
Abstract
The objective of the current study was to prepare microspheres of salmeterol (SM) using poly (lactide-co-glycolide) (PLGA) and assess its viability to enhance the oral bioavailability. Microspheres of SM were prepared by oil-in-water emulsion-solvent evaporation method. The formulations were characterized in encapsulation efficiency, particle size, zeta potential, and in vitro release. The prepared microspheres were found to be spherical in shape with smooth surface. The size of microspheres ranged from 14.7 to 16.5 µm. The polydispersity index (PDI) was 0.12 ± 0.05 and the zeta potential was −33.2 ± 1.4 mV. In vitro release profile, SM was graduated released from the microspheres as time lapsed, suggesting that SM was well entrapped in SM-loaded PLGA microspheres. The model that fitted best for SM released from the microspheres was Higuchi equation. In vivo study, SM-loaded PLGA microspheres are thought to have the potential to maintain SM concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency.
Introduction
Salmeterol (SM, ) is a new kind of selective long-acting β2 agonist, acting on adrenergic receptors, effective by expansion of bronchial smooth muscle to control asthma, with a long duration of action, good tolerance, ideal for the current drug treatment for asthma (Cates & Lasserson, Citation2012). Hospital clinical work in China, SM and inhaled corticosteroids can be used as maintenance therapy by patients with moderate to severe chronic obstructive pulmonary disease (Spencer et al., Citation2011). However, SM is currently in clinical applications of inhalation and aerosol formulations only, with short dosing interval (every 12 h), high doses (each 50 μg), adverse reactions, which is not conducive to the onset or during the night exacerbations of asthma patients (Daley-Yates & Parkins, Citation2011; Harrison et al., Citation2011). It has important practical significance to develop a new formulation with sustained release in order to reduce the adverse reactions and the number of medication caused by drug valley phenomenon.
Figure 1. Structure of salmeterol (SM) (A) and Bambuterol (B).
Oral delivery was the preferred route for delivery of drugs because it afforded easy handling, high patient compliance, less stringent production conditions, and lower costs (Yin et al., Citation2006). The major problem for oral delivery of SM is its low bioavailability, which may be caused by the excretion effect of P-glycoprotein (Chang et al., Citation2006). Microsphere is a new delivery system, the drug with a suitable carrier by the microsphere technique into a ball, the selectivity and bioavailability of drugs can be improved after oral administration, to maintain a high blood concentration (Islam et al., Citation2012; Gupta et al., Citation2013). Microspheres prepared by the carrier material that can be divided into natural polymer microspheres (e.g. starch microspheres, albumin microspheres, gelatin microspheres, chitosan, etc.) and synthetic polymer microspheres (De La Vega et al., Citation2013; Hu et al., Citation2013). Because synthetic polymer prepared microspheres have good biocompatibility and biodegradability, it has gradually become a hot topic for research in recent years.
Among the variety of materials available to polymeric microspheres, the most widely used are the aliphatic polyesters, specifically poly (lactic acid), poly (glycolic acid) and their copolymers, and poly (lactide-co-glycolide) (PLGA) (Huang et al., Citation1997; Fahmy et al., Citation2005). PLGA is a widely used carrier material in drug delivery systems due to its biocompatibility and degradability into glycolic acid and lactic acid in the body. Recent advances in the design of these types of drug deliverers have included modification of the particle surface to improve stability and circulation throughout the body (Moghimi et al., Citation2001), biospecific targeting against cellular ligands or extracellular matrix components (Takeuchi et al., Citation2001; Kim et al., Citation2005; Farokhzad et al., Citation2006), and incorporation of diagnostic imaging agents (Fahmy et al., Citation2007).
The aim of this study was to prepare the PLGA microspheres in order to improving the oral bioavailability of SM. SM-loaded PLGA microspheres were prepared from oil-in-water emulsion-solvent evaporation method. The formulations were characterized in encapsulation efficiency, particle size, zeta potential, and in vitro release. The main objective of the current study was to assess its viability to augment the oral bioavailability. To our knowledge this is the first study which investigates the potential of PLGA microspheres to enhance the oral bioavailability of SM.
Materials and methods
Materials
PLGA 502H (lactide/glycolide ratio 50:50) was obtained as a gift sample from Boehringer Ingelheim (China). SM was obtained from Zhejiang PuLuo Medical technology co., Ltd. Bambuterol (internal standard) was purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Polyvinyl alcohol (PVA) (degree of hydrolysis: 87.0–89.0% (mol/mol), CPS: 4.6–5.4 mPas) was purchased from Shanghai Jingchun Reagent Co., Ltd. All other chemicals were of commercially analytical grade.
Preparation of the microspheres
SM-loaded PLGA microspheres were prepared from oil-in-water emulsion-solvent evaporation method. Briefly, 1 mL of organic phase was prepared by completely dissolving both SM (20 mg) and PLGA (200 mg) in chloroform. Then, the organic phase was added to 5 mL of 1% (w/v) PVA solution dissolved in distilled water in a constant rate, stirring for 1 min at a high speed to form oil-in-water (O/W) initial emulsion. Subsequently, the initial emulsion was added to 20 mL of 1% (w/v) PVA aqueous solution used as dilution, and magnetically stirred for 3 h at room temperature to evaporate the organic solvent. The solidified SM-loaded PLGA microspheres were collected by centrifugation (12 min, 4000 rpm) and washed three times with distilled water. After 1 mL of aqueous mannitol (20%, w/v) was added to prevent the aggregation of microspheres, the SM-loaded PLGA microspheres were freeze dried.
Evaluation of microspheres
Surface morphology
The surface morphology of SM-loaded PLGA microspheres was observed under transmission electron microscope (CM-200 FEG; Philips, Eindhoven, Netherlands) after negative staining with phosphotungstic acid solution (2%, w/v). The size and zeta potential of SM-loaded PLGA microspheres were measured by the light scattering method. The analyses were performed with a helium-neon laser (632.8 nm) at a scattering angle of 90 degrees at 25 °C using a Nicomp™ 380 ZLS (Particle Sizing Systems, Port Richey, FL). The samples were diluted to the appropriate concentration using deionized water before measurement.
Drug content, entrapment efficiency and drug loading
The various batches of the microspheres were subjected for drug content analysis. Accurately weighed microsphere samples were mechanically powdered. The powdered microspheres were dissolved in adequate quantity of phosphate buffer pH 6.8 and then filtered. The drug content of SM-loaded PLGA microspheres was detected by high-performance liquid chromatography (HPLC) (Carter & Cápka, Citation2008). HPLC analysis was performed using a Hypersil™ ODS-2 column (4.6 mm × 250 mm, 5-μm particle size; Elite Analytical Instruments Co, Ltd, Dalian China) on a Shimadzu® HPLC system (LC-20A; Shimadzu Corporation, Tokyo, Japan) with an ultraviolet detector at room temperature. The wavelength of the ultraviolet detector was set at 225 nm. Acetonitrile and 0.5% triethylamine solution (68:32, v/v, pH = 4.0) was used as the mobile phase at a flow rate of 1 mL/min. The accurate weight of microspheres was detected after lyophilization. The drug entrapment efficiency and drug loading was calculated by the following equations:
WSM represents the amount of SM loaded in the microspheres, WTotal represents the total SM amount added during preparation of the microspheres, and WSM-MS represents the weight of the SM-PLGA microspheres.
In vitro drug release
The in vitro release of SM-loaded PLGA microspheres was carried out in phosphate buffer of pH 6.8 containing 0.02% NaN3 known as antimicrobial agent. SM-loaded PLGA microspheres (containing 10 mg SM) were added into 2 mL of release buffer in dialysis bags (Mol≈10 000), and then the bags were put into a tube containing 100 mL of pH 6.8 phosphate buffer (containing 0.02% NaN3). All the samples were under the sink condition and kept in a constant shaking bath at 100 rpm maintained at 37 °C. At time intervals (0.5, 1, 2, 4, 8, 12, 16, 24, 32, 48 and 72 h), 5 mL of dialysis solution was taken for analysis; next, 5 mL of fresh medium was added. After filtration and appropriate dilution, the concentration of SM in release medium was measured by HPLC. Meanwhile, the release of SM solution (2 mL, 5 mg/mL) was monitored as control. The accumulative release was calculated, and the results were shown as mean ± SD (n=3).
In vivo studies
In vivo experiments were carried out in male, 6–8 weeks Sprague-Dawley rats (220–250 g) which were maintained on a half light cycle in an animal facility with unlimited access to food and water (Hospital Animal Ethical Committee, Shanghai, China). The oral bioavailability of SM-loaded PLGA microspheres was determined by estimating the serum SM at predetermined time points. Three groups of rats (six animals in each group) fasted for 12 h with free access to water were used in the study. The diluted SM injection (50 μg/mL) was administered intravenously at doses of 100 μg/kg. The SM-loaded microspheres were diluted with physiological saline to a SM concentration of 50 μg/mL and SM suspensions were prepared with 0.5% HPMC for orally administered at a dose of 250 μg/kg. The blood samples were collected (∼200 μL) from orbital using dry heparinized tubes at predetermined time intervals (0.25, 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 h). The sample withdrawn at time zero served as the baseline value. The samples collected were centrifuged at 12 000 rpm for 10 min.
Totally 100 µL volume of the plasma sample was transferred to a 3 mL plastic test tube together with 20 µL of I.S. solution (100 µg/mL). After vortex shaking for 0.5 min, 0.5 mL of diethyl ether was added and the mixture was vortexed for 2 min. After centrifugation at 3500 × g for 10 min, the upper organic layer was quantitatively transferred to a 5-mL glass tube and evaporated to dryness using evaporator at 40 °C. The residue was reconstituted in 50 µL of the mobile phase, and then vortex-mixed. After centrifugation at 3500 × g for 5 min, 20 µL aliquot of the solution was injected into the HPLC system for analysis.
Pharmacokinetic parameters
The pharmacokinetic parameters were calculated using non-compartmental pharmacokinetic model. Pharmacokinetic variables of interest included area under the concentration–time curve from time 0 to ∞ (AUC0–∞), peak concentration (Cmax), terminal elimination rate constant (Ke), half-life of the terminal phase (T1/2) and time to peak concentration (Tmax). A linear-up/logdown method of estimation was used for the calculation of AUC0–∞ and was obtained by adding Clast/Ke to AUC0–t. The terminal elimination rate constant (Ke) was determined from the slope of terminal exponential phase of the logarithmic plasma concentration–time curve. The elimination half-life (T1/2) was calculated using 0.693/Ke.
Data analysis
The statistical analysis of data was done by performing unpaired Student's t-test to determine the level of significance between two groups. The mean values were presented with standard deviation. p < 0.05 was considered as the level of significance.
Results and discussion
Preparation and characteristics of the microspheres
The microspheres of SM using PLGA were prepared by emulsion-solvent evaporation method. This technique appears to be suitable method for the preparation of SM-loading PLGA microspheres. Emulsion-solvent evaporate process is rapid, easy and two steps process which leads to the formation of porous microparticles. Optimum parameters were drug:copolymers = 1:10; the concentration of PVA solution was 1%; the time to evaporate was 3 h. It was observed that as the PVA concentration increased, in relation to the drug concentration, the particle size of the microspheres also increased (data not shown).
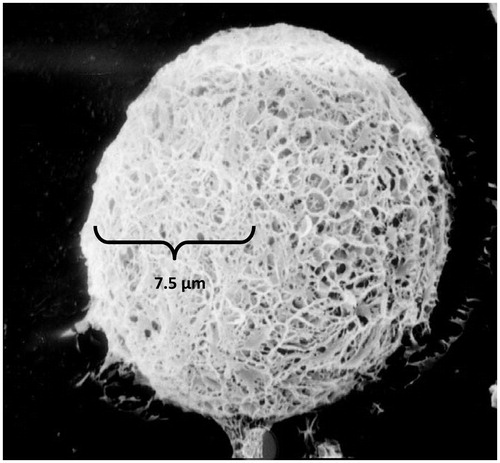
show transmission electron micrographs of the prepared microspheres. The size of microspheres ranged from 14.7 to 16.5 µm and the average size was 15.3 ± 1.45 µm. The polydispersity index (PDI) was 0.12 ± 0.05 and the zeta potential was −33.2 ± 1.4 mV. The particle size of the raw material (as obtained) was 83.8 ± 5.2 µm. The image showed that the microspheres have smooth surfaces and small holes. The main reason was because the organic solvent was extracted rapidly from the inner layers of the PLGA microspheres. The microspheres prepared were negatively charged, indicating the presence of PLGA at the surface of all the microspheres. Studies have mentioned that polymers with charged density can serve as good mucoadhesive agents.
Figure 2. Transmission electron microscope photograph of SM-loaded PLGA microspheres. × 5000.
The determination of drug content shows good uniformity. The drug content was found to be in the range of 39.5%. The percentage yield of SM-loading PLGA microspheres was determined by weighing the microspheres after collection from lyophilization. The percentage yield was found to be 25.2%.
The drug loading and encapsulation efficiency could be affected by several factors, such as the methods of preparation, the physicochemical characteristics of drug, the concentrations of drug and polymer, phase ratio, the species and concentrations of the stabilizer, solvent evaporation time and stirring speed (Yen et al., Citation2001; Zhang et al., Citation2011). In this study, it was an efficient method to increase the drug loading and encapsulation efficiency by adding SM to the diluent. Nonetheless, the concentration of SM in the diluent should be considered for the purpose of avoiding the burst release of the drug. An average drug loading of 7.6 ± 1.2% and encapsulation efficiency of 82.1 ± 6.2% of SM-loaded PLGA microspheres were obtained with the optimized preparation parameters, suggesting that the drug loading and encapsulation efficiency were high enough for further studies.
In vitro drug release
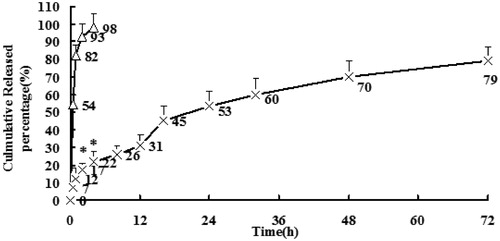
Drugs released from PLGA microspheres may occur through polymer erosion, diffusion or a combination of those two. Furthermore, polymer properties, including molecular weight, copolymer composition and crystallinity can alter polymer degradation and lead to the change of drug release profiles (Zolnik & Burgess, Citation2007). Besides, physicochemical properties of the drug and properties of the microspheres can also greatly affect the release mechanism and rate of the drug from PLGA microspheres (Schaefer & Singh, Citation2002). The in vitro drug release profiles of SM from SM-loaded PLGA microspheres and SM solution (5 mg/mL) in phosphate buffer (pH 6.8, containing 0.02% NaN3) are shown in . The function of NaN3 was preservative. While a burst release was observed in the initial 4 h in SM solution with 98% free SM liberated from the solution into the bulk phase, only 26% free SM was released from SM-loaded PLGA microspheres in the initial 8 h. SM was graduated released from the microspheres as time lapsed, suggesting that SM was well entrapped in SM-loaded PLGA microspheres. Under certain conditions, the in vitro release can be used for the assessment of bioequivalence (Costa & Sousa, Citation2001). Several models (Higuchi; Weibull; Neibergull; zero-order; first-order) describing drug release from immediate and modified release dosage forms were adopted. The model that fitted best for SM released from the microspheres was Higuchi equation expressed as Y = 13.76T1/2 + 2.76, r = 0.9976, revealing that SM could be controlled released from the microspheres. Whereas the fittest model for SM solution was first-order kinetics. In both accelerated and long-term stability test, the microspheres maintained good round shape and the data showed no significant change in SM microspheres.
Figure 3. In vitro release of SM from SM-loaded PLGA microspheres and SM solution. Number represent the percentage of release. (n = 3) *p < 0.05, SM-loaded PLGA microspheres versus SM solution.
In vivo studies
The plasma concentration–time profiles of SM after oral administration of SM solution, SM-loaded PLGA microspheres and intravenous administration of SM injection to rats are shown in and the pharmacokinetic parameters are summarized in . The area under the curve of SM in microspheres was 1.94-fold that of SM solution (p < 0.05). The relatively slower time to maximum plasma concentration of SM-loaded PLGA microspheres suggests a sustained-release profile of microparticles in vivo, which was consistent with the results of the in vitro release study. The increased area under the curve and maximum plasma concentration of SM-loaded PLGA microspheres demonstrates an increased oral absorption. Moreover, SM microspheres showed a significantly higher absolute oral bioavailability (65.7%) compared to that of orally administered SM solution (34.7%). The improved oral bioavailability of SM in the microspheres might be attributed to the sustained-release property of the microparticles increased the circulation time of SM, which prolonged the drug residence time in systematic circulation and resulted in better bioavailability (des Rieux et al., Citation2006). Hence, SM-loaded PLGA microspheres are thought to have the potential to maintain SM concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency.
Figure 4. Plasma concentration–time profiles of SM after intravenous or oral administration of SM suspensions, SM-loaded PLGA microspheres, and SM injection to rats. (n = 6) *p < 0.05, SM-loaded PLGA microspheres versus SM solution and SM suspensions.
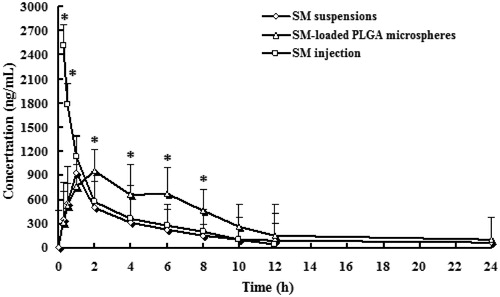
Table 1. Pharmacokinetic parameters after intravenous or oral administration of SM formulations to rats (n = 6).
Conclusions
SM microspheres were successfully formulated using PLGA as carrier by emulsion-solvent evaporation method. Physicochemical characteristics such as entrapment efficiency, particle size, zeta potential and the in vitro release results signified that the prepared microspheres were ideal for the delivery of SM by oral route. In vivo studies verify the potential of the prepared microparticulate system (SM-loaded PLGA microspheres), and promise to maintain SM concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency.
Declaration of interest
The authors report no conflicts of interest.
References
- Carter SJ, Cápka V. (2008). Investigation of interaction between salmeterol and fluticasone propionate and its effect on quantitative accuracy of an LC/MS/MS assay in human plasma at low pg/mL concentrations. J Chromatogr B Analyt Technol Biomed Life Sci 876:163–9
- Cates CJ, Lasserson TJ. (2012). Regular treatment with formoterol versus regular treatment with salmeterol for chronic asthma: serious adverse events. Cochrane Database Syst Rev 3:CD007695
- Chang C, Bahadduri PM, Polli JE, et al. (2006). Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos 34:1976–84
- Costa P, Sousa Lobo JM. (2001). Modeling and comparison of dissolution profiles. Eur J Pharm Sci 13:123–33
- Daley-Yates PT, Parkins DA. (2011). Systemic bioavailability of hydrofluoroalkane formulations containing fluticasone propionate and salmeterol. Br J Clin Pharmacol 72:353–4
- De La Vega JC, Elischer P, Schneider T, Häfeli UO. (2013). Uniform polymer microspheres: monodispersity criteria, methods of formation and applications. Nanomedicine (Lond) 8:265–85
- des Rieux A, Fievez V, Garinot M, et al. (2006). Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release 116:1–27
- Fahmy TM, Fong PM, Goyal A, Saltzman WM. (2005). Targeted for drug delivery. Mater Today 8:18–26
- Fahmy TM, Fong PM, Park J, et al. (2007). Nanosystems for simultaneous imaging and drug delivery to T cells. AAPS J 9:171–80
- Farokhzad OC, Cheng J, Teply BA, et al. (2006). Nanotechnology in cancer treatment and detection. PNAS 103:6315–20
- Gupta S, Jain A, Chakraborty M, et al. (2013). Oral delivery of therapeutic proteins and peptides: a review on recent developments. Drug Deliv 20:237–46
- Harrison LI, Novak CC, Needham MJ, Ratner P. (2011). Comparative pulmonary function and pharmacokinetics of fluticasone propionate and salmeterol xinafoate delivered by two dry powder inhalers to patients with asthma. J Aerosol Med Pulm Drug Deliv 24:245–52
- Hu L, Zhang H, Song W. (2013). An overview of preparation and evaluation sustained-release injectable microspheres. J Microencapsul 30:369–82
- Huang YY, Chung TW, Tzeng TW. (1997). Drug release from PLA/PEG microparticulates. Int J Pharm 156:9–15
- Islam MA, Firdous J, Choi YJ, Yun CH, Cho CS. (2012). Design and application of chitosan microspheres as oral and nasal vaccine carriers: an updated review. Int J Nanomedicine 7:6077–93
- Kim SH, Jeong JH, Chun KW, Park TG. (2005). Target-specific cellular uptake of PLGA nanoparticles coated with poly(l-lysine)–poly(ethylene glycol), folate conjugate. Langmuir 21:8852–7
- Moghimi SM, Hunter AC, Murray JC. (2001). Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 53:283–318
- Schaefer MJ, Singh J. (2002). Effect of tricaprin on the physical characteristics and in vitro release of etoposide from PLGA microspheres. Biomaterials 23:3465–71
- Spencer S, Karner C, Cates CJ, Evans DJ. (2011). Inhaled corticosteroids versus long-acting beta(2)-agonists for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 12:CD007033
- Takeuchi H, Yamamoto H, Kawashima Y. (2001). Mucoadhesive nanoparticulate systems for peptide drug delivery. Adv Drug Deliv Rev 47:39–54
- Yen SY, Sung KC, Wang JJ, et al. (2001). Controlled release of nalbuphine propionate from biodegradable microspheres: in vitro and in vivo studies. Int J Pharm 220:91–9
- Yin Y, Chen D, Qiao M, et al. (2006). Preparation and evaluation of lectin-conjugated PLGA nanoparticles for oral delivery of thymopentin. J Control Release 116:337–45
- Zhang Z, Bi X, Li H, et al. (2011). Enhanced targeting efficiency of PLGA microspheres loaded with Lornoxicam for intra-articular administration. Drug Deliv 18:536–44
- Zolnik BS, Burgess DJ. (2007). Effect of acidic pH on PLGA microsphere degradation and release. J Control Release 122:338–44