscopic studies showed permeation of the modified chitosan particles from the caco-2 cell lines. In vivo studies performed in diabetic rats indicated the reduction in blood glucose levels for ∼6 h when administered with lauryl succinyl chitosan nanoparticles. The modified nanoparticles with hydrophobic moieties were responsible for enhanced mucoadhesion and permeability as compared to unmodified chitosan-based nanoparticles (Rekha & Sharma, Citation2009).
In another attempt to confer acid stability to chitosan nanoparticles, cross linking with a pH sensitive polymer, hydroxypropyl methylcellulose phthalate was used. The peptide was stable and had significant biological activity in the acidic environment. Quantitative fluorescence analysis and confocal microscopy studies showed fluorescently-labeled chitosan-hydroxypropyl methylcellulose phthalate nanoparticles showed 2–4-fold increase in intestinal mucoadhesion and penetration compared to chitosan-tripolyphosphate nanoparticles. On oral administration of chitosan-hydroxypropyl methylcellulose phthalate nanoparticles, there was an increase in hypoglycemic effect when compared to oral insulin solution (9.8-folds) and insulin-loaded chitosan-tripolyphosphate nanoparticles (2.8-folds) (Makhlof et al., Citation2011b).
A polymeric protease inhibitor-based delivery system composed of chitosan–aprotinin conjugate complexed with negatively charged multilamellar vesicles was developed by Werle et al. This complex was compared with the chitosan-coated multilamellar vesicles. At different concentrations of chitosan, the conjugate had significant capability of inhibiting the enzyme trypsin. The chitosan–aprotinin-coated multilamellar vesicles and chitosan-coated multilamellar vesicles both have considerable mucoadhesive properties. The nanoparticles were tested for the delivery of calcitonin upon oral administration in rats and both the type of complexes showed a significant increase in the blood calcium concentration–time curve, i.e. 11-fold increase for calcitonin-loaded chitosan-coated multilamellar vesicles and 15-fold increase for calcitonin-loaded chitosan–aprotinin-coated multilamellar vesicles, respectively (Werle & Takeuchi, Citation2009).
Jin et al. formulated thymopentin-loaded poly (butyl cyanoacrylate) nanoparticles by using optimized emulsion polymerization and these nanoparticles were further coated with chitosan–glutathione conjugate utilizing electrostatic interactions. Their efficacies for oral delivery were evaluated before and after coating with chitosan or chitosan–glutathione conjugate. Results showed pH-dependent release profiles; in simulated gastric fluid, 20% of thymopentin was released from polymeric nanoparticles in 3 h as compared to ∼95% for thymopentin solution while at pH 6.8, 78.45 ± 2.3% of drug was released in 3 h from polymeric nanoparticles as compared to 94.63 ± 1.7% for drug solution. These results indicated the affinity of thymopentin with nanoparticles at low pH. Furthermore, chitosan-coated thymopentin-loaded poly (butyl cyanoacrylate) nanoparticles were able to restore the T lymphocyte counts in immune dysfunctioned rats. These results showed the potency of the nanoparticles to improve the bioavailability of orally administered peptides and thus a potential drug delivery system (Jin et al., Citation2011).
Hydrogels have also been loaded with nanoparticles in order to utilize their pH dependent characteristics. Polymeric nanoparticles consisting of the carboxylated chitosan-grafted nanoparticles were introduced into a bilaminated hydrogel made up with alginate-Ca2+ mucoadhesive layer and one hydrophobic layer. The model peptide used in this case was calcein which was entrapped within the above polymeric nanoparticles. The polymer showed good mucoahesion (∼6000 N/m2) and pH sensitivity. The release of peptide that was restricted in the acidic environment (4%) can be achieved even in the neutral environment (∼95%) due to the pH sensitivity of hydrogels (He et al., Citation2009).
Nanocomplexes
Nanocomplexes can be formed by the electrostatic interactions that generally occur between cationic drugs and anionic polymers, thereby further increasing the shelf life of the encapsulated agent (Yousefpour et al., Citation2011).
Nanocomplexes formed from salmon calcitonin and amphiphilic polyelectrolyte, poly(allyl)amine, grafted with palmitoyl and quaternary ammonium moieties have been characterized both in vivo and in vitro as a novel peptide delivery system. The complex consists of vesicular bilayer with an aqueous core. The physical stability of the nanocomplex can be maintained up to 7 days. The nanocomplexes were found to be more resistant to peptidases than free salmon calcitonin and retained 33% activity even after a period of 7 days. The nanocomplexes were also tested for both intravenous and intra-jejunal administration in rats. Free and complexed salmon calcitonin reduced serum levels over 120 min upon intravenous and over 240 min after intra-jejunal administration, respectively. Therefore, suggesting an improved efficacy of these nanocomplexes during oral administration (Cheng et al., Citation2010).
In a recent study, nanocomplexes were formed using poly(allylamine) and insulin. The complexes were further modified with either cetyl or cholesteryl groups to form quaternized derivatives. The complexation efficiency was found to be 86.5% at polymer:insulin ratio of 0.4:1 mg/ml and 44.9% for higher ratio of 2:1 mg/ml, respectively. Further, it was observed that the polymer architecture played a significant role in providing protection against α-chymotrypsin and pepsin. Cholesteryl polymers were effective against α-chymotrypsin while cetyl polymers provided protection from pepsin degradation of insulin (Thompson et al., Citation2010).
Owing to the increasing demand of salmon calcitonin used for osteoporosis, salmon calcitonin–sodium tripolyphosphate ionic complex was synthesized and characterized as an oral delivery vehicle. To optimize the ionic complex, the effect of incubation time and molar ratio of the two components was evaluated and was observed to be ranging in between 1:5 and 1:10. The complexation efficiency was about 95%. Using SEM images, it was observed that the freeze-dried ionic complex had rough morphology on their surface and the particle size in phosphate buffer saline (pH 7.4) was about 220 nm. Differential scanning calorimetry and FT-IR results provided evidences for ionic interaction between –NH2 groups and –P = O groups of salmon calcitonin and sodium tripolyphosphate, respectively. The release profile of salmon calcitonin depicted a sustained release for 3 weeks. In vivo studies performed on rats confirmed its ability to prevent enzymatic attack and maintain a continuous hypocalcemic effect. Hence, osteoporosis can be addressed effectively using this novel formulation (Lee et al., Citation2010).
Microparticles
Micro-particulate delivery system can provide the sustained and controlled release of drug for long period of time. They are also a beneficial way of delivering proteins or peptides that are otherwise difficult to deliver due to limited solubility in water. Microparticles are small particles of solid or small droplets of liquid surrounded by a natural/synthetic polymer and having diameter ranging up to 0.1–200 µm (Madhav & Kala, Citation2011).
Kilpelainen and associates tested the use of nanostructured porous silicon microparticles for the sustained and prolonged release of the model peptide melanotan II for both in vivo and in vitro conditions to evaluate its efficacy. A peptide-loading degree of 15% (w/w) was achieved by the subcutaneous administration of the drug. In vitro and in vivo studies showed sustained release of the drug from the microparticles. In vivo, melanotan II microparticles induced an increase in heart rate 2 h later which lasted for 1 h longer than melanotan II solution (Kilpelainen et al., Citation2011).
Suksamran et al. prepared bovine serum albumin (BSA)-loaded alginate microparticles by cross-linking alginate with calcium chloride solution using an electrohydrodynamic spraying technique. Initially, BSA of 5, 10, 20, 40 and 60% w/w to polymer was incorporated into these alginate microparticles. The results of entrapment efficiency revealed that with initial BSA 10% w/w showed the highest entrapment efficiency of 49.70 ± 0.01%. The result of BSA content revealed that with the initial BSA of 20% w/w showed the highest amount of BSA content of 3.92 ± 0.02 mg/g of particles. It was found that with the initial BSA of 40% w/w showed the slowest release rate (60% over a period of 24 h) and sustained release (Suksamran et al., Citation2009).
In order to prolong the bioavailability of insulin in GI tract, magnetite nanocrystals and insulin were coencapsulated into poly(lactide-co-glycolide) (PLGA) microparticles by Cheng et al. and their effects on hypoglycemia were evaluated in mice in the presence of a circumferentially applied external magnetic field. Results indicated that 100 U/kg of insulin-magnetite-PLGA microparticles when administered to fasted mice reduced the blood glucose levels up to 43.8% in the presence of an external magnetic field for 20 h. Bioavailability of insulin-magnetite-PLGA microparticles was found to be 2.77 ± 0.46 and 0.87 ± 0.29% based on glucose and ELISA assay, respectively, which was higher than similarly dosed mice without a magnetic field (0.66 ± 0.56 and 0.30 ± 0.06%). Therefore, a substantially improved hypoglycemic effect was observed in mice that were orally administered with insulin-magnetite-PLGA microparticles in the presence of an external magnetic field (Cheng et al., Citation2006).
A chitosan derivative, N-(2-hydroxyl) propyl-3-trimethyl ammonium chitosan for the oral delivery of insulin was synthesized by coupling glycidyl trimethylammonium chloride to chitosan in an aqueous medium. MTT (dimethyl thiazolyl diphenyl tetrazolium salt) assay employed to study its cytotoxicity showed its in vitro safety till the concentration of 2.5 mg/ml. Adhesion studies on mucin and freshly excised rat intestinal sections were carried and the results indicated that the different compositions of polymer (1:0.5 and 1:1) exhibited adhesion of 86% and 95%, respectively, as compared to 72% for native chitosan. Insulin loading efficiency of these polymeric microparticles was found to be 65%. All the above studies indicated that these polymeric microparticles can serve for the mucoadhesive and sustained oral release of encapsulated proteins and peptides (Sonia & Sharma, Citation2011).
Microspheres
Microspheres are discrete spherical particles ranging from 1 to 50 microns. In a recent study, an alginate-chitosan microsphere was developed by membrane emulsification using calcium ion and polymer solidification. The particle size distribution, surface morphology and the zeta potential of the microsphere was studied in detail. Due to the positive results, they were then successfully used as vectors for oral delivery of insulin peptide. The loading efficiency was observed to be 56.7%, while the immunological activity was a noteworthy 99.4%. An in vitro model having simulated GI conditions and another with that of the blood was used to test the release profile. While only 32% was found to be released during the transit time in stomach and intestine (2 h + 4 h), it was maintained for a longer period of time in the blood (14 days). Moreover, the structure remained stable during absorption. Therefore, the blood glucose level of diabetic rats was effectively reduced and kept at a stable level for about 60 h at a stretch (Zhang et al., Citation2011).
Lipid-based delivery systems
Liposomes
Liposomes consist of phospholipid bilayers (lamellas) which help to encapsulate the proteins or the peptides within its lipid core. The method of preparation of these liposomes regulates the size and shape of the liposomes. Liposomes are classified into large unilamellar, small unilamellar and multilamellar. Some of the clinically approved liposomal-based drugs are liposomal amphotericin, liposomal doxorubicin (Sharma & Sharma, Citation2011).
Liposomes are reported to protect the encapsulated drug/peptide from oxidation and deamidation. Moreover, liposomes encapsulating drug/peptide are known to be up taken by the lymphatic system which drains them directly into the systemic circulation thereby by-passing first pass metabolism (Porter & Charman, Citation1997).
The advances that have been reported in the recent past for oral delivery of peptides are the wheat germ agglutinin–carbopol-modified liposomes where the wheat germ agglutinin is covalently linked to the carbopol molecule. The uptake of modified liposomes was better as compared to the unmodified ones by caco-2 cells without causing any cytotoxicity. Extent of internalization of modified liposomes depends upon the concentration of wheat germ agglutinin, temperature and incubation period. Pharmacological efficacy of calcitonin-loaded wheat germ agglutinin–carbopol-modified liposomes upon oral administration was enhanced by 20-folds and 3-folds when compared to non-modified and carbopol-modified liposomes. Therefore, both in vitro and in vivo analyses support its use as an efficient oral peptide delivery system (Makhlof et al., Citation2011a).
Bilosomes are novel colloidal delivery systems (similar to niosomes) that are formed by incorporating bile salts into the vesicular lipid bilayer membrane. Bilosomes containing diphtheria toxoid were prepared by Shukla et al. using thin-film hydration method. Results of this study demonstrated that orally administered nano-bilosomes produced comparable anti-diphtheria toxoid IgG levels in serum to those induced by intra muscular (i.m.) alum-adsorbed diphtheria toxoid at a 4-fold higher dose without inducing tolerance. Therefore, this approach provided an effective immune protection against diphtheria with good patient compliance (Shukla et al., Citation2011).
Archaeosomes
Archaeosomes are based on a lipid-based delivery system and are made of a polar lipid fraction E extracted from Sulfolobus acidocaldarius. Archaeosomes provide protection to the peptides in the GI tract, and therefore allow the peptide to stay in the GI tract for a longer period of time (12 h) as confirmed by fluorescence imaging. In the artificial intra-intestinal fluid, the released amounts of insulin from conventional liposomes reached about 92% within 4 h as compared to ∼70% of insulin released from the archeosomes within the same incubation time. Archaeosomes containing insulin lowered the blood glucose levels to a greater extent as compared to the conventional liposome formulations. However, the transport of the insulin across the caco-2 cell line is not much affected by the use of archaeosomes (Li et al., Citation2010).
Emulsion system
An emulsion is a mixture of two or more liquids in which one is dispersed in the other as microscopic or ultramicroscopic droplets. Whether an emulsion turns into a water-in-oil emulsion or an oil-in-water emulsion depends on the volume fraction of both phases and on the type of emulsifier. These can be used to produce reverse micelles by maintaining optimum conditions. Using these aggregates present in oil, a drug molecule can be transported to the desired target site (Gupta et al., Citation2011).
A recent advance was made in this field by using water-in-oil emulsions to prepare phospholipid-based anhydrous reverse micelles for oral delivery of peptides. Using freeze-drying technique on water-in-oil emulsions, hydrophilic peptide-containing oily formulations were synthesized. An aqueous phase containing insulin and an oily phase containing phosphatidylcholine were emulsified and afterwards lyophilized. Dynamic light scattering showed that an anhydrous reverse micelle system of 20 nm insulin nanoparticles was formed. The lyophilates that were formed on addition of the oil to the solution contained these insulin nanoparticles. This was further confirmed by SEM, small angle X-ray scattering and DSC analysis. Administration of insulin-containing anhydrous reverse micelles to fasting diabetic rats significantly reduced the plasma glucose level. Moreover, drug release was slow and less than 12% was released after 24 h. This confirmed the bioactivity and the potential of phospholipid-based oily formulations (Wang et al., Citation2010).
Self-double-emulsifying drug delivery system (SDEDDS)
To address the major issue of concern related to the instability of the water-in-oil-in-water (w/o/w) double emulsions, a group recently developed novel SDEDDS by putting together a mixture of hydrophilic surfactants and water-in-oil (w/o) emulsions. This was used to increase the oral absorption of pidotimod (a synthetic dipeptide molecule) with high solubility and low permeability. The complex formed between pidotimod and SDEDDS was found to be stable up to 6 months under 25 °C. Plasma concentration–time profiles from pharmacokinetic studies in rats dosed with SDEDDS showed 2.56-fold increased absorption of pidotimod, compared to the pidotimod solution. Histopathological studies also showed that the drug was being absorbed without any side-effects (Qi et al., Citation2011).
Pulsatile devices
Pulsatile drug delivery systems (PDDS) provide a rapid and transient release of an active molecule within a short time period. The pulsatile effect, i.e. the release of the drug as a pulse after a lag time has to be designed in such a way that complete and rapid drug release should follow the lag time. This method can be highly effective in case of asthma, peptic ulcer, cardiovascular diseases, arthritis, attention deficit syndrome in children, and hypercholesterolemia. PDDS can be controlled by time, stimulus or external factors. The stimuli could be the pH or enzymes present in the intestinal tract or drug delivery system itself. In an externally regulated system, release is programmed by external stimuli like magnetism, ultrasound, electrical effect and irradiation.
PDDS has gained importance mainly due to a better understanding of a human body and the detailed pathophysiology of diseases. Degradation of peptides by the gastric enzymes, high molecular weight of the proteins, permeation and absorption are the major limitations that can be surmounted by the use of PDDS. Adjuvants can be timed in such a manner that they get released before the peptide. In this way, an environment complimenting to the needs of a peptide can be achieved.
Recently, a system of this kind was proposed for the oral delivery of insulin. The device consisted of a drug containing core and an inner swellable coating of hydroxypropyl methyl cellulose coating, an intermediate adjuvant layer, and an additional outer polymer coating. Using the technique of spray coating, this coat along with the protease inhibitor camostat mesilate/absorption enhancer sodium glycocholate was applied to the cores. The release performance was observed to be well correlated with the said polymer coating level. Hence, this system paved way for a two-pulse catatonic release of insulin along with a protease inhibitor and absorption enhancer (Del Curto et al., Citation2011).
Thioether formulation
After oral administration, the peptide needs to travel through different compartments of the GI-tract that result in their degradation. Therefore, to avoid the degradation of protein/peptide, one of the latest approaches is the modification of the peptide to make it resistant against breakdown or to facilitate its translocation or uptake.
A simple thioether group can be defined as a group of atoms containing single sulphur between two carbons.
Using thioether group as stabilizing agents in a reaction, an analog of the peptide hormone angiotensin was generated by de Vries et al. As proteolytic degradation is the main concern of oral delivery, this analog was made resistant to the degradation in circulation. In vitro, the peptide was subjected to the simulated oral ingestion conditions and it was observed that cAng was stable at pH 2.0. Furthermore, it showed resistance to the pancreatic proteases at a pH of 7.4 and biliary proteases at the lysosomal pH of 5.0. In vivo, it was detected that cAng can be delivered orally and via the pulmonary route, with bioavailability of 0.28 ± 0.05% and 28 ± 5%, respectively, whereas the drug uptake after a subcutaneous administration of the same formulation was remarkably higher (98 ± 6%). However, the therapeutic concentrations could be reached via all three routes of administration but oral route, being non invasive, is the most preferred route out of the three. Therefore, the introduction of a thioether bridge in the peptides can be utilized to improve their half-life (de Vries et al., Citation2010).
Cell penetrating peptide (CPP)
CPP helps in the intracellular delivery of the macromolecules. It can be used to deliver the therapeutic macromolecule across the intestinal mucosa, thus increasing its absorption and bioavailability. Some of these are HIV-1 Tat, penetratin and oligoarginine. One of the other important features of this delivery system is the easy co-administration of CPP with the drug through intermolecular interaction. Oral drug delivery system based on CPP thus can be used to make oral formulations for therapeutic peptides for their delivery through the oral route and increase its absorption through the intestinal mucosa. A further advantage of this promising strategy is that through intermolecular interaction between CPPs and drugs, successful non-invasive absorption could be achieved (Khafagy & Morishita, Citation2012).
Schwarze and co-workers evaluated the in vivo absorption of β-galactosidase conjugated to CPPs. The i.p. injection of β-galactosidase, fused to HIV TAT protein, resulted in delivery of the biologically active fusion protein to all tissues in mice. The injection of 1 mg of a TAT fusion protein per kilogram of body weight, daily to a mouse for 14 consecutive days produced no signs of neurological problems or systemic distress (Schwarze et al., Citation1999).
In another study performed by Liang et al. prepared FITC-labeled insulin covalently conjugated with TAT fusion protein and evaluated its absorption across the epithelial cell layer in the GI mucosa. The intestinal absorption of FITC-insulin and FITC-insulin/TAT conjugates was compared in vitro in a caco-2 cell monolayer. Results indicated that the transporting efficiency of the insulin/TAT conjugate was 5–8 times higher than that of free insulin (Liang & Yang, Citation2005).
Conclusion
Oral delivery of therapeutic proteins/peptides has always been a significant challenge for researchers due to their poor plasma levels. Owing to the related advantages including high patient compliance and ease of administration, oral delivery is far most favored route. In recent years, there has been a considerable growth in the development of non-invasive delivery systems for biopharmaceuticals. However, currently there are only a few oral peptide/protein formulations in the market but the ongoing research in this area promises a new array of techniques that could support the successful administration without any chemical/physical modification of active ingredient. As discussed in the review, nanotechnology offers a number of vehicles to encapsulate the peptide-based drug. Mucoadhesion-based nanoparticles and liposome are centered more towards increasing the absorption of the drug by attachment with the mucosal layers of the GI tract. However, permeation efficiency is improved with this technique but the protection of the drug from the proteases and low pH environment is still an issue that requires imperative consideration. The delivery systems based on polymeric nanoparticles and liposomes protect the therapeutic peptide from the action of proteases and often are based on the pH-based release of the protein/peptide so that it is released in a neutral pH environment and protected from degradation. Although, chitosan-based nanoparticles have good permeability and allow controlled release of the drugs but mainly efficient for the delivery of low molecular weight proteins. The chitosan derivatives have good mucoadhesive property and also help in maintaining the activity of the protein/peptide. SLNs and silicon-based nanoparticles show controlled and site specific delivery of the therapeutic peptides. Micelles show site specificity and help in maintaining the stability and bioavailability of the drugs. Archaesomes provide protection in the GI tract similar to that of a lipid-based delivery system. Microparticles allow the steady and prolong release of the drugs and the microspheres help in maintaining the activity of the drug and also allow the sustained release of the drug. The complex formed in the case of SDEDDs is stable for several months that allow achieving a good pharmacokinetic profile of the protein/peptide being delivered. Thioether formulations are resistant to low pH and are protected from pancreatic proteases. CPPs help in increasing the absorption of the drug across the intestinal mucosa leading to good bioavailability.
In conclusion, due to the progress in the development of oral drug delivery systems in recent years, several therapeutic peptides might be administered via the oral route in the near future. Further, there are many factors like the individual variations, toxicity, protein quality, site specific delivery that are required to be addressed. On a closer analysis of all the delivery systems, it is difficult to narrow down to few delivery mechanisms that may incorporate all aspects of improving the pharmacokinetic profile of the proteins/peptides. Therefore, incorporation of all the factors mentioned above in a single delivery system with efficient and commercial viability is still to be worked upon in the labs.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgements
We thank Department of Biotechnology, Jaypee Institute of Information Technology, Noida, U.P., India.
References
- Bernstein H, Morrel E, Mathiowitz E, et al.; Alkermes Controlled Therapeutics, Inc., assignee. Protein microspheres and methods of using them. United States patent US 5679377. October 21, 1997
- Blanco D, Alonso MJ. (1998). Protein encapsulation and release from poly(lactide-co-glycolide) microspheres: effect of the protein and polymer properties and of the co-encapsulation of surfactants. Eur J Phar Biopharm 45:285–94
- Carvalho FC, Bruschi ML, Evangelista RC, Gremião MPD. (2010). Mucoadhesive drug delivery systems. Braz J Pharm Sci 46:1–17
- Chan JM, Valencia PM, Zhang L, et al. (2010). Polymeric nanoparticles for drug delivery. Methods Mol Biol 624:163–75
- Cheng J, Teply BA, Jeong SY, et al. (2006). Magnetically responsive polymeric microparticles for oral delivery of protein drugs. Pharm Res 23:557–64
- Cheng WP, Thompson C, Ryan SM, et al. (2010). In vitro and in vivo characterisation of a novel peptide delivery system: amphiphilic polyelectrolyte–salmon calcitonin nanocomplexes. J Contr Rel 147:289–97
- de Vries L, Reitzema-Klein CE, Meter-Arkema A, et al. (2010). Oral and pulmonary delivery of thioether-bridged angiotensin-(1–7). Peptides 31:893–8
- Del Curto MD, Maroni A, Palugan L, et al. (2011). Oral delivery system for two-pulse colonic release of protein drugs and protease inhibitor/absorption enhancer compounds. J Pharm Sci 100:3251–9
- Dünnhaupta S, Barthelmesa J, Hombach J, et al. (2011). Distribution of thiolated mucoadhesive nanoparticles on intestinal mucosa. Int J Pharm 408:191–9
- Ferrari M, Dehlinger P, Martin FJ, et al., inventor; The Regents of the University of California, assignee. Particles for oral delivery of peptides and protein. United States patent US 6355270. 2002 March 12
- Florence AT, Hussain N. (2001). Transcytosis of nanoparticle and dendrimer delivery systems: evolving vistas. Adv Drug Deliv Rev 50:69–89
- Gupta S, Gabrani R, Ali J, Dang S. (2011). Exploring novel approaches to vaginal drug delivery. Recent Pat Drug Deliv Formul 5:82–94
- Gupta S, Vyas SP. (2010). Carbopol/chitosan based pH triggered in situ gelling system for ocular delivery of timolol maleate. Sci Pharm 78:959–76
- He C, Cui F, Yin L, et al. (2009). A polymeric composite carrier for oral delivery of peptide drugs: bilaminated hydrogel film loaded with nanoparticles. Eur Polymer Journal 45:368–76
- Hussain N, Florence AT. (1998). Utilizing bacterial mechanisms of epithelial cell entry: invasin-induced oral uptake of latex nanoparticles. Pharm Res 15:153–6
- Jin X, Xua Y, Shena J, et al. (2011). Chitosan-glutathione conjugate-coated poly (butyl cyanoacrylate) nanoparticles: Promising carriers for oral thymopentin delivery. Carbohydr Polym 86:51–7
- Kafka AP, McLeod BJ, Radesa T, McDowell A. (2011). Release and bioactivity of PACA nanoparticles containing D-Lys6-GnRH for brushtail possum fertility control. J Contr Rel 149:307–13
- Kaur IP, Smitha R. (2002). Penetration enhancer sand ocular bioadhesives: two new avenues for ophthalmic drug delivery. Drug Dev Ind Pharm 28:353–69
- Khafagy ES, Morishita M. (2012). Oral biodrug delivery using cell-penetrating peptide. Adv Drug Deliv Rev 64:531–9
- Kilpelainen M, Monkarea J, Vlasova MA, et al. (2011). Nanostructured porous silicon microparticles enable sustained peptide (Melanotan II) delivery. Eur J Pharm Biopharm 77:20–5
- Lee HE, Lee MJ, Park CR, et al. (2010). Preparation and characterization of salmon calcitonin–sodium triphosphate ionic complex for oral delivery. J Contr Rel 143:251–7
- Li Z, Chen J, Sun W, Xu Y. (2010). Investigation of archaeosomes as carriers for oral delivery of peptides. Biochem Biophys Res Commun 394:412–17
- Liang JF, Yang VC. (2005). Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem Biophys Res Commun 335:734–8
- Liu C, Tan Y, Liu C, et al. (2007). Preparations, characterizations and applications of chitosan-based nanoparticles. J Ocean Univ China 6:237–43
- Madhav NVS, Kala S. (2011). Review on microparticulate drug delivery system. Int J PharmTech Res 3:1242–54
- Makhlof A, Fujimoto S, Tozuka Y, Takeuchi H. (2011a). In vitro and in vivo evaluation of WGA–carbopol modified liposomes as carriers for oral peptide delivery. Eur J Pharm Biopharm 77:216–24
- Makhlof A, Tozuka Y, Takeuchi H. (2011b). Design and evaluation of novel pH-sensitive chitosan nanoparticles for oral insulin delivery. Eur J Pharm Sci 42:445–51
- Makhlof A, Werlea M, Tozukaa Y, Takeuchi H. (2011c). A mucoadhesive nanoparticulate system for the simultaneous delivery of macromolecules and permeation enhancers to the intestinal mucosa. J Contr Rel 149:81–8
- Marks FM, Lowman A. (2011). Enhanced mucoadhesive capacity of novel co-polymers for oral protein delivery. J Biomater Sci Polym Ed 22:2079–95
- Marttin E, Verhoef JC, Merkus FW. (1998). Efficacy, safety and mechanism of cyclodextrins as absorption enhancers in nasal delivery of peptide and protein drugs. J Drug Target 6:17–36
- Michael JC, Litwin A, inventor; The University of Cincinnati, assignee. Oral administration of therapeutic proteins. United States patent US 6613332. 2003 September 2
- Nomura H, Maruyama K, inventor; Kirin-Amgen, Inc., assignee. Oral dosage form of biologically active proteins. United States patent US 5597562. 1997 January 8
- Pather SI, Gupte SV, Khankari RK, et al., inventor; Cima Labs Inc., assignee. Microemulsions as solid dosage forms for oral administration. United States patent 6280770. 1996 August 28
- Peppas NA, Robinson JR. (1995). Bioadhesives for optimization of drug delivery. J Drug Target 3:183–4
- Plate NA, Valuev LI, Valueva TA, et al., inventor; Ores Pharmaceutical Development Corp., assignee. Polymer composition for oral administration of peptides and proteins. European Patent EP 0918543. 2004 April 21
- Porter CJH, Charman WN. (1997). Uptake of drugs into the intestinal lymphatics after oral administration. Adv Drug Del Rev 25:71–89
- Qi X, Wang L, Zhu J, et al. (2011). Self-double-emulsifying drug delivery system (SDEDDS): A new way for oral delivery of drugs with high solubility and low permeability. Int J Pharm 409:245–51
- Rabanel JM, Elkin V, Mokhtar M, Hildgen P. (2012). Drug-loaded nanocarriers: passive targeting and crossing of biological barriers. Curr Med Chem 19:3070–102
- Radhakrishnan B, Aggarwal D, Ferro M, et al.; Biocon Ltd., assignee. Fatty acid formulations for oral delivery of proteins and peptides, and uses thereof. United States patent 7605123. 2009 October 20
- Rekha MR, Sharma CP. (2009). Synthesis and evaluation of lauryl succinyl chitosan particles towards oral insulin delivery and absorption. J Contr Rel 135:144–51
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. (1999). In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569–72
- Senel S, Kremer MJ, Kas S, et al. (2000). Enhancing effect of chitosan on peptide drug delivery across buccal mucosa. Biomaterials 21:2067–71
- Serra L, Domenech J, Peppas NA. (2006). Design of poly(ethylene glycol)-tethered copolymers as novel mucoadhesive drug delivery systems. Eur J Pharm Biopharm 63:11–18
- Shantha KL, Thennarasu S, Krishnamurti N. (1989). Developments and applications of cyanoacrylate adhesives. J Adhes Sci Technol 3:237–60
- Sharma A, Sharma US. (2011). Liposomes in drug delivery: Progress and limitations. Int J Pharm 154:123–40
- Shimoni E, Ramon O, Kopelman IJ, et al., inventor; Technion Research and Development Foundation Ltd., assignee. Oral delivery of proteins and peptides. World Intellectual Property Organization WO/2008/132727. 2008 November 6
- Shukla A, Singh B, Katare OP. (2011). Significant systemic and mucosal immune response induced on oral delivery of diphtheria toxoid using nano-bilosomes. Br J Pharmacol 164:820–7
- Sonia TA, Sharma CP. (2011). In vitro evaluation of N-(2-hydroxy) propyl-3-trimethyl ammonium chitosan for oral insulin delivery. Carbohydr Polym 84:103–9
- Suksamran T, Opanasopit P, Rojanarata T, et al. (2009). Biodegradable alginate microparticles developed by electrohydrodynamic spraying techniques for oral delivery of proteins. J Microencapsul 26:563–70
- Suslick KS, Toublan FJ, Boppart SA, Marks DL, inventors; The Board of Trustees of the University of Illinois, assignee. Surface modified protein microparticles. United States patent US 7217410. 2007 May 15
- Takada K, inventor. Oral formulation for gastrointestinal drug delivery. United States patent 7097851. 2006 August 29
- Teng LL, inventor; Research Corporation, assignee. Orally administered biologically active peptides and proteins. United States patent US 4582820. 1986 April 15
- Thompson CJ, Tetley L, Cheng WP. (2010). The influence of polymer architecture on the protective effect of novel comb shaped amphiphilic poly(allylamine) against in vitro enzymatic degradation of insulin—towards oral insulin delivery. Int J Pharm 383:216–27
- Thompson CJ, Tetley L, Uchegbu IF, Cheng WP. (2009). The complexation between novel comb shaped amphiphilic polyallylamine and insulin—towards oral insulin delivery. Int J Pharm 376:46–55
- Trapani A, Lopedota A, Franco M, et al. (2010). A comparative study of chitosan and chitosan/cyclodextrin nanoparticles as potential carriers for the oral delivery of small peptides. Eur J Pharm Biopharm 75:26–32
- Vol A, Gribova O, inventor; Oshadi Drug Administration Ltd., assignee. Methods and composition for oral administration of protein and peptide therapeutic agents. World Intellectual Property Organization WO2009087633. 2009 July 16
- Wang T, Wang N, Hao A, et al. (2010). Lyophilization of water-in-oil emulsions to prepare phospholipid-based anhydrous reverse micelles for oral peptide delivery. Eur J Pharm Biopharm 39:373–9
- Werle M, Takeuchi H. (2009). Chitosan–aprotinin coated liposomes for oral peptide delivery: Development, characterisation and in vivo evaluation. Int J Pharm 370:26–32
- Yina L, Dinga J, He C, et al. (2009). Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 30:5691–700
- Yousefpour P, Atyabi F, Farhani EV, et al. (2011). Polyanionic carbohydrate doxorubicin-dextran nanocomplex as a delivery system for anticancer drugs: in vitro analysis and evaluations. Int J Nanomedicine 6:1487–96
- Zhang Y, Wei W, Lv P, et al. (2011). Preparation and evaluation of alginate–chitosan microspheres for oral delivery of insulin. Eur J Pharm Biopharm 77:11–19