
Abstract
Objective of the present investigation was to prepare and evaluate the potential of enteric coated dextran microspheres for colon targeting of 5-fluorouracil (5-FU). Dextran microspheres were prepared by emulsification-crosslinking method and the formulation variables studied included different molecular weights of dextran, drug:polymer ratio, volume of crosslinking agent, stirring speed and time. Enteric coating (Eudragit S-100) of dextran microspheres was performed by oil-in-oil solvent evaporation method using different coat:core ratios (4:1 or 8:1). Uncoated and coated dextran microspheres were characterized by particle size, surface morphology, entrapment efficiency, DSC, in vitro drug release in the presence of dextranase and 2% rat cecal contents. The release study of 5-FU from coated dextran microspheres was pH dependent. No release was observed at acidic pH; however, the drug was released quickly where Eudragit starts solublizing there was continuous release of drug from the microspheres. Organ distribution study was suggested that coated dextran microspheres retard the release of drug in gastric and intestinal pH environment and released of drug from microspheres in colon due to the degradation of dextran by colonic enzymes.
Introduction
After heart and infectious diseases, cancer is currently the third most common cause of death in the world. Despite of all available chemotherapy, radiotherapy and surgery, overall survival rate still has not significantly improved over the last 30 years. While the search for the efficacious therapeutics continues, it becomes increasingly clear that the accurate and informative visualization of anti-cancer agents following their administration can play an essential role to formulate effective treatment plans. Colorectal cancer is the second leading cause of cancer deaths in USA, and more than 66 000 cases of colon cancer are reported to occur in the Indian subcontinent every year (Paharia et al., Citation2007).
However, conventional chemotherapy is not as effective in colorectal cancer as it is in other cancers, as the drug does not reach the target site in effective concentrations (Krishnaiah & Satyanarayan, Citation2001; Michor et al., Citation2005). Therefore, the effective treatment of colon cancer requires relatively large doses to compensate for drug loss during passage through the upper gastrointestinal (GI) tract. These large doses may be associated with undue side effects. This can be overcome by site-specific delivery of the drug molecule to the colon. A particular challenge in the pharmaceutical field is the development of a site-specific delivery system that could control delivery time for the release of active ingredient in the lower part of the small intestine, or in the colon. Various approaches have been utilized colon specific drug delivery such as coating with pH-sensitive polymers, coating with enzymatic degradable polymers, pro-drugs, timed-release systems and fabrication of biodegradable matrices and hydrogels (Rubinstein, Citation1990). Among the different approaches to achieve colon-selective drug delivery, the use of polymers, specifically biodegraded by colonic bacteria, appears to have a promising prospective. The pH-dependent systems exploit the generally accepted view that pH of the human GI tract increases progressively from the stomach (pH 2–3) to the small intestine (pH 6.5–7.0) to the colon (7.0–8.0) (Ashford & Fell, Citation1994). Most commonly used pH-dependent coating polymers are methacrylic acid copolymer (i.e. Eudragit-L-100, and Eudragit-S-100), which dissolve above pH 6.0 and 7.0, respectively.
Dextran is an exo-cellular bacterial polysaccharide consisting a linear linkages polymer backbone having mainly 1,6-α-d-glucopyranosidic units with some degrees of 1,3-branching. Enzyme (dextranase) from molds such as Penicillium and Verticillium has been shown to degrade dextran (Tsuchiya et al., Citation1952). Dextranase activity in the colon is shown by anaerobic gram-negative intestinal bacteria, especially the Bacteroides (Hehre & Sery, Citation1952). Clinically, dextran has been used for plasma volume replacement, prophylaxis of postoperative and post-traumatic thromboembolism (Mehvar, Citation2000). Dextran polymers have also been evaluated for colon drug delivery (Sinha & Kumria, Citation2001). In particular, some authors have investigated the stability of dextran hydrogels in vitro models, simulating human small intestinal and colonic environments to evaluate the suitability of dextran matrices as carriers for colonic specific drug delivery (Simonsen et al., Citation1995).
There are also some studies related with the oral colon delivery of 5-fluorouracil (5-FU) using non-biodegradable polymers including blend microspheres of poly(3-hydroxybutyrate) and cellulose acetate phthalate (Chaturvedi et al., Citation2011, 2013). Polyethylene glycol crosslinked chitosan microspheres enteric coated with cellulose acetate phthalate and bile acid conjugated PHB–PEG block copolymeric nanoparticles were also used as possible colon delivery system for therapeutic agents (Ganguly et al., Citation2011; Chaturvedi et al., 2013).
In our earlier work, we have reported the use of chitosan hydrochloride and Eudragit-coated pectin microspheres for colon specific drug delivery of albendazole and 5-FU, respectively (Rai et al., Citation2005; Paharia et al., Citation2007). In continuation, we attempted to prepare Eudragit-coated dextran microspheres for colon targeting of 5-FU. The objective of the present investigation was to design a multiparticulate delivery system for site-specific delivery of 5-FU using pH-sensitive polymer (Eudragit-S-100) and natural polysaccharides (dextran) for the treatment of colon cancer. This system is anticipated to protect the drug loss in the upper GI tract, which results from the inherent property of Eudragit, and deliver 5-FU in the colon only. The use of enteric polymers as protective coating on the microspheres makes them able to release the drug at the particular pH of colonic fluid. A combined mechanism of release is proposed, which combines specific biodegradability of polymer and pH-dependent drug release from the coated microspheres.
Materials and methods
Materials
5-FU was obtained as gift sample from M/S Dabur India Pvt. Ltd., Delhi, India. Dextran of different molecular weights, 40 000, 70 000 and 200 000 Da, were purchased from Sigma Chemicals Co. (St. Louis, MO). Eudragit-S-100 was procured as a gift sample from Rohm Pharma, Darmstadt, Germany. Span 80, glutaraldehyde (GA 25% w/v), castor oil and isopropanol were purchased from Himedia Laboratories (Mumbai, India). Dextranase (Chaetomium erraticum) was generously supplied as gift sample by M/S Amano Enzymes Inc. (Nagoya, Japan). All other chemicals were of analytical grade and used as received.
Preparation of GA crosslinked dextran microspheres
Dextran microspheres were prepared by using a combined method involving water-in-oil (w/o) emulsification and crosslinking method. Dextran (150 mg) was dissolved in deionized water and 1 mL MgCl2 (5% w/v) solution was added. 5-FU (50 mg) was added to the dextran solution with stirring. The emulsion was first prepared by dispersing the dextran solution through syringe into a continuous oil phase consisting of 75 mL castor oil, 25 mL isopropanol and 2% (w/v) Span 80 in a 250 mL beaker at 50 °C. The dispersion was stirred using a stainless steel stirrer with a half-moon paddle at 1500 rpm for 10 min and thereafter 3 mL of GA was added into the beaker under stirring. The crosslinking reaction was allowed to proceed for a total time of 3 h. Hardened microspheres were filtered, washed repeatedly with isopropanol and water to remove castor oil and unreacted GA, dried under vacuum at 40 °C overnight and kept in a desiccator until further use.
Preparation of Eudragit-coated crosslinked dextran microspheres
Eudragit coating of GA crosslinked dextran microspheres containing 5-FU was performed using oil-in-oil solvent evaporation method (Rodriguez et al., Citation1998). Eudragit-S-100 (400 mg) was dissolved in 10 mL of organic solvent (2:1, ethanol:acetone) to give either 4:1 or 8:1 ratio (coat:core ratio) and dextran microspheres (100 or 50 mg) were added. This organic phase was then poured in 100 mL of light liquid paraffin containing 2% w/v Span 80. The system was maintained under agitation speed of 1000 rpm using double blade mechanical stirrer (Remi, Mumbai, India) at 40 °C for 3 h to allow evaporation of the solvent. Finally, the coated microspheres were filtered, washed with n-hexane and dried over night in the vacuum desiccator.
Surface morphology and particle size
The shape and surface morphology of the uncoated and coated dextran microspheres were investigated using scanning electron microscopy. The microspheres were coated with gold palladium under an argon atmosphere for 150 s to achieve a 20 nm film (sputter coater, SCD004, BAL-TEC Balzers, Furstentum, Lienchestein). The coated samples were examined in a scanning electron microscope (Jeol JSM-1600, Tokyo, Japan). The particle size of microspheres was determined by an optical microscope using a calibrated ocular micrometer (Leica, Wetzlar, Germany).
Drug entrapment efficiency
The amount of 5-FU entrapped in the uncoated and coated dextran microspheres were determined by digesting a weighed quantity of microspheres (50 mg) in PBS (pH 7.4) and methanol, respectively, for 24 h. These were centrifuged (Remi) at 5000 rpm for 30 min and the supernatants were analyzed by HPLC. The HPLC system (LC-10AT VP, Shimadzu liquid chromatograph, Shimadzu, Kyoto, Japan) consisted of SPDM10AVP Shimadzu diode array detector, and a Luna 5µ C18 column (250 mm × 4.60 mm) was used for the analysis of drug. The mobile phase was water:methanol (95:5 v/v) pumped at a flow rate of 1 mL/min at 25 °C. The mobile phase was out-gassed under vacuum before use (Alsarra & Alarifi, Citation2004). The detection wavelength was 266 nm. The drug content in different parts of the GI tract at different time intervals was calculated.
Swelling behavior
A known weight (50 mg) of uncoated dextran microspheres were placed separately in simulated gastric fluid (pH 1.2) or simulated intestinal fluid (pH 7.5) and allowed to swell for the required period of time at 37 ± 0.5 °C in the dissolution apparatus (United States Pharmacopeia [USP] XXIII, model DT-06, Erweka, Heusenstamm, Germany). The microspheres were periodically removed and blotted with filter paper; then their change in weight (after correcting for drug loss) was maintain until attainment of equilibrium. The swelling ratio (SR) was then calculated using the following formula:
(1)
where SR indicates swelling ratio; W0 is the initial weight of microspheres and Wg is the final weight of microspheres.
In vitro enzymatic degradation
Enzymatic degradation of the uncoated dextran microspheres was carried out using dextranase and 2% rat cecal contents (Hovgaard & Brondsted, Citation1995). Weighed amount of microspheres (100 mg) was suspended separately in 5 mL of phosphate buffer, pH 7.4 containing 0.7 IU/mL dextranase or 2% rat cecal contents or without dextranase and 2% rat cecal contents and agitated on the shaker at 37 ± 0.5 °C at 50 rpm for 24 h. The residue was washed with water, centrifuged and dried in oven at 37 ± 0.5° C until the constant weight was reached. The weight loss of the microspheres after enzymatic degradation was quantified as the difference between the initial weight of the microspheres and that of the dried residue.
DSC study
Thermal analysis was performed on plain 5-FU, dextran and uncoated and coated dextran microspheres using a Mettler system with a differential scanning calorimeter equipped with a computerized data station (DSC 30, Toledo AG, Schwerzenbach, Switzerland). All samples were weighed (5 mg) and heated at a scanning rate of 10 °C/min between 0 and 300 °C. Aluminum pans and lids were used and the temperature calibrations were performed periodically using indium as standard.
Enteric nature of coated dextran microspheres
This test was performed to determine whether the drug would be released in the acidic environment of the stomach (i.e. pH between 1 and 3). USP recommends the use of 0.1 N HCl for testing of enteric coated formulations, accordingly the same was used in this study (Jain et al., Citation2006). A known weight (50 mg) of coated dextran microspheres were placed in 0.1 N HCl (pH 1.2) for 2 h at 37 ± 0.5 °C in the dissolution test apparatus (TDT-08L dissolution tester (USP), Electro Lab, Mumbai, India). After 2 h, the sample was withdrawn and centrifuged at 4000 rpm for 10 min and the drug content of supernatant was analyzed by HPLC method at 266 nm.
In vitro drug release study
An accurately weighed amount of microspheres, equivalent to 100 mg of 5-FU, was added to 900 mL of dissolution medium and the release of 5-FU form microspheres was investigated using USP XXI rotating paddle dissolution test apparatus (TDT-08L dissolution tester (USP), Electro Lab) at 100 rpm and 37 ± 0.5 °C. The simulation of GI transit conditions was achieved by altering the pH of dissolution medium. Initially it was kept at pH 1.2 for 2 h with 0.1 N HCl. Then KH2PO4 (1.7 g) and Na2HPO4 · 2H2O (2.225 g) were added to the dissolution medium, adjusting the pH 4.5 for third and fourth hours and pH 6.8 for fifth hour with 1.0 N NaOH. After 5 h, the pH of dissolution medium was adjusted to 7.4 and maintained up to 8 h. The samples were withdrawn from dissolution medium at various time intervals using a pipette fitted with a microfilter at its tip and analyzed HPLC at 266 nm.
In vitro drug release in the presence of dextranase and rat cecal contents from microspheres
In vitro 5-FU release was determined in the presence of dextranase and rat cecal contents for the enzymatic degradability of the microspheres by colonic bacteria. The in vitro drug release in presence of rat cecal contents was performed in accordance with the protocol approved by the Institutional Animals Ethical Committee of Dr. Hari Singh Gour University, Sagar, India, following the guidelines approved by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Empowerment, Government of India. Rat cecal content was prepared by the method reported by Van Den Mooter et al. (Citation1994). Albino rats of uniform body weight (150–200 g) with no prior drug treatment were used for the present in vitro study. The rats were weighed, maintained on normal diet and administered 1 mL of 2% solution of dextran in water and this treatment was continued for 7 d for polymer induction to animals. Thirty minutes before starting the study, the rat was sacrificed, the abdomen was opened, cecal was traced, legated at both ends, dissected and immediately transferred into PBS (pH 7.4), which was previously bubbled with CO2. The cecal bag was opened; contents were weighed, homogenized and then suspended in PBS (pH 7.4) to give the desired concentration (2%) of cecal contents, which was used as simulated colonic fluid. The suspension was filtered through cotton wool and ultrasonicated for 10 min in an ice bath at 40% voltage frequency using a probe sonicator (Soniweld, Imeco Ultrasonics, Mumbai, India) at 4 °C to disrupt the bacterial cells. After sonication, the mixture was centrifuged (Remi) at 2000 rpm for 20 min. Drug release rate studies for the initial 5 h were performed as described above. From sixth hour and onwards it was carried out presence of dextranase and rat cecal contents. The weighed amount of coated microspheres (100 mg) was placed in 200 mL of dissolution medium (PBS pH 7.4) containing 2% w/v rat cecal contents. The experiment was carried out with continuous CO2 supply into the dissolution medium. At different time intervals the samples were withdrawn and replaced with fresh PBS. The experiment was continued up to 24 h. The in vitro drug release study was also performed in the presence of dextranase enzyme. A required amount of dextranase was added to the aqueous buffer (pH 7.4) to produce the concentration of 0.7 IU/mL. The withdrawn sample was pipetted into 10 mL volumetric flask and the volume made up to the mark with PBS. It was then transferred to the centrifuged tubes. The supernatant was filtered through 0.45 µm membrane filter (Millipore) and the filtrate was analyzed by HPLC for 5-FU content at 266 nm.
In vivo studies
These studies were carried out according to the guidelines of the CPCSEA, Ministry of Social Justice and Empowerment, Government of India and prior approval from the Institutional Animal Ethical Committee of Dr. H.S. Gour University, Sagar, MP, India.
Pharmacokinetic study
The pharmacokinetic profile of plain 5-FU and 5-FU loaded coated dextran microspheres was investigated on albino rats of either sex (150–200 g). The animals were divided into three groups comprising of six animals in each. The first group served as control and was administered 0.5 mL PBS (pH 7.4). The II group received plain 5-FU solution (20 mg/kg) by oral gavage and the animals of III group were orally given coated dextran microspheres containing 5-FU is same equivalent dose as given for plain drug solution. Blood samples were collected from retro orbital plexus of eye in heparinized tubes and plasma concentration of the drug was determined at various time points of sampling, i.e. 0.5, 1.0, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 20 and 24 h after drug administration. Blood samples were centrifuged (Remi) at 3000 rpm for 10 min and plasma was separated and stored at −20 °C for later determination of 5-FU concentration by HPLC as reported above.
Organ distribution study
Albino rats (150–200 g) were divided into three groups with 48 animals in each. The first group served as control, which was maintained on the same regular diet throughout study period. The second group was orally administered 1 mL plain 5-FU solution (20 mg/kg) and the animals of third group was orally given coated dextran microspheres, containing equivalent amount of drug with the help of feeding tube, followed by sufficient volume of drinking water. Three animals from each group were killed by deep ether anesthesia at 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 20 and 24 h after drug administration. The GI tract was removed; stomach, small intestine and colon were isolated. The luminal contents were removed by applying gentle pressure with wet scissors to the tissues. Organs and luminal contents were weighed. The organs were cut open longitudinally and rinsed with saline solution (0.9% NaCl) to remove any remaining luminal contents. The organs were cut into small pieces and homogenized by Micro Tissue Homogenizer (Mac, Mumbai, India) at 4 °C along with a small amount of HPLC grade water. One milliliter of methanol was added to tissue homogenate and kept for 30 min. The contents were centrifuged and the supernatant liquid was separated. After appropriate dilution of supernatants, the drug content was determined by HPLC method. The drug content in different parts of the GI tract at different time intervals was calculated.
Results and discussion
Particle size and entrapment efficiency of crosslinked dextran microspheres
In the present work, the crosslinked dextran microspheres were successfully prepared by the emulsification and crosslinking method. The effect of the formulation variables on particle size and entrapment efficiency of 5-FU-loaded crosslinked dextran microspheres is shown in . The microspheres had a smoother surface and were found to be discreet and spherical in shape ().
Figure 1. Scanning electron photomicrographs of crosslinked dextran microspheres.

Table 1. Effect of fabrication variables on mean diameter, entrapment efficiency and in vitro drug release of uncoated GA crosslinked dextran microspheres.
Effect of molecular weight
With increasing dextran molecular weight from 40 000 to 200 000 Da, particle size of the microspheres was increased from 8.54 ± 0.25 to 23.17 ± 0.74 μm (). This is because at constant solvent volume, the viscosity of the polymeric solution is proportional to the molecular weight of the polymer. Low viscosity polymer solution could be dispersed in the external aqueous phase to a greater extent than high viscosity polymer solution and hence reduced the microspheres to smaller size. The entrapment efficiency was increased with increasing molecular weight of dextran (). The high viscosity of the higher molecular weight dextran may have restricted drug diffusion to the oil phase during the formation of microspheres resulting in higher entrapment efficiency (Youan et al., Citation2001).
Effect of drug:polymer ratio
Upon increasing the drug polymer ratio from 1:2 to 1:4, the particle size of microspheres was increased from 09.45 ± 0.34 to 20.71 ± 0.84 μm (). The increase in the size of microspheres may be attributed to an increase in the viscosity of polymer with increasing concentration, which resulted in larger emulsion droplets and finally greater size. The drug: polymer ratio was increased from 1:2 to 1:4; the entrapment efficiency was increased from 79.41 ± 1.73% to 86.53 ± 1.71% ().
Effect of crosslinking agent
Dextran microspheres prepared without the addition of crosslinking agent were found to be fragile. Crosslinking of dextran with GA was previously shown to be necessary for the preparation of controlled release capsule for colon-specific delivery. GA was added with MgCl2 to make it insoluble. MgCl2 is known to act as a catalyst for the crosslinking reaction (Brondsted et al., Citation1998). As the volume of crosslinking agent was increased from 2 to 4 mL, the particle size of microspheres decreased from 19.37 ± 0.77 to 11.51 ± 0.29 μm. Possibly due to increase in the volume of crosslinking agent, formation of more rigid polymer network and shrinkage of particles might have decreased the particle size (Rokhade et al., Citation2007). As the volume of crosslinking agent was increased, entrapment efficiency was decreased (). This was due to higher degree of crosslinking, since microspheres are denser and the free volume space within the matrix would decrease, resulting in reduced encapsulation efficiency.
Effect of stirring speed and time
The stirring speed increased from 1000 to 3000 rpm; the particle size and entrapment efficiency of microspheres was decreased from 19.63 ± 0.51 to 10.45 ± 0.24 µm and 85.34 ± 2.35% to 78.89 ± 2.65%, respectively (). This is due to the higher stirring speed would produce small size of droplet with increased surface area, such that diffusion of drug from such microspheres will be fast, resulting in the loss of drug with a consequent lowering in entrapment efficiency (Mateovic et al., Citation2002). However, it was necessary to find out the required stirring time and how it may affect the microspheres characteristics. The effects of stirring time on the characteristics of the microspheres are shown in . Stirring time is related to be mainly the time for crosslinking reaction. The dextran microspheres were shown to be morphologically stable and crosslink at 3 h reaction time. The higher drug entrapment efficiency as well as smaller particle size was obtained on stirring for 3 h as compared to stirring for 2 h. However, it was clear that lower drug entrapment efficiency was obtained on stirring for 4 h as compared to stirring for 3 h. This may be due to long stirring responsible for some more drug to be leached out in the oil phase during the emulsification process. This concluded that stirring for 3 h is sufficient for producing the required microspheres of small particle size and high drug entrapment. On the other hand, prolonged stirring did not have any significant effect on particle size (Bodmeier & Wang, Citation1993).
Swelling ratio
The swelling ratio decreased with increase in volume of crosslinking agent. A possible reason for this result was denser the crosslinking between the dextran macromolecules, which probably allow the formation of more packed structures. Such a structure can be characterized by a lower and slower penetration of the solvent through the chain structure of the polymer (Orienti & Zecchi, Citation1993).
In vitro enzymatic degradation
In vitro degradation study in the presence of dextranase and rat cecal contents was performed in order to investigate whether the dextran microspheres are degradable in human colonic contents. Degradation behavior of microspheres was evaluated as the difference between the initial weight of the microspheres and remaining dried residue. The weight loss of crosslinked microspheres in the presence of dextranase and rat cecal contents was significantly greater than that of control microspheres (without dextranase and rat cecal contents). Increasing the volume of crosslinking agent from 2 to 4 mL led to a decrease in weight loss from 20.66 ± 1.36% to 8.80 ± 1.60%, 40.57 ± 3.12% to 29.67 ± 3.07% and 83.69 ± 2.79% to 72.94 ± 1.77% in the case of control microspheres, presence of dextranase and 2% rat cecal contents, respectively. This indicates that the developed microspheres were biodegradable, but the rate of degradation decreased upon increasing the degree of crosslinking. The presence of rat cecal contents caused a substantial increase in the percentage of weight loss of microspheres in comparison to that of respective control microspheres. Their effect on the percentage of weight loss was greater than presence of dextranase enzyme. Since the percentage of weight loss of microspheres after degrading enzyme treatment due to the cleave dextran chains, by degrading glucosidic linkages, with enzymatic hydrolysis into the oligosaccharides and then weight loss occurs via diffusion of these oligosaccharides from the microspheres (Basan et al., Citation2007).
In vitro drug release from crosslinked dextran microspheres
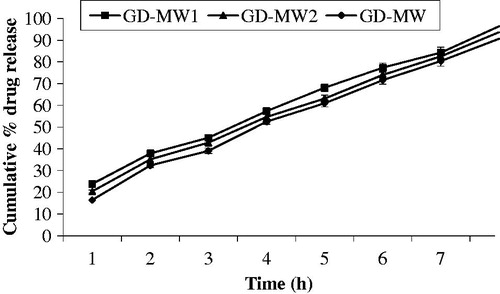
In vitro drug release studies were performed in GI fluids of different medium pH at 37 ± 0.5 ° C. The effect of molecular weight, drug:polymer ratio, amount of crosslinking agent, stirring speed and time was observed on in vitro drug release. Microspheres which were prepared by using the 40 000 Da molecular weight dextran showed a higher drug release rate (99.40 ± 1.47%) than those prepared using a 200 000 Da molecular weight dextran (90.63 ± 2.95%) after 8 h (). This may be due to the longer chain length between crosslinks in the higher molecular weight polymer, whereas the lower molecular weight polymer possesses smaller free chains. Thus, in the particles with the longer chain segments between crosslinks, chain entanglements may occur to a larger extent, resulting in an increase of the crosslinking density. These increased chain entanglements may be held responsible for the slower release rate of drug from the microspheres of higher molecular weight dextran. As the drug:polymer ratio was increased, the in vitro drug release was decreased. Drug release after 8 h was found to be 97.78 ± 2.82% in case of microspheres prepared of 1:2 drug:polymer ratio, while it was 89.32 ± 3.31% for microspheres prepared with 1:4 drug:polymer ratio. The drug release after 8 h was decreased (97.70 ± 2.09% to 89.03 ± 3.91%) when the concentration of GA, was increased (2–4 mL), which is due to the fact that as the amount of GA was increased it produced microspheres with pronounced crosslinking between polymer chains that retarded the release of drug (). Microspheres, which were prepared at agitation speed of 3000 rpm, did not exhibit good shape and released 96.41 ± 3.25% of 5-FU, while those that were produced at 2000 rpm released 92.34 ± 2.80% of 5-FU after 8 h (). Microspheres which were prepared at 2 h stirring time released 97.43 ± 3.21% of drug while those with 4 h stirring time released 95.79 ± 3.74% of drug after 8 h. This due to the incomplete crosslinking between polymer chains of microspheres at 2 h stirring time however gave faster drug release in comparison to those prepared at 3 and 4 h stirring time. All the formulations released more than 89% of the drug during dissolution studies for 8 h.
Figure 2. Effect of molecular weight on drug release from crosslinked dextran microspheres (bars represent mean ± SD, n = 3).
Eudragit-coated crosslinked dextran microspheres
About 80–90% of drug was released from uncoated dextran microspheres within 8 h, suggesting the fast release from uncoated microspheres, which indicated the drug loaded in uncoated microspheres, released before they reached colon. Brondsted et al. (Citation1998) have also been reported that GA crosslinked dextran capsules showed some instability in acidic medium. The release of maximum amount of drug in the upper part of GI tract necessitated manipulation in the formulation to control the drug release according to need. Hence, crosslinked dextran microspheres were coated with Eudragit-S-100, to retard the release of the drug in an acidic environment.
In the second part of this investigation GA crosslinked dextran microspheres were coated with Eudragit-S-100 in different coating:core ratio by oil-in-oil solvent evaporation method. The shape of coated dextran microspheres was spherical with a smooth surface area (). The particle size was considerably increased on coating and varied between 157.85 ± 5.57 and 184.40 ± 5.36 μm and the entrapment efficiency was found to be 75.79 ± 2.73% and 71.55 ± 3.63% in the case of 4:1 and 8:1 coating:core ratio, respectively (). The enteric nature of the coating for all coated dextran microspheres was evaluated and no drug was released in 0.1 N HCl medium (pH 1.2) in 2 h.
Figure 3. Scanning electron photomicrographs of Eudragit-S-100-coated crosslinked dextran microspheres.

Table 2. Particle size and entrapment efficiency of various Eudragit-coated crosslinked dextran microspheres.
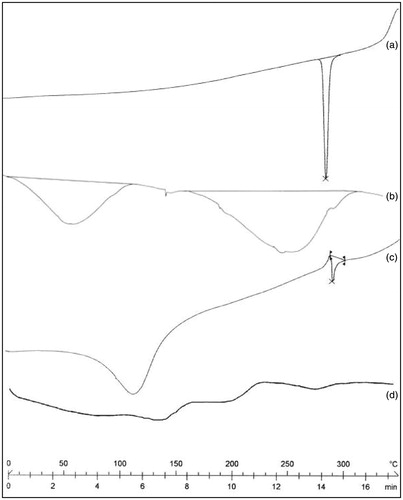
Differential scanning calorimetry is very useful in the characterization of the thermal properties of polymer and drug within the microspheres (Dubernet, Citation1995). In the present study, DSC thermograms of 5-FU, dextran, uncoated dextran microspheres and coated crosslinked dextran microspheres were taken (). A sharp endotherm was observed for 5-FU at 280 °C, corresponding to its melting transition temperature (). In case of dextran, broad endothermic peak observed between 25 and 120 ° C is due to the evaporation of residual water and peak was observed at 230 °C, corresponding to its glass transition temperature () (Zumreoglu-Karan et al., Citation2002). However, the melting endotherm also appeared in the DSC thermogram of uncoated dextran microspheres although less intense (). This is due to the small amount of drug present in the surface of dextran micrsopheres. In the case of coated dextran microspheres, indicating that the drug was dissolved in the polymer matrix and its own melting endotherm peak had disappeared ().
Figure 4. DSC thermogram of (a) 5-FU (b) dextran (c) GA crosslinked dextran microspheres (d) Eudragit-S-100 (8:1) coated 5-FU-loaded crosslinked dextran microspheres.
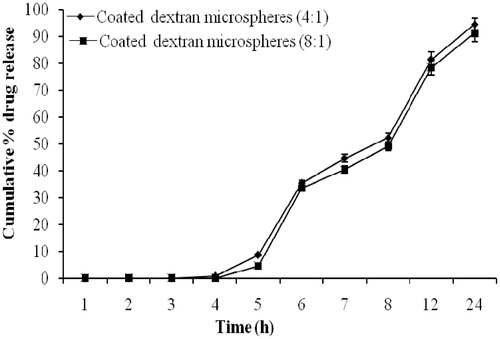
In vitro release study of coated dextran microspheres revealed that no drug was released during first 4 h. Drug release after 12 h with coating:core ratio of 4:1 and 8:1 was found to be 49.34 ± 1.67% and 44.47 ± 1.25%, respectively (). Drug release was decreased with increasing of Eudragit coating ratio but release pattern was unchanged. This may be due to increased coating thickness on dextran micrsopheres. These results are in good agreement with the previous study (Chourasia & Jain, Citation2003). The coated dextran microspheres successfully delayed and hindered the release of drug until it enters into the colon. As dextran microspheres were prepared to release the drug in colon by degradation of the dextran in the presence of anaerobic colonic bacteria present in the colon, in vitro drug release study in the presence of dextranase and rat cecal contents were performed. A significant difference was observed in the amount of 5-FU released at the end of 8 h in the dissolution medium having dextranase (0.7 IU/mL) when compared to the study conducted without using dextranase. In vitro drug release studies of coated dextran microspheres after 8 h without dextranase released 30.57 ± 2.60% and 27.62 ± 1.53% of drug in case of microspheres of 4:1 and 8:1 coating:core, respectively. Whereas the presence of dextranase increased the drug release by 46.28 ± 3.44% and 41.35 ± 2.27% with the same coating:core ratio, respectively ().
Figure 5. In vitro drug release from coated dextran microspheres (bars represent mean ± SD, n = 3).
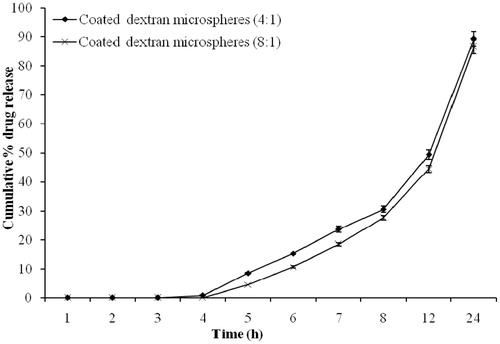
Figure 6. In vitro drug release from coated dextran microspheres in the presence of dextranase (bars represent mean ± SD, n = 3).
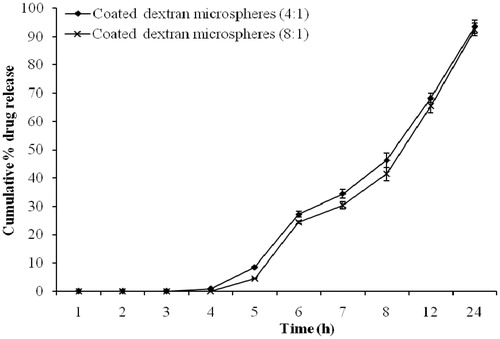
In vitro drug release study was also performed in SCF (pH 7.4) with and without using rat cecal contents. The presence of 2% rat cecal contents in the dissolution medium resulted in improved drug release at different time intervals in comparison to that carried out without using rat cecal contents. In vitro drug release study of coated dextran microspheres after 12 h without rat cecal content released 49.34 ± 2.67% and 44.47 ± 3.25% of drug with coating:core ratio of 4:1 and 8:1, respectively, while the presence of 2% rat cecal contents in the dissolution medium showed a catalytic effect on drug release, with the same polymer ratio it released up to 81.52 ± 2.81% and 78.33 ± 2.64% after 12 h. (). The interpretation of results of in vitro release study in all the formulation revealed a good deal of increment in terms of drug release in rat cecal contents, when compared to study of incorporating no cecal contents. The drug release from crosslinked dextran microspheres involves the following processes: (1) swelling/gelling of dextran microspheres and dissolution of the drug, (2) diffusion of the drug through the microspheres and (3) degradation of dextran by anaerobic colonic bacteria.
Figure 7. In vitro drug release from coated dextran microspheres in the presence of 2% rat cecal contents (bars represent mean ± SD, n = 3).
In vivo studies
Pharmacokinetic study
Following oral administration of plain 5-FU solution, the drug was detected rapidly in plasma. The Cmax of 5-FU was 30.61 ± 1.56 μg/mL after about 1 h. Thereafter, the plasma concentration decreased quickly and the drug was not detectable after 6 h, which has a large surface area available for absorption so that most of the drug was absorbed in the upper part of the GI tract. In contrast, the observed Cmax from coated dextran microspheres (14.27 ± 1.03 μg/mL) was lower than that of the aqueous solution of 5-FU (30.61 ± 1.56 μg/mL). The tmax after the administration of plain 5-FU solution was 1.0 ± 0.15 h, which was significantly different (p < 0.05) from the tmax, i.e. 12.0 ± 1.65 h obtained with coated microspheres microspheres (). This long tmax indicated that the coated dextran microspheres effectively prevented drug release in the upper part of the GI tract. Degradation of polysaccharides by enzymes of the colon is a slow process and usually requires 12 h for complete degradation (Yang et al., Citation2000); release is thus further modulated by slow degradation of dextran. These results were in agreement with previous report (Rahman et al., Citation2008).
Table 3. Pharmacokinetic parameters of 5-FU after administration of plain 5-FU solution and 5-FU-loaded coated dextran microspheres in rats.
Pharmacokinetics of 5-FU was greatly altered when loaded in coated dextran microspheres (). The Mean residence time (MRT) values of 5-FU from the coated dextran microspheres were about 10.06 ± 1.75 h, i.e. 10-fold higher than those for the respective plain 5-FU solution (1.69 ± 0.30 h). This indicated that the average residence time of 5-FU after oral dosing of coated microspheres is longer than that of the plain 5-FU solution. The longer residence time of released drug in colon was supplemented with sustained drug release in the vicinity of target site as is very obvious from serum plasma concentration of drug. These results were in agreement with the previous report (Wei et al., Citation2008). The colon, like a homogeneous reservoir, elicited slow and constant drug input into systemic circulation similar to that observed with continuous infusion, which is beneficial to the cancer therapy due to its short plasma half-life of 10–20 min. There was a statistically significant difference (p < 0.05) in the Area under the curve (AUC) values between the plain 5-FU solution (42.24 ± 3.29 μg h/mL) and the coated dextran microspheres (113.41 ± 6.84 μg h/mL). Further, Kel of 5-FU loaded in coated dextran microspheres was about 13 times less than plain 5-FU solution, exhibiting slower clearance of drug from the body upon encapsulation in microspheres. t1/2 values are also in agreement to decreased clearance of drug upon encapsulation. The relative bioavailability of the coated dextran microspheres when compared to the plain 5-FU solution was 2.26.
Organ distribution study
Organ distribution of coated dextran microspheres was investigated after oral administration to albino rats. Plain 5-FU was included in the study to compare the results. The results indicated that maximum (80.34 ± 3.28%) drug concentration was observed in stomach 1 h after oral administration of plain 5-FU solution. Moreover, a minimal drug concentration was observed in the small intestine (5.23 ± 0.30%) and no drug concentration was detected in the colon. At the end of 4 h, 36.81 ± 1.84% drug concentration was observed in small intestine while the colon was still devoid of drug. The relatively sharp decrease in the drug concentration in stomach at time intervals 2–4 h may be attributed to drug absorption through the stomach, systemic distribution and intestinal drug transit. After 6 h, maximum drug concentration in colon was 5.29±0.40% ().
Figure 8. Organ distribution of plain 5-FU solution and 5-FU-loaded coated dextran microspheres after oral administration (bars represent mean ± SD, n = 3).
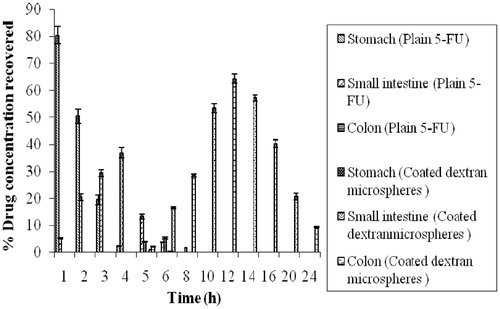
In case of dextran microspheres enterically protected with Eudragit-S-100 (8:1) coating, no drug concentration was recovered from small intestine even after 4 h (). However, the drug concentration was recovered only in small intestine after 5 h from coated microspheres. This may be due to pH-dependent solubility of Eudragit-S-100, which dissolves as the pH moves towards alkaline range. The formulation might have entered into ileum after 5 h and due to which 1.08 ± 0.01%, drug concentration was recovered from small intestine after 5 h. In the colon, abundant microflora and extended transit time were exploited for targeted drug release and prolonged drug exposure. After entry of microspheres in colon, the release of drug was continuously increased. These results are in well agreement with those we have observed during in vitro drug release studies.
The peak drug concentration in colon observed with coated dextran microspheres was 64.34 ± 1.67%. There was significant difference in the drug concentration after 12 h from coated microspheres in comparison to uncoated microspheres. However, the drug concentration was continuously declined and after 20 h, 20.68 ± 1.26%, drug concentration was recovered in colon ().
Thus, higher drug concentration was achieved with these colon-specific microspheres at all time points. The drug concentration was still detectable in the colon after 24 h in the case of colon-specific microspheres. The drug concentration recovered in colon was 9.34 ± 0.33% from coated dextran microspheres, after 24 h. This high concentration in the colon could be attributed to protection of the core dextran microspheres from the environment of the upper part of GI tract by the Eudragit-S-100 coating, thereby preventing drug release in the stomach and small intestine.
Conclusions
This research concludes that the Eudragit-S-100-coated dextran microspheres can be explored for effective site-specific delivery of 5-FU. Moreover, the release of the entrapped 5-FU in these microspheres can be controlled for longer period of time. The pharmacokinetic and organ distribution studies reveal that the drug concentration was found higher in the colon tissue, with low systemic exposure to the drug. However, proposed system was able to reduce the side effects of the drug caused by its absorption from the upper part of the GI tract.
Acknowledgements
The authors gratefully acknowledge Head, Dept. Pharm. Sci., Dr. H.S. Gour University, Sagar, India, for providing the necessary facilities to carry out the research work. The authors thank Dr. Rajesh Pathania, All India Institute of Medical Sciences, New Delhi, India, for providing SEM facility. The authors also thank Dr. S. Dhaneshwar, Bharati Vidyapeeth Deemed University, Pune, India, for providing DSC facility.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Alsarra IA, Alarifi MN. (2004). Validated liquid chromatographic determination of 5-Fluorouracil in human plasma. J Chromatogr B 804:435–39
- Ashford M, Fell T. (1994). Targeting drugs to colon: delivery system for oral administration. J Drug Target 2:241–58
- Basan H, Gumusderelioglu M, Tevfik Orbey M. (2007). Release characteristics of salmon calcitonin from dextran hydrogels for colon-specific delivery. Eur J Pharm Biopharm 65:39–46
- Bodmeier R, Wang J. (1993). Microencapsulation of drugs with aqueous colloidal polymer dispersions. J Pharm Sci 82:191–4
- Brondsted H, Andersen C, Hovgaard L. (1998). Crosslinked dextran—a new capsule material for colon targeting of drugs. J Control Rel 53:7–13
- Chaturvedi K, Ganguly K, Kulkarni AR, et al. (2013). Ultra-small fluorescent bile acid conjugated PHB–PEG block copolymeric nanoparticles: synthesis, characterization and cellular uptake. RSC Adv 3:7064–70
- Chaturvedi K, Kulkarni AR, Aminabhavi TM. (2011). Blend microspheres of poly(3-hydroxybutyrate) and cellulose acetate phthalate for colon delivery of 5-fluorouracil. Ind Eng Chem Res 50:10414–23
- Chaturvedi K, Tripathi SK, Kulkarni AR, Aminabhavi TM. (2013). Cytotoxicity and antitumour activity of 5-fluorouracil-loaded polyhydroxybutyrate and cellulose acetate phthalate blend microspheres. J Microencapsul 30:356–68
- Chourasia MK, Jain SK. (2003). Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Pharm Sci 6:33–66
- Dubernet C. (1995). Thermo analysis of microspheres. Thermochim Acta 248:259–69
- Ganguly K, Aminabhavi TM, Kulkarni AR. (2011). Colon targeting of 5-fluorouracil using polyethylene glycol cross-linked chitosan microspheres enteric coated with cellulose acetate phthalate. Ind Eng Chem Res 50:11797–807
- Hehre EJ, Sery TW. (1952). Dextran splitting anaerobic bacteria from the human intestine. J Bacteriol 63:424–6
- Hovgaard L, Brondsted H. (1995). Dextran hydrogels for colon-specific drug delivery. J Control Release 36:159–66
- Jain D, Majumdar DK, Panda AK. (2006). Insulin loaded eudragit L100 microspheres for oral delivery: preliminary in vitro studies. J Biomater Appl 21:195–211
- Krishnaiah YSR, Satyanarayan S. (2001). Colon-specific drug delivery systems. In: Jain NK, ed. Advances in controlled and novel drug delivery. : CBS Publishers and Distributors, 89–119
- Mateovic T, Kriznar B, Bogataj M, Mrhar A. (2002). The influence of stirring rate on biopharmaceutical properties of Eudragit RS microspheres. J Microencapsul 19:29–36
- Mehvar R. (2000). Dextrans for targeted and sustained delivery of therapeutic and imaging agents. J Control Release 69:1–25
- Michor F, Iwasa Y, Lengauer C, Nowak MA. (2005). Dynamics of colorectal cancer. Semin Cancer Biol 15:484–93
- Orienti I, Zecchi V. (1993). Progesterone-loaded albumin microparticles. J Control Release 21:1–7
- Paharia A, Yadav AK, Rai G, et al. (2007). Eudragit-coated pectin microspheres of 5-fluorouracil for colon targeting. AAPS Pharm Sci Technol 8:E1–7
- Rahman Z, Kohli K, Zhang SQ, et al. (2008). In vivo evaluation in rats of colon specific microspheres containing 5-fluorouracil. J Pharm Pharmacol 60:615–23
- Rai G, Jain SK, Agrawal S, et al. (2005). Chitosan hydrochloride based microspheres of albendazole for colonic drug delivery. Pharmazie 60:131–4
- Rodriguez M, Vila-Jato JL, Torres D. (1998). Design of a new multiparticulate system for potential site-specific and controlled drug delivery to the colonic region. J Control Release 55:67–77
- Rokhade AP, Shelke NB, Patil SA, Aminabhavi TM. (2007). Novel hydrogel microspheres of chitosan and pluronic F-127 for controlled release of 5-fluorouracil. J Microencapsul 24:274–88
- Rubinstein A. (1990). Microbially controlled drug delivery to the colon. Biopharm Drug Dispos 11:465–75
- Simonsen L, Hovgaard L, Mortensen PB, Brøndsted H. (1995). Dextran hydrogels for colon-specific drug delivery. V. Degradation in human intestinal incubation models. Eur J Pharm Sci 3:329–37
- Sinha VR, Kumria R. (2001). Polysaccharides in colon-specific drug delivery. Int J Pharm 224:19–38
- Tsuchiya HM, Jeanes A, Bricker HM, Wilham CA. (1952). Dextran-degrading enzymes from molds. J Bacteriol 64:513–9
- Van den Mooter G, Samyn C, Kinget R. (1994). The relation between swelling properties and enzymatic degradation of azo polymers designed for colon-specific drug delivery. Pharm Res 11:1737–41
- Wei H, Qing D, De-Ying C, et al. (2008). Study on colon-specific pectin/ethylcellulose film-coated 5-fluorouracil pellets in rats. Int J Pharm 348:35–45
- Yang L, Chu JS, Fix JA. (2002). Colon-specific drug delivery: new approaches and in vitro/in vivo evaluation. Int J Pharm 235:1–15
- Youan BBC, Jacson TL, Dickens L, et al. (2001). Protein release profiles and morphology of biodegradable microcapsules containing an oily core. J Control Release 76:313–26
- Zumreoglu-Karan B, Mazi H, Guner A. (2002). Dextran–rare earth ion lnteractions. II. Solid-state characteristics. J Appl Polym Sci 83:2168–74