
Abstract
Context: Applicability of natural polymers in pharmaceutical drug delivery.
Objective: The objective of the present investigation was to evaluate the applicability of badam gum (BG) obtained from Terminalia catappa LINN, belongs to the family combretaceae as a buccoadhesive polymer using metoprolol succinate as a model drug.
Methods: Tablets were prepared by wet granulation technique. Compression coating technique was employed for the preparation of unidirectional release buccal tablets using cellulose acetate as an impermeable backing layer.
Results: Muco/buccoadhesive properties of the BG were increased with the increase in the concentration of polymer which was evident form the detachment force measurement, ex vivo residence time, and swelling studies. MBG 2 was found to be the optimized formulation based on drug dissolution studies and bioadhesion studies. FTIR and DSC studies performed on the optimized formulation indicated no drug–polymer interaction. MBG 2 was found to be stable after accelerated stability testing for 6 months as per ICH guidelines. Pharmacokinetic studies of the optimized formulation were performed in six healthy human volunteers in comparison with that of the commercial extended release oral tablet GUDPRESS XL-25 by estimating pharmacokinetic parameters and mean residence time (MRT). It was found that there is a significant increase in the bioavailability of metoprolol succinate from BG formulation which was evident from the high AUC and MRT values compared with the commercial formulation.
Conclusion: The above results clearly indicated that badam gum can be used as a mucoadhesive polymer for buccal drug delivery.
Introduction
Due to advances in the drug delivery systems, there is a continuous need for the development of new excipients. Development or synthesis of new excipients is a cumbersome, time consuming, expensive process, and has some environmental related issues. Hence, exploring the new excipients from the abundantly available natural sources is economic and safer.
Natural polymers have certain advantages over synthetic polymers like they are economical, abundant, readily available, capable of chemical modification, non-toxic, potentially bio-degradable, and with a few exceptions also bio-compatible (Amelia et al., Citation2011). Despite the advantages, they have certain disadvantages like microbial contamination, batch-to-batch variation, isolation and purification, and possibility of metal contamination (Shirwaikar et al., Citation2008).
Natural polymers have many applications as excipients in dosage form design and manufacture of solid matrix systems, implants, films, beads, microparticles, nanoparticles, inhalable, injectable systems, as well as liquid formulations (Ikoni et al., Citation2011; Kulkarni et al., Citation2012).
Many plants that grow under semi-arid conditions produce exudates/gums in copious quantities when their bark is damaged which serves to seal the wound and prevent dehydration of plant. Gums are mixture of long-chain polysaccharides that become mucilaginous when mixed with water and can be hydrophobic and hydrophilic in nature. These are amorphous translucent solids insoluble in alcohol and, in most organic solvents, but soluble in water to yield viscous, adhesive solutions, or form gels by retaining the absorbed water. Plant species of many families can produce gums, when grown under adverse conditions like high elevation, heat, and lack of moisture. This clearly suggests that the secretion of gum is favorable under extreme conditions. Sometimes they are considered to be the result of an infection of the plant by microorganisms. Gum production has also been attributed to fungus growth of plants with the liberation of enzymes that synthesize the complex polysaccharides (Tyler et al., Citation1988).
Extensive research was carried out in the exploration of these plant gums as pharmaceutical excipients. Badam gum (BG), a plant exudate obtained from Terminalia catappa LINN, belongs to the family combretaceae. As BG is a novel polysaccharide, physico-chemical characterization was performed earlier in our laboratory by Srikanth et al. (Citation2012) in order to establish its suitability as pharmaceutical excipient.
Srikanth et al. developed gastric floating matrix tablets of propranolol hydrochloride and concluded that the gum has good swelling nature with drug release retarding property and can be used as a controlled release polymer. This was the only work reported on applicability of BG as a pharmaceutical excipient.
As the BG has sufficient swelling and sticking property, in the present investigation for the first time, we are trying to evaluate the buccoadhesive nature of BG using metoprolol succinate as a model drug. Buccal route of administration has certain advantages over other routes as it bypasses first pass metabolism and gastro-intestinal degradation of drugs by delivering directly into systemic circulation.
Metoprolol succinate is β-adrenergic receptors’ antagonist (Frishman & Alwarshetty, Citation2002) used in the treatment of hypertension. Metoprolol succinate is a BCS class I drug, with a short biological half-life and suffers first pass metabolism with bio-availability of 50%. An increased frequency of administration is required to achieve desired therapeutic effect. Hence, it was selected as the model drug in the design of buccoadhesive controlled release dosage forms.
The objective of the present study is to evaluate the suitability of the BG as a buccoadhesive polymer. Unidirectional release metoprolol succinate tablets were prepared by wet granulation technique with cellulose acetate as a backing layer to avoid the drug release from the three surfaces that are exposed to non-mucosal area. Optimized buccoadhesive formulation was further studied for its pharmacokinetic behavior in comparison with the orally administered commercial metoprolol succinate-extended release tablet (GUDPRESS XL-25) in healthy human volunteers.
Materials and methods
Metoprolol succinate was obtained as a gift sample from Dr. Reddy’s laboratories Ltd (Hyderabad, India). BG samples were obtained from the Palaniappa Chettiar Traders, Rasappa Chetty Street, Park Town, Chennai, Tamil Nadu, India. An analytical grade of acid orange dye was procured from a local vendor. All other reagents and chemicals used were of analytical grade.
Preparation of unidirectional release BG-based metoprolol succinate buccoadhesive tablets
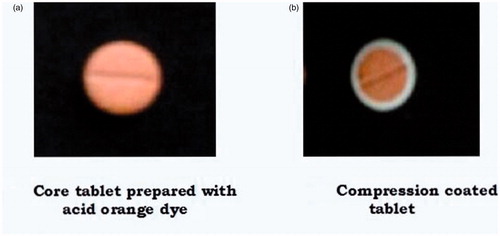
Development of unidirectional release buccoadhesive tablets involves preparation of core tablets and coating of the core tablets with water impermeable polymer ().
Table 1. Formulation of BG-based buccoadhesive tablets of metoprolol succinate.
Preparation of core tablet
Core tablets were prepared by wet granulation technique. According to the formulae presented in , all the ingredients sufficient for a batch of 200 tablets were weighed and passed through the sieve 40 (420 µm). The drug was geometrically mixed with BG and diluent in a poly bag until a homogenous blend was achieved. Polyvinyl pyrrolidone (PVP) 5% w/v solution in isopropyl alcohol (IPA) was used as a binder and the dough mass was passed through sieve 18 (1 mm) and dried at 60 °C in a hot air oven. The dried granules were sifted through sieve 30 (600 µm) to break down into smaller uniform granules and lubricated with magnesium stearate and talc. Lubricated blend was compressed into tablets on a 12-station rotary tablet punching machine (Karnavati Engineering Ltd., Nani Kadi Part, Gujarat, India) using 6 mm round concave punches with compression force to obtain a hardness of 2–3 kg/cm2.
Compression coating of the core tablet
Core tablets were coated on three surfaces except the surface that will be in contact with the mucosal membrane using cellulose acetate by compression-coating technique. Minimum amount of the coating material required for uniform coating of the three surfaces of core tablet was determined by the trial-and-error method and it was fixed as 75 mg. Cellulose acetate was used as a coating material in the present investigation which retards the permeability of the water across the coat and hinders the drug release from the coated surface.
Core tablet was placed in the center of the die cavity of 9 mm punches 75 mg of cellulose acetate was added on to the core and compressed with a compressional force to obtain a hardness of 3–5 kg/cm2. According to Higuchi et al., there exist a liner relationship between tablet hardness and logarithm of compressional force except at higher forces (Higuchi, Citation1954).
Compression-coated tablets with differentiation between core and coat are shown in . Differentiation between the coat and the core tablet became difficult in the cross section depicted in without color and hence acid orange dye was used in the coat for differentiating the coat from core tablet for presentation in figure for showing uniformity of compression coating. In regular studies, no color was used.
Figure 1. Buccoadhesive tablets of metoprolol succinate.
Evaluation of the prepared tablets
Prepared buccoadhesive tablets were evaluated for physico-chemical properties like hardness, friability, thickness, uniformity of weight, drug content uniformity (Indian Pharmacopoeia, Citation2010). In vitro dissolution, buccoadhesion strength, ex vivo residence time, swelling, and surface pH studies were also performed.
In vitro dissolution
In vitro dissolution of the prepared buccoadhesive tablets was studied using USP XXIV dissolution rate test Apparatus-II (Model: DISSO 2000, M/s. Lab India, Andhra Pradesh, India) employing the paddle stirrer. Uncoated surface of the tablet was exposed to dissolution medium by the adhesion of the tablet to a 2 × 2 cm glass slide with cyanoacrylate glue. About 500 ml of pH 6.6 phosphate buffer was used as a dissolution medium maintained at a temperature of 37 ± 0.5 °C and the paddle was rotated at 50 rpm (The United States Pharmacopoeia, Citation2007). Aliquots (5 ml each) were withdrawn at predetermined time intervals by means of a syringe fitted with a 0.45 µm prefilter and immediately replaced with 5 ml of a fresh medium maintained at 37 ± 0.5 °C. The absorbance of the samples was measured using a UV–visible spectrophotometer at 222 nm after suitable dilution with the medium.
Measurement of buccoadhesion strength
Buccoadhesive strength of the tablet was calculated by measuring the force required to detach the tablet from the mucosal surface (Aditya et al., Citation2010).
The detachment force was measured on a modified balance in which the right pan was removed, a plastic beaker was kept in left pan and both the sides were balanced by weights. The buccal mucosal tissue of swine was collected from the local slaughterhouse and underlying connective tissue was removed and washed with pH 6.6 phosphate buffer. The mucosal membrane was cut and attached to the glass slide with the mucosal side facing outwards using cyanoacrylate glue, the slide was attached to the petri plate, and it was placed on the right side of the balance. The tablet was attached to the glass slide facing the core outwards using cyanoacrylate glue. Glass slide was suspended on the right-hand side of the balance using a non-elastic thread and the height of thread was adjusted. The mucus layer and the tablet were wetted with pH 6.6 phosphate buffer, and the tablet was fixed to the mucus layer by applying a little pressure with the thumb. This was kept undisturbed for 5 min. On the left-hand side, water was added slowly into a plastic beaker with the help of a burette till the tablet just separated from the membrane surface. Weight of water (grams) required to detach the tablet was measured and the detachment force was calculated in milli Newtons which corresponds to mucoadhesive strength of the tablet. Experiment was performed in triplicate (n = 3), and the mean value was calculated:
(1)
Determination of the ex vivo residence time
The ex vivo residence time of the tablet was determined using a modified USP disintegration apparatus in which baskets were removed and the buccal mucosa which was previously washed was attached to the glass slide with the mucosal side facing outwards using cyanoacrylate glue (Nakamura et al., Citation1996). This was vertically fixed to shaft of the disintegration apparatus with the help of glass rod. Core side of tablet was wetted with 0.5 ml of pH 6.6 phosphate buffer and attached to the mucus tissue with a little pressure to develop initial contact. About 500 ml of pH 6.6 phosphate buffer maintained at 37 °C was used as a medium. The apparatus was allowed to run in such a way that the tablet was completely immersed in the buffer solution at the lowest point and was out at the highest point. The time taken for the complete erosion or detachment of the tablet from the mucosal surface was recorded. The experiments were performed in triplicate (n = 3) and the mean was determined.
Surface pH study
The buccoadhesive tablet was allowed to swell by placing the core in contact with 1 ml of pH 6.6 phosphate buffer for 2 h at room temperature (Bottenberg et al., Citation1991). The pH was measured by bringing the pH paper (range 2–14) in contact with the surface of the tablet and the values are measured.
Swelling studies
About 2% agar gel was prepared and poured into a petri dish and allowed to solidify (Garima Sharma et al., Citation2006). Buccoadhesive tablets were weighed individually (W1) and placed in petri dish with the core facing agar gel and allowed to swell. Tablets were removed at time intervals of 1, 2, 3, 4, 5, 6, 7, and 8 h from the petri dish and the excess surface water was removed carefully with the tissue paper. The swollen tablets were then reweighed (W2). This experiment was performed in triplicate. The swelling index was calculated according to the following equation:
(2)
Release kinetics
To analyze the drug release from the dosage form, by model dependent approach dissolution data was fitted in to popular models.
The order of drug release was established by analyzing zero-order and first-order release profiles (Wagner, Citation1969) and the mechanism of drug release was established from Higuchi (Citation1963), Hixon–Crowell cube root model (Hixson & Crowell, Citation1931), and Korsemeyer Peppas (Korsmeyer et al., Citation1983) equations.
Characterization of the drug interaction studies of the optimized formulation
Interaction between the drug and the polymer plays an important role with respect to drug release from the formulation. These interactions can be determined by FTIR spectroscopy, DSC, and XRD studies.
Fourier transform infrared spectroscopy (FTIR)
FTIR spectra can be used to detect drug–excipient interactions by following the shift in vibrational or stretching bands of key functional groups. FTIR spectra were obtained by using an Alpha FTIR spectrophotometer (Bruker Optik GmbH, Ettlingen, Germany). All the spectra were analyzed using OPUS 6.5 software (Bruker Optics Inc., Billerica, MA). Samples were prepared by the KBr pellet method, in which samples are prepared by gently mixing 1 mg of the sample with 200 mg of KBr. The spectra were scanned over a wave number range of 4000–500 cm−1.
Differential scanning calorimetry
DSC is a frequently used thermo-analytical technique that generates data on melting endotherms and glass transitions. DSC was performed utilizing Mettler DSC 821 (Mettler-Toledo, Greifensee, Switzerland). Samples of 3–4 mg were encapsulated and hermetically sealed in flat-bottomed aluminum pan with crimped on lid. The pans were positioned on a sample pan holder. Samples were allowed to equilibrate for 1 min and then heated in an atmosphere of nitrogen over a temperature ranging from 0 to 240 °C with a heating rate of 10 °C/min. An empty aluminum pan is served as a reference. Nitrogen was used as a purge gas, at the flow rate of 20 ml/min for all the studies. Thermograms were obtained by using the STARe SW 9.10 software (Mettler Toledo, Columbus, OH).
X-ray diffractometry (XRD)
XRD is a powerful tool in detecting crystallinity. The samples were recorded on XRD (PW 1729, Philips, Amsterdam, The Netherlands). XRD patterns were recorded using monochromatic Cu Kα radiation with a Ni filter at a voltage of 45 kV and a current of 40 mA between 5° and 50° 2θ values. The data were processed with the software Diffrac Plus V1.01 (Vernier Software & Technology, Beaverton, OR).
Stability studies
Stability studies of the optimized formulation were carried out as per the ICH guidelines to assess its stability (World Health Organization, Citation2003). The stability studies were performed by using stability chambers, Kemi-KHO-3A (Kadavil Electro Mechanical Industries, Kerala, India). Tablets were packed in a HDPE screw-capped containers and stored at 40 ± 2 °C/75 ± 5% (RH) for accelerated studies. Samples were removed at 3 and 6 months and analyzed for physico-chemical and mucoadhesive properties.
In vivo studies
In vivo residence time studies
Six healthy human volunteers with a mean age of 25 ± 3 years were selected to participate in the study. Buccal tablets were attached to the cheek by pressing them with a little pressure for 30 s without moistening before application. Time taken for the dislodgement/erosion of the tablet was noted and the mean residence time was calculated. After completion of the study, a questionnaire was given to the volunteers to score the parameters such as irritancy, comfort, taste, dry mouth, salivation, and dislodgement of the system during the study.
In vivo pharmacokinetic study
Pharmacokinetic evaluation of the optimized formulation MBG 2 was performed in comparison with a commercial oral extended released tablet (GUDPRESS XL-25, Batch no. GXL 2502, manufactured by Mankind Pharma Ltd., Gurgaon, Haryana, India) on six healthy human male volunteers with a mean age of 25 ± 3 years and a mean body weight of 72 ± 8 kg, after getting an approval from the institutional ethics committee, A.U. College of Pharmaceutical Sciences, Andhra University, Visakhapatnam, India (IEC/29/AUCOPS/2010). Experiment was conducted in an open label, balanced, randomized, two-period, two-treatment, and single-dose crossover study design in which six volunteers are divided into two groups and they receive one treatment (product) each for every 7 d such that two products are tested in all the six volunteers during the study after getting written informed consent from the volunteers. The volunteers participated in the study abstained from other drug during the study and the studies were carried out under the supervision of a physician. The subjects were fasted overnight at least 10 h prior to dosing in each period of the study and were given standardized breakfast, lunch, and dinner after collecting the zero hour sample. During each period, 3 ml venous blood samples were collected from the forearm veins of each subject in AcCuvet tubes (Quantum Biologicals Pvt Ltd., Chennai, India) containing K3EDTA. Blood samples were collected at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 18, and 24 h. Plasma was immediately separated by centrifugation at 7500 rpm for 15 min from the blood samples and stored in frozen conditions at −20 °C with appropriate labeling of subject code number, study date, and collection time prior to analysis. The concentration of metoprolol succinate in human plasma samples was measured by the reverse phase high-performance liquid chromatography (HPLC).
Analysis of the metoprolol succinate in human plasma by HPLC
Prior to injection of the sample, metoprolol succinate was extracted from the plasma by precipitating the plasma proteins as per the following procedure 0.1 ml of plasma was transferred in to the Eppendorf micro centrifuge tubes. To this, 0.1 ml of internal standard (metformin hydrochloride) and 0.8 ml of methanol were added and vortexed for 3 min in Remi cyclo mixer (R-24, Rimek, Mumbai, India) followed by centrifugation for 20 min at 7500 rpm. After centrifugation, supernatant was collected and 20 µl of the supernatant was injected in to the HPLC by Hamilton microsyringe (RZ-07939-01, Mumbai, India). Peak area and height were computed and the concentration of the metoprolol succinate in the sample was determined by the standard graph (peak area ratios of metoprolol to metformin versus concentration of metoprolol succinate).
A sensitive HPLC method developed by Venkateswarlu et al. (Citation2010) was used in the present study to estimate the plasma concentration of drug. Samples were analyzed using reversed phase C-18, Pack Pro C18 and YMC (150 × 4.6 mm, particle size 3 µm, and pore size of 12 nm) column. The mobile phase consisting of acetonitrile:pH 6.2 phosphate buffer (20:80) at a flow rate of 1.0 ml/min and maintained at a pressure of 120–180 kg/cm2. The mobile phase was filtered through a 0.45 µm-membrane filter before its use. The UV absorption was measured at 223 nm using a Shimadzu SPD M10 AT VP model Photo Diode Array (PDA) detector (Shimadzu Corporation, Kyoto, Japan).
Pharmacokinetic parameters of the each subject peak plasma concentration (Cmax), time at which Cmax occurred (Tmax), absorption rate constant (Ka) (Wagner & Nelson, Citation1964) area under the curve (AUC) (Shargel & Yu, Citation1999), biological half-life (t1/2), elimination rate constant (Kel), and mean residence time (MRT) were estimated by KINETICA 4.4.1 software (Thermo Electron Corporation, Reading, Berkshire, UK).
Pharmacokinetic parameters were statistically analyzed using paired sample t test. A value of p < 0.05 was considered to be significant and the results were expressed as mean ± standard deviation (SD).
Results and discussion
Evaluation of tablets
Metoprolol succinate buccoadhesive tablets were evaluated both for their tableting properties like hardness, friability, thickness, uniformity of weight, and drug content uniformity, in vitro dissolution and mucoadhesive properties like buccoadhesion strength, ex vivo residence time, swelling, and surface pH study. Hardness of all the formulations was found to be in the range of 3–5 kg/cm2. The thickness of the tablets was measured using Vernier callipers and was found to be 2.5 ± 0.2 mm. Friability, uniformity of weight, and drug content uniformity were within the limits as per the Indian Pharmacopoeia (IP). Results of the corresponding studies are shown in .
Table 2. Tableting characteristics of metoprolol succinate buccoadhesive tablets.
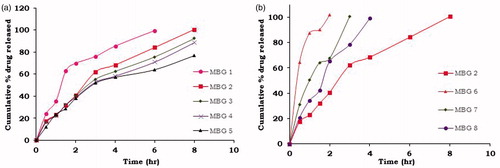
Buccoadhesive tablets were formulated to release 100% of drug up to 8 h. In vitro dissolution studies were performed in pH 6.6 phosphate buffer and dissolution profiles are shown in . From the dissolution studies, it was observed that as the concentration of BG increases drug release was prolonged indicating the drug release retarding ability of the BG. Formulation MBG 2 containing drug to BG in the ratio of 1:1 and Pharmatose as a diluent extended the drug release up to 8 h and was considered as preliminary optimized formulation. To study the effect of different diluents on drug release, further formulations were prepared at same drug to BG ratio (1:1) with varying diluents MBG 6 (MCC), MBG 7 (DCP), and MBG 8 (mannitol). Formulation MBG 8 controlled the drug release up to 4 h which was not desirable. Burst release was observed for the formulations containing microcrystalline cellulose (MCC) and di basic calcium phosphate (DCP) as diluents as they released 100% of drug within 2 and 3 h, respectively. This might be due to the elastic nature of the MCC and hydrophobic nature of the DCP which hinder the release of highly soluble drug from the matrix tablet resulting in the development of excess pressure in tablet as tablet was coated with impermeable cellulose acetate layer on three sides where there is less/no chance for the drug release. From the above observations, MBG 2 was considered as optimized formulation.
Figure 2. Dissolution profiles of BG-metoprolol succinate buccoadhesive tablets with (a) varying concentrations of BG (b) varying diluents.
Drug release kinetics
The dissolution data was fitted to kinetic parameters and the correlation coefficient values (‘r’ values) of release kinetics of all the formulations of metoprolol succinate are shown in . All the BG-based formulations followed first-order rate kinetics with the non-Fickian diffusion mechanism.
Table 3. Correlation coefficient values and release kinetics of BG-based metoprolol succinate buccoadhesive tablets.
Mucoadhesive characteristics
Results of the mucoadhesive properties of the prepared formulations are shown in . Mucoadhesive strength and ex vivo residence time of the formulations were found be in the range of 133–224 mN and 7–12 h, respectively. While the maximum mucoadhesive strength 224 mN and ex vivo residence time of 12 h were observed for the formulation MBG 5 containing drug to BG in the ratio of 1:2, the optimized formulation MBG 2 (drug to BG 1:1) has shown a mucoadhesive strength of 139 mN and ex vivo residence time of 8 h.
Table 4. Buccoadhesive strength, ex vivo residence time, and surface pH values of BG-based metoprolol succinate tablets.
Surface pH of the tablets was found to be in the range of 6–7 indicating the suitability of BG gum for buccal administration as the pH of the formulation was with in the buccal pH range.
All the prepared formulations were evaluated for their swelling ability. Swelling index was measured up to 8 h and results are given in . Swelling index values were found to be increased with the increase in concentration of BG.
Table 5. Swelling index (%) of BG based metoprolol succinate tablets (mean ± s.d., n = 3).
From the obtained results, it can be concluded that mucoadhesive property of BG is directly proportional to its concentration in the formulations.
Drug–polymer interaction studies
FTIR studies
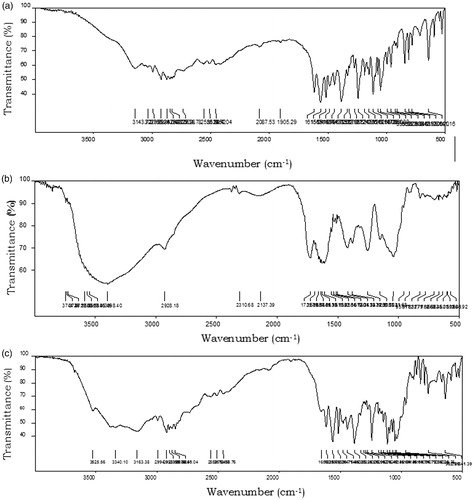
FTIR spectrum of metoprolol succinate showed –NH symmetric stretching of the 2°-amine at 3194 cm−1, –C = O stretching of succinic acid at 1615 cm−1, –C = C– ring stretching at 1487 cm−1, and –C–O–C– asymmetric stretching at 1242 cm−1. FTIR spectra of the optimized formulation MBG 2 showed all the characteristic peaks of metoprolol succinate with minor shifts. The FTIR spectrum of MBG 2 showed –NH stretching (2°-amine), –C = O stretching of succinic acid, and –C = C ring stretching of metoprolol succinate at 3155, 1616, and 1475 cm−1, respectively. The above data clearly indicated the absence of any chemical interaction between the drug and the BG used in the present study. The FTIR spectra of metoprolol succinate and optimized formulation MBG 2 are shown in .
Figure 3. FTIR spectra of (a) metoprolol succinate, (b) badam gum, and (c) MBG 2.
XRD studies
X-ray diffraction studies were carried out to disclose the crystalline modifications of drug during the preparation. The X-ray diffractograms of metoprolol succinate and optimized formulation MBG 2 are shown in . The diffractograms of the metoprolol succinate showed sharp peaks at an angle of 7.05, 14.10, 14.38, 20.04, 21.26 23.16, 24.23, and 26.20 (°2θ). The natural polymer BG showed no peaks indicating its amorphous nature.
Figure 4. XRD spectra of (a) metoprolol succinate, (b) badam gum, and (c) MBG 2.
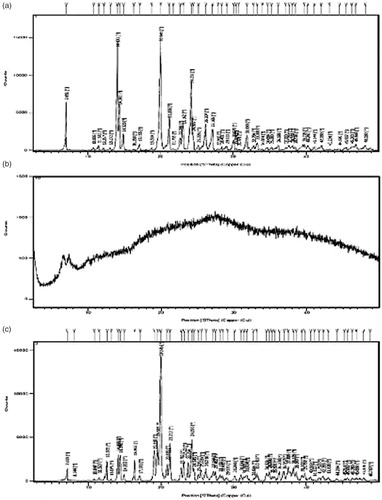
The diffractograms of optimized formulation MBG 2 showed decreased intensity of the drug peaks indicating a decrease in the crystallinity of the drug due to the compression force applied during the preparation of tablets.
DSC studies
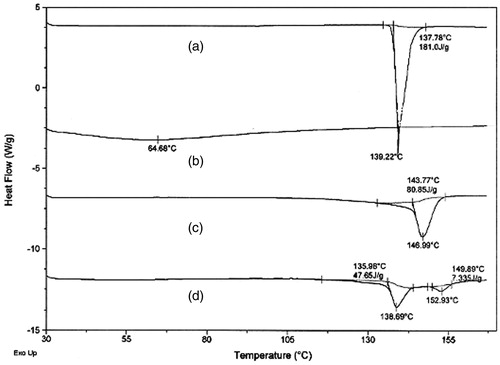
The DSC thermograms of pure metoprolol succinate exhibited sharp endothermic peak at a temperature of 139.22 °C. In the case of optimized formulation, characteristic drug peak was observed at 138.69 °C as shown in . This minor or negligible shift in the drug peak indicated that there was no physical or chemical interaction between the drug and the polymer used in the study.
Figure 5. DSC thermograms of (a) metoprolol succinate, (b) badam gum, (c) pharmatose, and (d) MBG 2.
Stability studies
Accelerated stability studies were conducted for the optimized formulation by packing tablets in a HDPE screw-capped containers and stored at 40 ± 2 °C/75 ± 5% (RH) for 6 months. Samples were removed at 3 and 6 months and analyzed for physical appearance, average weight, assay, hardness, buccoadhesive strength, ex vivo residence time, surface pH, and in vitro dissolution studies.
No visible physical changes were observed in all the formulations withdrawn from the humidity chambers. The average weight, hardness, and assay of all the formulations complied with the compendia standards for tablets and the results are given in . There was no significant difference in the buccoadhesion strength, ex vivo residence time, and surface pH of the optimized formulation. Since these formulations are under developmental stage, test samples for stability studies were packed in screw-capped HDPE bottles instead of strip and/or blister packing.
Table 6. Stability data of MBG 2.
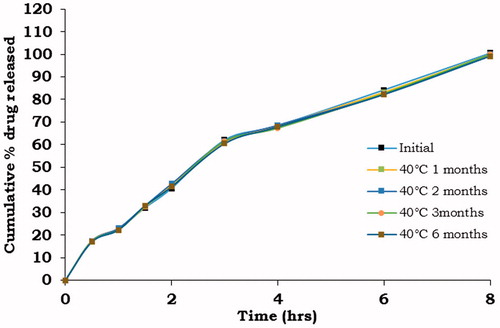
The dissolution profiles of metoprolol succinate from the MBG 2 before and after storage are given in . The drug-release profiles of all the formulations did not change significantly after storage at 40 ± 2 °C/75 ± 5 %RH for a period of 6 months.
Figure 6. Comparative dissolution profiles of MBG 2 before and after storage at 40 ± 2 °C/75 ± 5% RH.
In vivo studies
In vivo residence time study
Optimized formulation was studied for in vivo residence time in healthy human volunteers along with the residence time several parameters such as irritancy, comfort, taste, dry mouth, salivation, and dislodgement of the system during the study was observed.
Pharmacokinetic studies
Pharmacokinetics of the metoprolol succinate from optimized formulation MBG 2 in comparison with the commercial oral tablet (GUDPRESS XL-25) were studied in six healthy human volunteers. Mean plasma concentration of the metoprolol succinate following the administration MBG 2 and GUDPRESS XL-25 are shown in . And calculated parameters are given in .
Figure 7. Comparative mean plasma drug concentrations of MBG 2 and GUDPRESS XL-25.
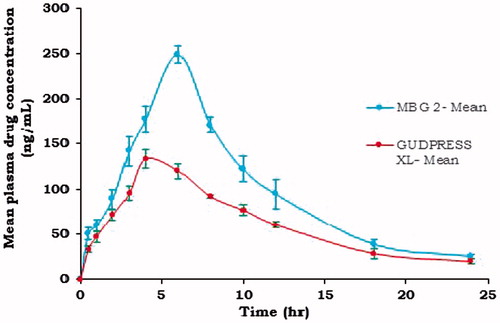
Table 7. Pharmacokinetic parameters (mean ± s.d.) of metoprolol succinate obtained from MBG2 and GUDPRESS XL-25.
Cmax of metoprolol succinate from MBG 2 and GUDPRESS XL-25 was found to be 248.53 ± 9.27 and 133.12 ± 9.43 ng/ml, respectively. The Tmax values were found to be 6 h for MBG 2 and 4 h for GUDPRESS XL-25. The Kel values calculated from the slope of the terminal portion of log plasma concentration of metoprolol succinate versus time graph as shown in were found to be 0.103667 ± 0.01 and 0.092857 ± 0.02 h−1 for MBG 2 and GUDPRESS XL-25, respectively. The corresponding t1/2 values were found to be 6.69 ± 0.95, and 7.46 ± 1.32 h for MBG 2 and GUDPRESS XL-25, respectively. The Ka values obtained by the application of the Wagner–Nelson method to the plasma concentration data as shown in were found to be 0.303 ± 0.02 h − 1 for MBG 2 and 0.384 ± 0.03 h−1 for GUDPRESS XL-25. The values of AUC0–24 and MRT0–24 for MBG 2 were 2329.72 ± 115.40 ngh ml − 1 and 8.73 ± 0.22 h and for GUDPRESS XL-25, they were 1463.46 ± 97.52 ngh ml−1 and 8.91 ± 0.18 h, respectively.
Figure 8. Log mean plasma concentration–time curves of MBG 2 and (GUDPRESS XL-25).
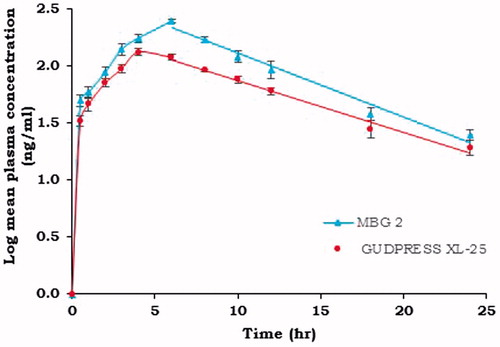
Figure 9. Log % drug unabsorbed versus time plot (Wagner–Nelson plot) of (a) MBG 2 and (b) GUDPRESS XL-25.
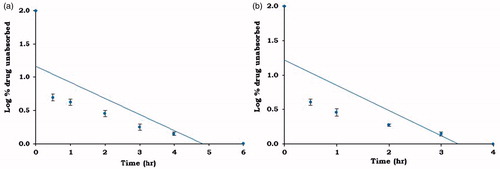
The in vivo data demonstrated that the experimental formulation MBG 2 exhibited larger AUC values than marketed formulation. To test the significant differences if any between the pharmacokinetic parameters of experimental formulation and commercial formulation, the pharmacokinetic data were subjected to statistical analysis, and the results are given in . The results of statistical analysis indicated no significant difference in Ka and t1/2 but significant difference was observed in Cmax, Tmax, AUC0–24, and MRT0–24 values of MBG 2 and GUDPRESS XL-25 (p < 0.05).
Table 8. Statistical summary of pharmacokinetic parameters (p < 0.05) of metoprolol succinate tablets (n = 6).
The relative bioavailability obtained with optimized tablets MBG 2 is 159.19% compared with those of the commercial formulation, indicating the improvement in bioavailability with this formulation. This further indicated that the developed buccoadhesive tablets are more effective in improving the bioavailability of metoprolol succinate. This may be due to the reason that the buccal route of administration can bypass the first pass metabolism of drug which resulted in the improved bioavailability. Increased AUC and MRT values of prepared buccoadhesive tablets reflected that effective plasma concentrations were maintained for longer times compared to marketed formulation.
In vitro–in vivo correlation
An IVIVC should be evaluated to demonstrate the predictability of in vivo performance of a drug product from its in vitro dissolution characteristics.
The main objective of developing and evaluating an IVIVC is to establish the dissolution test as a surrogate for human bioequivalence studies, which may reduce the number of bioequivalence studies performed during the initial approval process as well as with certain scale-up and post-approval changes.
Even though Level B utilizes all the in vitro and in vivo data, it cannot considered as it being a point-to-point correlation, it does not reflect the actual in vivo plasma level curve and hence one cannot rely up on a Level B correlation. Similarly Level C correlation represents a single point correlation and it does not reflect the complete shape of the plasma level which is a critical factor that defines the performance of modified release products.
A significant correlation between the in vitro and in vivo parameters indicated that the in vitro release procedure is capable of discriminating between formulations having different bioavailabilities. Level A IVIVC is considered to be the most informative and is recommended for regulatory process.
The data for linear correlation plots for percentage of drug absorbed in vivo and percent drug released in vitro at different time intervals are shown in . A very good correlation between the in vivo and the in vitro data was obtained for all the three formulations tested with correlation coefficients of 0.9827 and 0.9843 for MBG 2 and GUDPRESS XL 25, respectively. This kind of Level A correlation is quite important since it represents a point-to-point relationship between the in vitro dissolution and the in vivo input rate of the drug from the dosage form. Thus, an in vitro dissolution curve can serve as a surrogate for in vivo performance.
Figure 10. IVIV Level A correlation of (a) MBG 2 and (b) GUDPRESS XL-25.
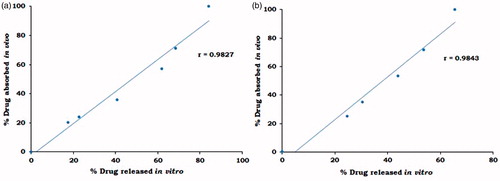
It can be concluded from the results of the present study that developed buccoadhesive tablets could achieve the required Level A correlation.
Conclusion
The present study indicated the applicability of natural polymer BG as a buccoadhesive polymer. In vitro drug-release studies indicated that BG can be used as a control-release polymer. Mucoadhesive characteristics of the gum was found to be good and increased with an increase in the concentration of the polymer indicating its applicability in buccal drug delivery.
BG was found to have good in vivo performance without any signs of irritation and damage to the buccal tissue. Pharmacokinetic evaluation of the optimized formulation and GUDPRESS XL-25 in healthy human volunteers indicated an increase in the bioavailability of the metoprolol succinate from MBG 2 as evident from the higher AUC and MRT values with % relative bioavailability of 159.19.
Thus the above results clearly indicated that BG can be used as a mucoadhesive polymer for buccal drug delivery.
Acknowledgements
We are thankful to Venkata Srikanth Meka, Sunil, S. A. and Srinivas N. for providing valuable information to carry out this work.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. We are very much grateful to Ministry of Earth Sciences (MoES) for awarding us research fellowship and financially supporting this project.
References
- Amelia MA, Rakesh RD, Shilpa NS. (2011). Recent investigations of plant based natural gums, mucilages and resins in novel drug delivery systems. Indian J Pharm Educ Res 45:86–99
- Aditya G, Ganeshkumar, Manasa G, et al. (2010). Design and evaluation of controlled release mucoadhesive buccal tablets of lisinopril. Int J Curr Pharm Res 2:24–7
- Bottenberg P, Cleymaet R, Muynek CD, et al. (1991). Development and testing of bioadhesive, fluoride-containing slow-release tablets for oral use. J Pharm Pharmacol 43:457–64
- Frishman WH, Alwarshetty M. (2002). β-Adrenergic blockers in systemic hypertension: pharmacokinetic considerations related to the current guidelines. Clin Pharmacokinet 41:505–16
- Sharma G, Jain S, Tiwary AK, Kaur G. (2006). Once daily bioadhesive vaginal clotrimazole tablets: design and evaluation. Acta Pharm 56:337–45
- Higuchi T. (1963). Mechanism of sustained action medication: theoretical analysis of rate release of solid drugs dispersed in solid matrices. J Pharm Sci 52:1145–9
- Higuchi T, Elowe LN, Busse LW. (1954). J Am Pharm Assoc, Sci Ed 43:685
- Hixson AW, Crowell JH. (1931). Dependence of reaction velocity upon surface and agitation (I) theoretical consideration. Ind Eng Chem 23:923–31
- Ikoni J Ogaji, Elijah I Nep, Jennifer D Audu-Peter. (2011). Advances in Natural Polymers as Pharmaceutical Excipients. Available from: http://dx.doi.org/10.4172/2153-2435.1000146. [last accessed 18 Jan 2012]
- Indian Pharmacopoeia. (2010). Government of India, Ministry of Health and Family Welfare. 6th ed. : The Indian Pharmacopoeia commission, Central Indian Pharmacopoeia Laboratory, 185
- Korsmeyer R, Gurny R, Peppas N. (1983). Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35
- Kulkarni VS, Butte KD, Rathod SS. (2012). Natural Polymers – a comprehensive review. Int J Res Pharm Biomed Sci 3:1597–613
- Nakamura F, Ohta R, Machida Y, Nagai T. (1996). In vitro and in vivo nasal mucoadhesion of soluble polymers. Int J Pharm 134:173–81
- Shargel L, Yu ABC. (1999). Pharmacokinetics of oral absorption. In: Shargel L, ed. Applied biopharmaceutics and pharmacokinetics. 4th ed. : Prentice-Hall International, 234–54
- Shirwaikar A, Prabu SL, Kumar GA. (2008). Herbal excipients in novel drug delivery systems. Indian J Pharm Sci 70:415–22
- Srikanth MV, Sreenivasa Rao N, Sunil SA, Ramana Murthy KV. (2012). Characterization and in vitro drug release studies of a natural polysaccharide Terminalia catappa Gum (Badam Gum). AAPS Pharm Sci Tech 13:1451–64
- Tyler VE, Brady LR, Robbers JE. (1988). Pharmacognosy. 9th ed. : K.M. Verghese Company, Lea and Febiger, 45
- The United States Pharmacopoeia. (2007). Metoprolol succinate extended release tablets. USP30-NF25. : The United States Pharmacopeial Convention, Inc, National Publishing, 2648
- Venkateswarlu P, Kumar BN, Seshaiah K, Prasad VVV. (2010). Selective and sensitive method for the determination of metoprolol in human plasma using liquid chromatography coupled with tandem mass spectrometry. Acta Pharm 60:177–84
- Wagner JG, Nelson E. (1964). Kinetic analysis of blood levels and urinary excretion in the absorption phase after single doses of drugs. J Pharm Sci 53:1392–403
- Wagner JG. (1969). Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J Pharm Sci 58:1253–7
- World Health Organization. (2003). ICH harmonized tripartite guideline on stability testing of new drug substances and products. Q1A (R2), World Health Organization, Geneva