
Abstract
The present study examined the effects of lipid vehicle and intestinally based efflux processes on intestinal lymphatic transport of paclitaxel (PTX) in the mesenteric lymph duct-cannulated anesthetized rat model. PTX solution alone, PTX solution pretreated with the P-glycoprotein (P-gp) inhibitor verapamil and/or PTX and a 2:1 (w/w) mixture of linoleic acid:glycerol monooleate were administered intraduodenally to anesthetized rats. Coadministration of a mixture of linoleic acid–monoolein significantly increased the extent of intestinal lymphatic transport of PTX, but it had little impact on the absolute oral bioavailability of PTX. In contrast, pretreatment with verapamil increased both the extent of lymphatic transport (3.5-fold) and absolute oral bioavailability (1.8-fold). Further increase in the lymphatic transport (6.5-fold) and absolute oral bioavailability (1.8-fold) was achieved by the combination of pretreatment with verapamil and coadministration with the linoleic acid–monoolein mixture. These data indicate that the application of lipid vehicle holds promise for selectively targeted lymphatic delivery of PTX. P-gp inhibition can result in both increased intestinal lymphatic levels and absolute oral bioavailability of PTX.
Introduction
In recent years, considerable academic attention has been focused on the lymphatic transport of orally administered highly lipophilic drugs. Lymphatic absorption is a specialized absorption pathway for dietary lipids, fat-soluble vitamins and lipophilic drugs (Porter & Charman, Citation1997). Drugs transported via the intestinal lymph first migrate to the lymphatic vessels and drain into lymph nodes before entering the systemic circulation (Cai et al., Citation2011). Therefore, drugs transported to the systemic circulation via the intestinal lymph can bypass the first-pass metabolism and have greater total systemic bioavailability. Furthermore, it has been reported that lymphatic transport increases drug targeting to the lymphocytes, which may potentially increase the efficacy of orally administered drugs such as immunomodulators, anticancer agents or anti-infectives (Trevaskis et al., Citation2010a). Since tumor cells spread along the lymphatic pathways and metastasize in the lymph nodes, lymphatic chemotherapy may be a promising novel therapeutic approach for patients with metastatic cancers (Garzonaburbeh et al., Citation1983; Cai et al., Citation2011).
Paclitaxel (PTX), an effective antineoplastic drug, is widely prescribed for the treatment of various cancers, including ovarian, breast and non-small-cell lung tumors and AIDS-related Kaposi sarcoma (Rowinsky & Donehower, Citation1995; Malingre et al., Citation2001a). Currently, PTX formulations available for clinical use (6 mg/mL) are formulated in a mixture of ethanol and polyoxyethylated castor oil (Cremophor EL) (1:1 by volume) and are administered through intravenous (i.v.) infusion. However, the vehicle used in formulation, Cremophor EL, may contribute to the hypersensitivity reaction and neurotoxicity in patients, which are notorious side effects associated with the use of i.v. PTX (van Zuylen et al., Citation2001). Thus, much research is being carried out to develop oral formulations of PTX to minimize the i.v. route-related adverse effects (Gao et al., Citation2003; Nornoo et al., Citation2009; Agueros et al., Citation2010). On the other hand, oral administration is hampered by the low bioavailability due to PTX's low solubility in water, cytochrome P-450 enzymes in the gut and liver, and high affinity to the multidrug efflux pump P-glycoprotein (P-gp) (Fisher & Sikic, Citation1995; Park et al., Citation2012).
The change of formulations would offer an opportunity to overcome some drawbacks related to oral administration, such as poor water solubility. Lipid formulations, such as self-microemulsifying drug delivery systems (SMEDDS) (Yang et al., Citation2004; Lo et al., Citation2010; Oostendorp et al., Citation2011) and lipid nanocapsules (LNC) (Peltier et al., Citation2006) have been reported to enhance the oral bioavailability of PTX. SMEDDS and LNC formulations of PTX have shown 2-fold and 3-fold higher oral bioavailability, respectively, compared to that of an orally administered Taxol formulation (Yang et al., Citation2004; Peltier et al., Citation2006). Mechanisms by which lipids improve the bioavailability of PTX may include enhanced solubilization of PTX in the gastrointestinal lumen and increased absorption via selective lymphatic uptake (Yang et al., Citation2004; Porter & et al., Citation2007). Lipid-based formulations have been widely applied for highly lipophilic drugs to promote their intestinal lymphatic transport (Ling et al., Citation2006; Murota et al., Citation2013). The majority of lipophilic drugs absorbed via lymphatic transport are either solubilized by lipids or are associated with the triglyceride core of chylomicrons in the enterocyte and are secreted into the mesenteric lymphatics (Charman & Stella, Citation1986a; Porter & Charman, Citation2001). It is suggested that lipid excipients may improve the lymphatic transport of lipophilic drugs by stimulating the production of chylomicrons (O'Driscoll, Citation2002). Despite the expected influences of lipid formulation on the intestinal lymphatic transport of PTX, the degree of intestinal lymphatic transport of PTX and the impact of lipid vehicles on the intestinal lymphatic transport of PTX remain unknown.
PTX, as has been mentioned, is a substrate for P-gp efflux in intestinal cells (Fisher & Sikic, Citation1995; Sparreboom et al., Citation1997). Previous studies have indicated that 54% of a PTX oral dose is extruded to the gut lumen by P-gp, when taken orally (Woo et al., Citation2003). The oral bioavailability is greatly improved by inhibiting the function and/or expression of P-gp by administering verapamil or verapamil analogs in both the patients and animals (Woo et al., Citation2003; Choi & Li, Citation2005). Although much effort has been put into the identification of the effects of P-gp efflux process on the portal and systemic blood levels of PTX, much less is known about the relative role of P-gp efflux processes in the lymph uptake of PTX.
The current study has therefore evaluated the contribution of the lymphatic route to the intestinal absorption of PTX and examined the effects of a lipid vehicle and a P-gp inhibitor on intestinal lymph transport processing. The effects of coadministration of PTX with a mixture of linoleic acid (FA) and monoglyceride (MG)-based vehicles and/or pretreated with a P-gp inhibitor (verapamil) on the lymphatic transport of PTX were studied using an anesthetized lymph duct-cannulated rat model.
Materials and methods
Materials
PTX was purchased from Shanghai Ronghe Pharmaceutical Technology Development Co., Ltd. (Shanghai, China) and diazepam was bought from Beijing Pharmaceutical Factory (Beijing, China). Methanol and acetonitrile were chromatography grade (Promptar Company Ltd., Georgetown, Canada). Glycerol monooleate was a gift from Danisco Ingredients, Copenhagen, Denmark. Linoleic acid was from URChem Chemicals (Shanghai, China). Water was obtained via a Millipore Milli-Q water purification system (Milford, MA). All other chemicals and solvents were of analytical grade. Specific pathogen free-grade Sprague–Dawley rats (body weight, 260–310 g) were purchased from Sino-British SIIPPR/BK Lab. Animal Ltd. (Shanghai, China).
Surgical procedure
All experiments were conducted in accordance with Fudan University Animal Care and Use Committee. Male Sprague-Dawley rats were fasted overnight prior to an experiment and given free access to water before surgery. For exposing the mesenteric lymphatic duct, the rats were given 0.5 mL of peanut oil by oral gavage 1 h before cannulation of the mesenteric lymph duct. The animals were anesthetized for the duration of the surgeries by intraperitoneal injection of 2% pentobarbital sodium salt (2.5 mL/kg), and additional injections were given if required. The mesenteric lymph duct was cannulated in accordance with a previously described method (Caliph et al., Citation2000) with some modification. A two-part cannula, consisting of a 3-cm-long silicone tube connected to an 8-cm-long polyethylene tube (PE50), was inserted into the mesenteric lymph duct and secured with medical adhesive bandage. An incision was also centered approximately 0.5 cm from the right neck midline; the jugular vein was cannulated with polyethylene tubing (PE50) to allow blood sampling. The duodenum was cannulated for intraduodenal infusion and rehydration.
For i.v. administration of PTX, the rats were sham operated in a manner similar to that used for cannulation of the mesenteric lymph duct. The right jugular vein was cannulated with polyethylene tubing (PE50) to allow blood sampling. The right femoral vein was cannulated for i.v. administration.
Experimental procedure
After the completion of the surgical procedures, the rats were transferred to a heated pad maintained at 37 °C. Normal saline was intraduodenally infused at a rate of 1.4 mL/h through a constant-rate infusion pump (Shanghai Angel Electronic Equipment Co., Ltd., Shanghai, China) to maintain hydration and lymph flow.
The rats were randomly assigned to one of five treatment groups: Group A (control group), intraduodenal PTX 20 mg/kg; Group B, intraduodenal PTX 20 mg/kg coadministered with 50 μL of lipid solution of a 2:1 (w/w) mixture of linoleic acid and glycerol monooleate; Group C, intraduodenal verapamil 20 mg/kg administered 30 min prior to 20 mg/kg intraduodenal PTX; Group D, intraduodenal verapamil 20 mg/kg administered 30 min prior to 20 mg/kg intraduodenal administration of PTX and coadministered with 50 μL of lipid solution of a 2:1 (w/w) mixture of linoleic acid and glycerol monooleate and Group E, i.v. dose of 2 mg/kg PTX.
The PTX solution was prepared by dissolving PTX in an equal-volume mixture of Cremophor EL and ethanol (1:1, v/v; 20 mg PTX/mL for intraduodenal route and 6 mg PTX/mL for the i.v. route). For the intraduodenal route, the PTX solution was intraduodenally infused at a rate of 2 mL/h over 30 min, whereas the i.v. PTX solution was diluted with saline immediately prior to injection and injected through the femoral vein within 10 s (0.3 mL/rat).
Lymph samples were collected 1 h pre-dose and hourly into a pre-weighted Eppendorf tube containing 1% EDTA-2Na for 8 h after dosing, the mean (±SD) total volumes of the lymph that were collected within the first 8 h were 6.559 ± 0.893 mL. Blood samples (0.3 mL) were collected into heparinized tubes via the jugular vein at 0, 0.33, 0.63, 1, 1.5, 2, 3, 4, 6 and 8 h after intraduodenal administration of the drug and at 0, 0.083, 0.16, 0.25, 0.5, 1, 2, 3, 4, 6 and 8 h after i.v. injection. Plasma samples were immediately harvested by centrifugation (4500 g, 3 min). Lymph and plasma samples were stored at −40 °C until analysis.
Assay of PTX in plasma and lymph
Preparation of plasma samples for HPLC analysis
Plasma samples (100 μL) were mixed with 10 μL of internal standard (IS) solution (1 μg/mL diazepam in acetonitrile) in polypropylene Eppendorf tubes, and vortexed for 30 s. Acetonitrile (1 mL) was then added to precipitate proteins; the samples were vortexed for another 1 min and centrifuged at 9100 g for 10 min. The supernatant was transferred into a polypropylene tube and evaporated to dryness under a stream of high-purity nitrogen in a water bath maintained at 40 °C. The dry residues were then dissolved in 100 μL of the mobile phase, and 50 μL was used for high-performance liquid chromatography (HPLC) analysis.
Preparation of lymph samples for HPLC analysis
A volume of 100 µL of lymph sample was spiked with 10 μL of the IS (1 μg/mL diazepam in acetonitrile) and vortexed for 30 s. Acetonitrile (1 mL) was then added; the sample was then vortexed for 1 min and centrifuged at 9100g for 10 min. A portion of the upper acetonitrile layer (800 μL) was then transferred to a clean polypropylene tube. The residue was reextracted repeatedly with 800 µL of aliquots of acetonitrile. The combined supernatants were evaporated to dryness in the presence of nitrogen in a water bath maintained at 40 °C. The residues were reconstituted with 100 μL of acetonitrile, and 20 μL was injected in the HPLC system.
Liquid chromatography
The HPLC system consisted of a Waters 2695 Alliance module and Waters 2487 dual λ absorption detector. A Waters Symmetry® C18 analytical column (150 mm × 4.6 mm, 5 μm) and a C18 guard column (Security Guard, C18, 4 mm × 3 mm i.d.; Phenomenex, USA) were used for separation. The mobile phase consisted of acetonitrile:Milli-Q water (48:52, v/v) at a flow rate of 1 mL/min. The temperature of the column was set at 30 °C, and the UV detector was set at 227 nm for detection. The specimen chamber was maintained at 4 °C, and the injection volumes of the lymph and plasma sample were 20 and 50 µL, respectively. Retention times for PTX and IS were 9.8 and 7.2 min, respectively. The detection limit of PTX was 20 ng/mL, the range of linear response for the lymph sample was 0.1–10 µg/mL (r2 > 0.9998), and the range of linear response for the plasma sample was 0.02–5 µg/mL (r2 > 0.9998). The recoveries of PTX at three different concentrations were similar both in plasma and in lymph fluid. Recoveries of PTX in plasma at concentrations of 0.05, 0.5 and 5 μg/mL were 77.3 ± 5.6%, 77.0 ± 2.3% and 85.3 ± 1.7%, respectively. Recoveries of PTX in lymph fluid at concentrations of 0.1, 1 and 10 μg/mL were 71.3 ± 6.48%, 79.5 ± 4.12%, 78.4 ± 2.3%, respectively.
Analysis of lymph TGs
Lymph triglyceride (TG) concentrations were analyzed using a KEHUA auto clinical chemistry analyzer by using a commercial kit (Shanghai Kehua Bioengineering Co., Ltd, Shanghai, China). Endogenous lymphatic transport, which was of zero order, was calculated to be 1.05 ± 0.32 mg/h from the slope of the regression of mean cumulative lymphatic TG transport measured over 8 h (r2 = 0.9911) in three rats; this finding was consistent with that reported by Holm et al. (Citation2001).
Pharmacokinetic data analysis
Data related to changes in plasma drug concentration versus time were analyzed by WinNonlin 4.0.1[q] program for the purpose of calculating pharmacokinetics parameters. Cmax was measured; AUC0–8 h was calculated using the trapezoidal rule. The absolute bioavailability of PTX after intraduodenal administration of 20 mg/kg compared to that after i.v. administration of 2 mg/kg was calculated using the following formula:
Statistical analysis
One-way analysis of variance (for multiple comparisons) was used to determine the statistical significance of the results calculated for the groups. To determine statistically significant differences between two groups, the Student's t-test was used when appropriate. Differences were considered to be significant at p < 0.05.
Results
Intestinal cumulative lymphatic transport of PTX
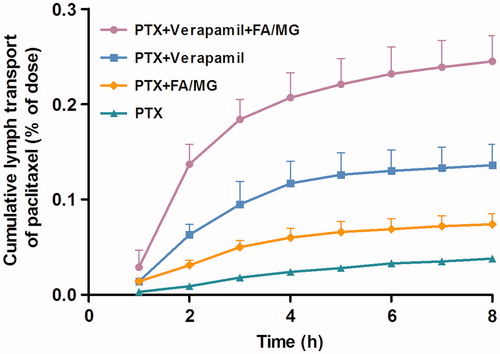
The cumulative extent (the percentage of dose administered) of lymphatic transport of PTX into mesenteric lymph after intraduodenal administration of 20 mg/kg PTX is given in . The extent of lymphatic transport of PTX over 8 h was 0.038 ± 0.003% (mean % dose ± SE, n ≥ 4 for all the groups) after intraduodenal administration of PTX solution. The cumulative lymphatic transport increased to 0.074 ± 0.011% when PTX was coadministered with 50 μL lipid solution of 2:1 (w/w) mixture of linoleic acid–monoolein (p < 0.05). When PTX (20 mg/kg) was pretreated with verapamil (20 mg/kg), the cumulative lymphatic transport of PTX (0.136 ± 0.022%) increased significantly (p < 0.01) compared to that in the control group. The most significant improvements in PTX levels were observed when PTX was pretreated with verapamil and coadministered with the linoleic acid–monoolein mixture. The cumulative lymphatic transport of PTX reached 0.245 ± 0.027%, which was nearly 6.5 times higher than when PTX solution was administered alone (p < 0.01).
Figure 1. Cumulative percentage dose of PTX (mean ± SE) collected in the mesentery lymph as a function of time. PTX (20 mg/kg) was administered intraduodenally as PTX solution alone (n = 4), coadministered with either FA/MG (n = 6) or pretreated with verapamil (n = 4), or pretreated with verapamil and coadministered with FA/MG (n = 4).
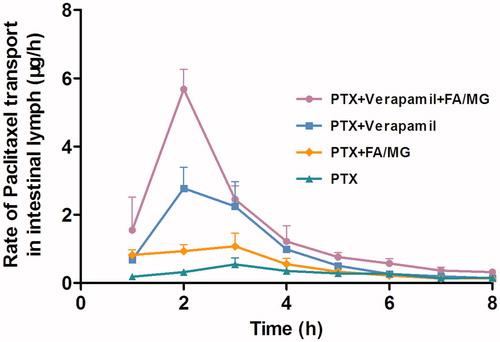
The rate profiles describing lymphatic transport of PTX as a function of time are presented in . The peak rates of lymph transport of PTX in animals intraduodenally administered with PTX solution alone and PTX coadministered with lipid (FA/MG) were 1.073 ± 0.389 and 0.547 ± 0.191 μg/h, respectively, indicating of a greater than 2-fold increase in the maximum rate of lymphatic drug transport in the presence of lipids (p < 0.05). When PTX was pretreated with verapamil, the maximum lymph transport rate of PTX (2.775 ± 0.723 μg/h) increased significantly (p < 0.05) compared to that in the control group. PTX lymph transport rate increased when it was pretreated with verapamil and coadministered with FA/MG solution. In these cases, the peak transport rate of PTX was 10-fold higher than that in the control group (p < 0.05), and 2-fold higher than that in the group that received PTX pretreated with verapamil alone (p < 0.05).
Figure 2. Concentration–time profiles of PTX in mesenteric lymph fluid following intraduodenally administered 20 mg/kg PTX solution alone (n = 4), coadministered with either FA/MG (n = 6) or pretreated with verapamil (n = 4), or pretreated with verapamil and coadministered with FA/MG (n = 4). Bars represent the SE.
Intestinal lymphatic transport of TGs
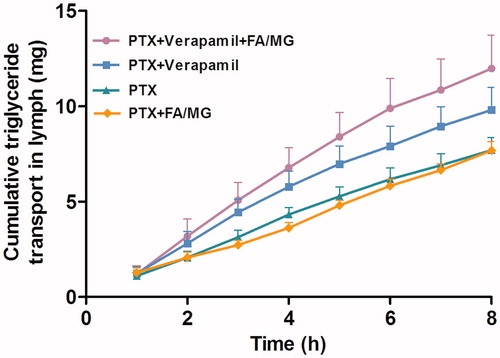
The cumulative transport of TGs into the lymph over 8 h after intraduodenal administration of PTX solution alone, PTX pretreated with verapamil and/or PTX coadministered with lipid is shown in . No significant difference in the mean value of the cumulative TG transport over 8 h was found among the groups treated with each modulator. The lipid turnover for the groups that were administered FA/MG solution and PTX or PTX pretreated with verapamil was similar to the lipid turnover in the absence of a modulator. However, there is a trend toward higher TG turnover when PTX was pretreated with verapamil and when PTX was coadministered with FA/MG; however, the difference was not significant.
Figure 3. Cumulative transport of TG into the lymph over 8 h following intraduodenal administration of 20 mg/kg PTX solution alone (n = 4), coadministered with either FA/MG (n = 6) or pretreated with verapamil (n = 4), or pretreated with verapamil and coadministered with FA/MG (n = 4). Bars represent the SE.
Pharmacokinetics of PTX
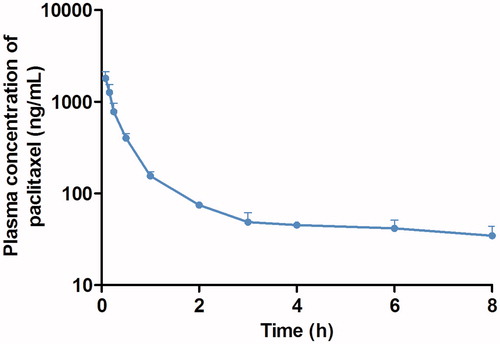
The plasma concentration–time profile for PTX obtained after the i.v. administration of PTX solution (2 mg/kg) is shown in . The i.v. blood concentration versus time data showed that the plasma AUC0–8 h of PTX was 1008.61 ± 308.07 ng/h/mL, and clearance calculated as dose/AUC was 521.40 ± 169.50 mL/h (n = 3, mean ± SD). The mean(±SD) apparent biological half-life was 1.95 ± 1.07 h, which is comparable with that reported by Park et al. (Citation2012).
Figure 4. Mean plasma concentration–time profiles of PTX following i.v. injection of PTX solution (2 mg/kg) to rats (n = 3). Bars represent the SE.
The mean plasma concentration–time profiles of PTX following intraduodenal administration of PTX (20 mg/kg) alone (control), in combination with FA/MG solution (50 μL), pretreated with verapamil (20 mg/kg), or pretreated with verapamil and coadministered with FA/MG (n ≥ 4 for each groups) are presented in . The bioavailability and the pharmacokinetic parameters of PTX after coadministration with FA/MG and (or) pretreatment with verapamil are shown in . The absolute bioavailability (AB%) of the drug was only 3.01% when the PTX solution was administered; this poor exposure of PTX from the PTX solution in rats was similar to the values reported in the literature (Choi & Li, Citation2005; Lee & Choi, Citation2010). Mean AUC0–8 h and Cmax of PTX following introduodenal administration of PTX in combination with FA/MG solution were similar to the corresponding values obtained for the controls. The Cmax and absolute oral bioavailability of PTX were 2.1- and 1.8-fold higher, respectively, when pretreated with verapamil. A further increase in PTX plasma concentration was observed after intraduodenal administration of PTX with FA/MG when pretreated with verapamil. The Cmax and absolute oral bioavailability of PTX pretreated with verapamil and administered with FA/MG were 2.3-fold and 1.75-fold higher than the corresponding values obtained for PTX administered alone.
Figure 5. Mean plasma concentration–time profiles of PTX following intraduodenal administration of 20 mg/kg PTX solution alone (n = 4), coadministered with either FA/MG (n = 6) or pretreated with verapamil (n = 4), or pretreated with verapamil and coadministered with FA/MG (n = 4). Bars represent the SE.
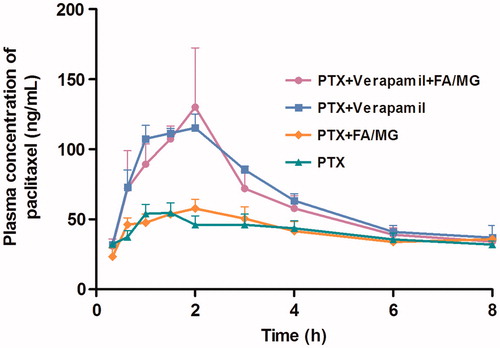
Table 1. Pharmacokinetic parameters of PTX (mean ± SD) after intraduodenal administration of PTX solution with or without FA/MG or verapamil in rats.
Discussion
The spread of tumor cells via lymphatic vessels to the regional lymph nodes and metastasis in the lymph nodes are important indicators of tumor aggressiveness and metastasis for most human malignancies (Skobe et al., Citation2001). The lymphatic system is the major route for metastasis of solid tumors (Garzonaburbeh et al., Citation1983; Trevaskis et al., Citation2008; Krishnaiah & Khan, Citation2012). From the site of administration, anticancer agents must be delivered at tumoricidal concentrations at lymph nodes and lymphatics where tumor cells potentially dwell in order to control lymphatic metastasis (Liu et al., Citation2009). Thus, a possible therapeutic benefit may be gained by increasing the level of PTX in the lymphatic system and in lymphoid tissue in general.
The extent of intestinal lymphatic transport of PTX (expressed as % dose administered) from PTX solution is low, which may partly be due to its unique physicochemical properties. PTX has a clog P value of 4.2 (Ge et al., Citation2008), its solubility in peanut oil is 0.55 mg/g. Previous studies have suggested that highly lipophilic compounds with a log p > 4.7 and long-chain TG solubility > 50 mg/g may well provide the prerequisite physicochemical properties for significant intestinal lymphatic drug transport in order to facilitate drug association with the TG-rich core of lipoproteins in the enterocyte (Charman & Stella, Citation1986b). However, Trevaskis et al. later reported that significant lymphatic transport can be achieved by the affinity for the colloidal interface of lymph lipoproteins rather than simple solubilization in core lipids (Trevaskis et al., Citation2010b). PTX is 95–98% bound to plasma proteins (Rowinsky & Donehower, Citation1995), this observation indicates a high affinity of PTX to the lipoprotein interface in the chylomicron, which may facilitate the intestinal lymph transport of PTX.
In addition to the physicochemical properties of PTX that limits the extent of its transport in lymph, P-gp efflux processes may also have a potential effect on the lymph transport of PTX. Hypothetically, the susceptibility of PTX to the P-gp counter transport system greatly limits the extent of its intestinal lymphatic transport, and it follows that the extent of lymphatic transport of PTX would significantly increase with the coadministration of P-gp inhibitors. In fact, the cumulative lymphatic transport of PTX pretreated with verapamil was 3.5-fold higher compared to control. Such a result suggests a role for enterocyte-based P-gp efflux processes as key factors of intestinal lymphatic transport of PTX, consistent with an earlier study by Griffin & O'Driscoll (Citation2008). The inhibition of P-gp may increase intracellular concentration and residence time of the intact drug in the cell, which results in increased concentration of drug available for partitioning into the lymphatic (O'Driscoll, Citation2002). It is, therefore, reasonable to expect that P-gp-mediated efflux plays an important role in the intestinal lymphatic transport of PTX.
It is also generally believed that the coadministered lipid vehicles can influence the degree of lymph targeting. Lipid vehicles may enhance lymphatic transport of lipophilic compounds by stimulating the production of chylomicrons. However, only fatty acids of chain length of 14 and above are considered to be resynthesized into TGs within the enterocyte prior to incorporation into chylomicrons (Charman & Porter, Citation1996). In the present study, 50 μL of lipid solution of a 2:1 (w/w) mixture of linoleic acid–monoolein was applied to each rat as exogenous lipid to trigger the biochemical processes of chylomicrons. Exogenous lipids are postulated to undergo the same chylomicron formation or lipid progress as to other lipid formulations. Not surprisingly, the coadministration of PTX with FA/MG introduced a lymphatic transport 2-fold higher than PTX solution, which leads to the hypothesis that lymphatic transport of PTX can be improved by employing lipid systems. The increased cumulative lymph transport of PTX after the coadministration with FA/MG may be due to the elevated levels of chylomicrons with increased binding of PTX to the colloidal interface of lymph lipoproteins, which in turn would promote the absorption of PTX into the lymph. The maximum lymphatic transport rate of PTX in the groups coadministered with FA/MG was also 2-fold higher than groups without coadministration with FA/MG, which further confirms that lipids are essential for increased lymphatic transport of PTX. Increased lymphatic transport of PTX is related to an increased drug transfer rate through enterocyte cells as a result of lipid coadministration.
However, no significant increase in the extent of TG turnover was observed following introduodenally administered verapamil given 30 min before administration with PTX solution (). It is expected that verapamil at that concentration would not have an impact on the production of chylomicrons, consistent with a previous study by Griffin & O'Driscoll (Citation2008). The extent of TG turnover also appears to be unaffected by the coadministration of FA/MG, at the concentrations used in this study. The cumulative transport of TG for control group and group coadministered with FA/MG are similar to the cumulative transport of endogenous TG. A dose of 50 μL of exogenous lipid may be relatively less and may be only marginally more than the amount of endogenous lipid transported during the experimental period. In a similar study, by Porter et al. suggest that the variation in endogenous lipid turnover between animals could affect the values obtained for exogenous lipid transport (Porter et al., Citation1996). A higher dose of lipid might be required to produce a more significant increase in the cumulative transport of TG in rats, as can be inferred from an article by Trevaskis et al. (Citation2010b). The increase in the lymphatic transport of TG in groups pretreated with verapamil and coadministered with FA/MG was comparable to the increase in lymphatic transport of PTX, but the difference was not significant.
The administration of FA/MG is not able to increase the lymph independent bioavailability of PTX significantly compared with control, as estimated from the similar plasma concentration profiles () and plasma AUC0–8 h (). Application of a 2:1 (w/w) mixture of linoleic acid–monoolein (50 μL) may be insufficient to provide suitable solubilization conditions to improve the absorption of PTX into the blood circulation. It appears that the FA/MG mixture increases the lymph absorption but not the blood absorption of PTX.
As shown in , P-gp inhibition significantly increased the absorption of PTX independent of the lymphatic administration route. Plasma concentration and oral bioavailability are prominently increased by pretreatment with verapamil, indicating that inhibition of P-gp markedly boosts the extent of PTX absorption via the portal blood. These results are consistent with the results reported by Choi & Li (Citation2005), Woo et al. (Citation2003) and Park et al. (Citation2012). In these studies, P-gp inhibitors such as verapamil, verapamil analog KR30031 and silymarin were used to increase the bioavailability of PTX by inhibiting either the P-gp efflux pump or cytochrome P-450 (CYP3A). It can be concluded that either the P-gp efflux pump or cytochrome P-450 (CYP3A) would comprise a major barrier for both lymph and blood absorption of PTX besides its unique physicochemical property. The application of a P-gp inhibitor can remarkably increase both the lymph transport and the bioavailability of PTX.
As known, P-gp is also extensively distributed and expressed in various organs, including the liver, brain, kidney, pancreas and colon (Thiebaut et al., Citation1987). Coadministration of P-gp inhibitor may lead to global inhibition, which may result in the accumulation or uptake of more PTX in normal organs where P-gp is also extensively distributed (Kemper et al., Citation2003). It is theoretically possible that the risk of side effects will increase owing to the global inhibition. However, we are satisfied with the finding that an oral combination therapy of PTX and the P-gp inhibitor, cyclosporine A, effectively increases the bioavailability of PTX without causing significant adverse events. The lack of adverse events is clearly related to the coadministered dose of cyclosporin A (Malingre et al., Citation2001b). Thus, whether oral coadministration of PTX with P-gp inhibitors leads to global inhibition or side effects requires further investigation.
Conclusion
In summary, the results clearly support a role for exogenous lipids in promoting PTX transport via the lymph route. Application of lipid vehicles appears to hold promise for selectively targeted lymphatic delivery of PTX. In addition, intestinal P-gp efflux processes also play a significant role in the intestinal lymphatic transportation of PTX. P-gp inhibition can result in both increased intestinal lymphatic levels and increased oral absolute oral bioavailability of PTX. It seems that lipid formulations coupled with the coadministration of a P-gp inhibitor may be a promising approach for enhancement of oral exposure and intestinal lymphatic transport of PTX.
Declaration of interest
Part of this work was supported by a Grant-in-Aid from the National Natural Science Foundation of China (81072594).
References
- Agueros M, Zabaleta V, Espuelas S, et al. (2010). Increased oral bioavailability of paclitaxel by its encapsulation through complex formation with cyclodextrins in poly(anhydride) nanoparticles. J Control Release 145:2–8
- Cai S, Yang Q, Bagby TR, et al. (2011). Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev 63:901–8
- Caliph SM, Charman WN, Porter CJ. (2000). Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J Pharm Sci 89:1073–84
- Charman WN, Porter CJ. (1996). Lipophilic prodrugs designed for intestinal lymphatic transport. Adv Drug Deliv Rev 19:149–69
- Charman WNA, Stella VJ. (1986a). Effects of lipid class and lipid vehicle volume on the intestinal lymphatic transport DDT. Int J Pharm 33:165–72
- Charman WNA, Stella VJ. (1986b). Estimating the maximal potential for intestinal lymphatic transport of lipophilic drug molecules. Int J Pharm 34:175–8
- Choi JS, Li XG. (2005). The effect of verapamil on the pharmacokinetics of paclitaxel in rats. Eur J Pharm Sci 24:95–100
- Fisher GA, Sikic BI. (1995). Clinical-studies with modulators of multidrug-resistance. Hematol Oncol Clin N Am 9:363–82
- Gao P, Rush BD, Pfund WP, et al. (2003). Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci 92:2386–98
- Garzonaburbeh A, Poupaert JH, Claesen M, et al. (1983). 1,3-Dipalmitoylglycerolester of chlorambucil as a lymphotropic, orally administrable anti-neoplastic agent. J Med Chem 26:1200–3
- Ge H, Wang J, Kayser MM, et al. (2008). Synthesis, tubulin assembly, and antiproliferative activity against MCF7 and NCI/ADR-RES cancer cells of 10-O-acetyl-5′-hydroxybutitaxel. Bioorg Med Chem Lett 18:6165–7
- Griffin BT, O'Driscoll CM. (2008). An examination of the effect of intestinal first pass extraction on intestinal lymphatic transport of saquinavir in the rat. Pharm Res 25:1125–33
- Holm R, Mullertz A, Pedersen GP, et al. (2001). Comparison of the lymphatic transport of halofantrine administered in disperse systems containing three different unsaturated fatty acids. Pharm Res 18:1299–304
- Kemper EM, van Zandbergen AE, Cleypool C, et al. (2003). Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. Clin Cancer Res 9:2849–55
- Krishnaiah YSR, Khan MA. (2012). Strategies of targeting oral drug delivery systems to the colon and their potential use for the treatment of colorectal cancer. Pharm Dev Technol 17:521–40
- Lee CK, Choi JS. (2010). Effects of silibinin, inhibitor of CYP3A4 and P-glycoprotein in vitro, on the pharmacokinetics of paclitaxel after oral and intravenous administration in rats. Pharmacology 85:350–6
- Ling SSN, Magosso E, Khan NAK, et al. (2006). Enhanced oral bioavailability and intestinal lymphatic transport of a hydrophilic drug using liposomes. Drug Dev Ind Pharm 32:335–45
- Liu J, Meisner D, Kwong E, et al. (2009). Translymphatic chemotherapy by intrapleural placement of gelatin sponge containing biodegradable paclitaxel colloids controls lymphatic metastasis in lung cancer. Cancer Res 69:1174–81
- Lo J, Chen B, Lee T, et al. (2010). Self-emulsifying O/W formulations of paclitaxel prepared from mixed nonionic surfactants. J Pharm Sci 99:2320–32
- Malingre MM, Beijnen JH, Rosing H, et al. (2001a). The effect of different doses of cyclosporin A on the systemic exposure of orally administered paclitaxel. Anti-Cancer Drugs 12:351–8
- Malingre MM, Beijnen JH, Schellens JHM. (2001b). Oral delivery of taxanes. Investig New Drugs 19:155–62
- Murota K, Cermak R, Terao J, et al. (2013). Influence of fatty acid patterns on the intestinal absorption pathway of quercetin in thoracic lymph duct-cannulated rats. Br J Nutr 109:2147–53
- Nornoo AO, Zheng H, Lopes LB, et al. (2009). Oral microemulsions of paclitaxel: in situ and pharmacokinetic studies. Eur J Pharm Biopharm 71:310–7
- O'Driscoll CM. (2002). Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci 15:405–15
- Oostendorp RL, Buckle T, Lambert G, et al. (2011). Paclitaxel in self-micro emulsifying formulations: oral bioavailability study in mice. Investig New Drugs 29:768–76
- Park JH, Park JH, Hur HJ, et al. (2012). Effects of silymarin and formulation on the oral bioavailability of paclitaxel in rats. Eur J Pharm Sci 45:296–301
- Peltier S, Oger J M, Lagarce F, et al. (2006). Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res 23:1243–50
- Porter CJ, Charman SA, Charman WN. (1996). Lymphatic transport of halofantrine in the triple-cannulated anesthetized rat model: effect of lipid vehicle dispersion. J Pharm Sci 85:351–6
- Porter CJ, Charman WN. (1997). Uptake of drugs into the intestinal lymphatics after oral administration. Adv Drug Deliv Rev 25:71–89
- Porter CJ, Charman WN. (2001). Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev 50:61–80
- Porter CJ, Trevaskis NL, Charman WN. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–48
- Rowinsky EK, Donehower RC. (1995). Drug–therapy–paclitaxel (Taxol). N Engl J Med 332:1004–14
- Skobe M, Hawighorst T, Jackson DG, et al. (2001). Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 7:192–8
- Sparreboom A, vanAsperen J, Mayer U, et al. (1997). Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA 94:2031–5
- Thiebaut F, Tsuruo T, Hamada H, et al. (1987). Cellular-localization of the multidrug-resistance gene-product P-glycoprotein in norma human-tissues. Proc Natl Acad Sci USA 84:7735–8
- Trevaskis NL, Charman WN, Porter CJ. (2008). Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 60:702–16
- Trevaskis NL, Charman WN, Porter CJ. (2010a). Targeted drug delivery to lymphocytes: a route to site-specific immunomodulation? Mol Pharm 7:2297–309
- Trevaskis NL, Shanker RM, Charman WN, et al. (2010b). The mechanism of lymphatic access of two cholesteryl ester transfer protein inhibitors (CP524,515 and CP532,623) and evaluation of their impact on lymph lipoprotein profiles. Pharm Res 27:1949–64
- van Zuylen L, Verweij J, Sparreboom A. (2001). Role of formulation vehicles in taxane pharmacology. Investig New Drugs 19:125–41
- Woo JS, Lee CH, Shim CK, et al. (2003). Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res 20:24–30
- Yang SC, Gursoy RN, Lambert G, et al. (2004). Enhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitors. Pharm Res 21:261–70