
Abstract
Atazanavir (ATV) is a HIV protease inhibitor. Due to its intense lipophilicity, the oral delivery of ATV encounters several problems such as poor aqueous solubility, pH-dependent dissolution, rapid first-pass metabolism in liver by CYP3A5, which result in low bioavailability. To overcome afore mentioned limitations, ATV-loaded Eudragit RL100 nanoparticles (ATV NPs) were prepared to enhance oral bioavailability. ATV NPs were prepared by nanoprecipitation method. The ATV NPs were systematically optimized (OPT) using 32 central composite design (CCD) and the OPT formulation located using overlay plot. The pharmacokinetic study of OPT formulation was investigated in male Wistar rats, and in-vitro/in-vivo correlation level was established. Intestinal permeability of OPT formulation was determined using in situ single pass perfusion (SPIP) technique. Transmission electron microscopy studies on OPT formulation demonstrated uniform shape and size of particles. Augmentation in the values of Ka (2.35-fold) and AUC0-24 (2.91-fold) indicated significant enhancement in the rate and extent of bioavailability by the OPT formulation compared to pure drug. Successful establishment of in vitro/in vivo correlation (IVIVC) Level A substantiated the judicious choice of the in vitro dissolution milieu for simulating the in vivo conditions. In situ SPIP studies ascribed the significant enhancement in absorptivity and permeability parameters of OPT formulation transport through the Peyer's patches. The studies, therefore, indicate the successful formulation development of NPs with distinctly improved bioavailability potential and can be used as drug carrier for sustained or prolonged drug release.
Introduction
The majority of recently discovered chemical entities are poorly soluble in water (Kawabata et al., Citation2011; Merisko-Liversidge & Liversidge, Citation2011; Beloqui et al., Citation2013). Enhancing the oral bioavailability of these poorly water-soluble compounds is of immense attention to the scientific community and a key area of pharmaceutical research. One of the most extensively studied strategies in this regard is nanotechnology (Dahan & Hoffma, Citation2007; Merisko-Liversidge & Liversidge, Citation2008; Shi et al., Citation2010) because of the ability of NPs to pass multiple biological barriers and to release a therapeutic compound within the optimal dosage range. Polymeric NPs appear to be promising tools for delivery of poorly soluble drugs, yet few have been commercialized (Mora-Huertas et al., Citation2010).
Polymeric NPs provide the possibility of achieving high drug-loading capacity, sustained release, enhanced drug stability and absorption, as well as targeted deposition (Al-Qadi et al., Citation2012). NPs protect the entrapped drug from gastrointestinal interferences, prolong the systemic circulation time and control the release of drug in blood (Singh & Pai, Citation2013). All these benefits can contribute to reduction of dose and dosing frequency, thereby reducing the side effects and ameliorating the patient compliance.
Antiviral therapy requires high, prolonged plasma and tissue drug levels, where viral replication could be controlled. NPs potentially provide more durable control of viral replication in plasma, therefore maintaining long-term efficacy of the regimen (Dodiya et al., Citation2011). ATV is an azapeptide compound and the seventh addition to the family of HIV protease inhibitor (PIs) and is the most important drug among the other PIs recommended by World Health Organization (WHO) for the management of all forms of HIV (Fukushima et al., Citation2007). ATV has been successfully used in treatment-native and experienced HIV patients (Mastan & Kumar, Citation2010). ATV belongs to Biopharmaceutics Classification System (BCS) class II drug with poor water-solubility and high permeability (log P of 4.11) (Benet et al., Citation2011 Singh & Pai, Citation2014c). In adults, ATV recommended therapeutic dose is 400 mg once a day (QD).
Besides these, it undergoes rapid first-pass metabolism and P-gp efflux, leading eventually to marked reduction in the drug oral bioavailable fraction (i.e. 60%) in humans and animals like rats, etc (Fukushima et al., Citation2007). To circumvent the afore-mentioned limitations various formulation approaches of ATV have been reported like, nanocrystals, (Balkundi et al., Citation2011) tablets and capsules (Simpson et al., Citation2007) but all with limited fruition.
It has been well reported in the literature that daily dose of ATV causes serious liver problems, specifically hepatotoxicity (Devi & Pai, Citation2006). As a shortfall for all the conventional oral dosage forms, the duration of action is limited since the absorption of the drug depends on the resident time of the drug in the gastrointestinal tract. In order to fulfill the need of a long-term treatment with anti-HIV agents, where most of them suffer from the shortcoming of frequent administration and plasma concentration fluctuation, it is desirable to have novel drug-delivery carriers such as NPs (Bender et al., Citation1996).
Various polymers are being utilized for preparation of NPs (Baba et al., Citation2011). Eudragit RL 100 polymer is a copolymer of poly(ethylacrylate, methyl-methacrylate) containing an amount of quaternary ammonium groups between 8.8% and 12%. It is insoluble at physiologic pH values and capable of limited swelling, thus representing a good material for the dispersion of drugs. Eudragit RL 100 polymer is commonly used for enteric coating and also for preparation of NPs of ATV for maximizing the oral bioavailability and prolonging the residence time in order to obtain sustain drug release. The NPs were characterized and evaluated for in vitro and in vivo performance in animals, and in vitro/in vivo correlation was established.
Materials and methods
ATV was provided ex-gratis by M/s Hetero Labs, Hyderabad, India. The purities of ATV were >99.7%. Eudragit RL 100 was received as gift sample from M/s Evonik Industries, Mumbai, India. Tetradecyl trimethyl ammonium bromide (TTAB) and acetone were purchased from M/s SD Fine-Chem Limited, Mumbai. The solvents, i.e. acetonitrile, and other chemicals employed for liquid chromatographic studies were all of HPLC grade. All other materials employed during the studies were of analytical grade, and were used as obtained. Double-filtered (0.45 µm pore size, Millipore) deionized water was used in all sample preparations to avoid any possible contaminations.
HPLC method development
The chromatographic separation was performed on a Phenomenex C18 column (250 mm × 4.6 mm I.D., 5 mm particle size), under isocratic conditions using UV detection at 249 nm. The mobile phase was composed of potassium dihydrogen phosphate (pH 3.5) and acetonitrile in a ratio of (58:42, v/v). The flow rate was set at 1 ml/min in isocratic elution and the injected sample volume was 20 µl. The assay was linear over the ATV concentration range of 0.050–2.0 µg/ml. The limits of detection and quantification of ATV were 0.004 µg/ml and 0.012 µg/ml, respectively.
Preparation of ATV NPs as per experimental design
ATV-loaded Eudragit RL 100 NPs were prepared based on the method of nanoprecipitation as previously described, using Ultra Turrax IKA T25 digital high shear homogenizer (Quevedo et al., Citation2011; Singh & Pai, Citation2014a&Citationb). In brief, 100 mg Eudragit RL 100 and weighed amount of ATV were dissolved in 30 ml acetone to form the organic phase. The resulting organic phase was poured drop wise into 50 ml aqueous phase containing 100 mg of TTAB while homogenizing at 12,000g for 30 min. The resulting emulsion was placed on the magnetic stirrer plate and continuously stirred at room temperature to evaporate acetone for 5 h. The NPs were collected by centrifugation at 12,000g for 30 min and washed four times with distilled water. The NPs were then lyophilized and stored at 4 °C until further analysis. Blank (without drug) NPs were also prepared using the same method. The yield of the polymeric NPs was 97.3% with this protocol.
For further formulation optimization work, Eudragit RL 100 (X1) and TTAB (X2), were chosen and finally selected as the two critical influential factors. A CCD was employed, where the amounts of Eudragit RL 100 (X1) and TTAB (X2), were studied at three levels each. Overall, a set of 13 experimental runs each were studied as per the experimental design matrix as depicted in . The formulation at the intermediate coded factor levels (i.e. 0,0) was studied in quintuplicate. The response variables considered for the current Design of Experiments (DoE) optimization studies encompassed, particle size (Dnm), drug encapsulation efficiency (DEE), and mean percentage drug release in 24 h (%Rel24h).
Table 1. Preparation of ATV NPs as per the experimental design.
Characterization of NPs
Particle size and Zeta (ζ) potential
The Dnm of the ATV NPs was determined using Malvern Zetasizer Nano S90 (Malvern Instruments Ltd., UK) and the zeta potential was measured using Malvern Zetasizer Nano ZS (Malvern Instruments Ltd). Samples were diluted in MilliQ water before measurement.
Drug encapsulation efficiency
The DEE of ATV NPs was calculated by determining the amount of pure drug using a filtration technique. The DEE was determined by the separation of drug-loaded NPs from the aqueous medium containing non-associated ATV NPs by ultracentrifugation (REMI high speed, cofor preparation of controlled-releaseoling centrifuge; REMI Corporation, India) at 12,000g for 30 min, at 4 °C. The un-encapsulated ATV NPs was determined using HPLC. The total drug content in the ATV NPs was determined by dissolving the ATV NPs in methanol to release entrapped ATV. The resulting solution was analyzed using HPLC. The drug-loading content was the ratio of incorporated drug to polymer (w/w).
In vitro drug release studies
Dialysis membrane method was used to determine the release of ATV from the NPs formulations. Freshly made ATV NPs were separated from the aqueous NPs suspension medium through ultra centrifugation. These NPs were dried at room temperature for 12 h. NPs equivalent to one dose of ATV (400 mg) were then redispersed in 1ml of purified water and placed in a dialysis bag (molecular weight cut off 10,000–12,000 Da; Hi-Media, India), which was tied and placed into 200 ml of dissolution media. The entire system was kept at 37 ± 0.5 °C with continuous magnetic stirring (25 rpm) and the study was carried out in an adequate dissolution medium phosphate buffered saline (PBS) pH 7.4 for 24 h. At appropriate time intervals, aliquots of 1 ml were collected and filtered by 0.22 µm membranes to remove NPs in suspension and replaced with 1 ml of fresh buffer. The amount of ATV in the resulting samples was determined by the described HPLC method described above.
Optimization data analysis and validation of optimization model
The response variables, which were considered for systematic DoE optimization studies, included Dnm, DEE and %Rel24h. The polynomial regression results were demonstrated for the studied responses. Finally, the prognosis of OPT formulation was conducted using overlay plots, drawn using the Design Expert software (M/s Stat-Ease, MN) (Singh et al., Citation2012a, Citationb). Overlay plot was obtained by superimposing the values of various response variables in order to locate the OPT formulation. The observed and predicted responses were critically compared and the percent bias (i.e. prediction error) was also calculated with respect to the observed responses.
Transmission electron microscopy
The morphology of the OPT formulation was observed using transmission electron microscopy (TEM) attached with a mega view II digital camera (H 7500, Hitachi, Tokyo, Japan). A drop of sample diluted with water was placed on a copper grid and the excess was drawn off with a filter paper. Samples were subsequently stained with 1% of uranyl acetate solution for 30 s. The image was magnified and focused on a layer of photographic film.
In vivo pharmacokinetic studies in rat
The pharmacokinetic studies were carried out in healthy male Wistar rats (250–300 g). The animals were fasted overnight before dosing with free access to water. The animals were acclimatized to laboratory conditions over the week before experiments and fed with standard rat diet, under controlled conditions of a 12:12 h light:dark cycle, with a temperature of 22 ± 3 °C and a relative humidity (RH) of 50 ± 5%. The experimental protocol was approved by the Institutional Animal Ethical Committee (AACP/IAEC/Jun-2012-02). Twelve male Wistar rats were randomly separated into two groups (six animals each group). The grouping of animals was as follows:
Group I: Administered with pure drug (as solution).
Group II: Administered with OPT formulation (as dispersion in 1ml of water and then administered orally using oral gavage needle (No. 18).
All the animal groups received a dose equivalent to 7.2 mg/kg/rat of ATV (Fukushima et al., Citation2007). Following oral drug administration, the rats kept in cages were allowed access to food and water ad libitum. Serial aliquots of the blood samples (100 µl each) were withdrawn from the retro-orbital plexus under mild ether anesthesia at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16, 20 and 24 h in the heparinized microcentrifuge tubes (50 units heparin/ml of blood). Plasma was harvested by centrifugation at 15,000g for 15 min and stored at −20 °C until analyzed. Acetonitrile was added to precipitate the plasma proteins. Thereafter, samples were vortexed and centrifuged at 15,000g for 15 min.
Plasma samples were deproteinated with 1 ml of acetonitrile, vortexed for 30 s and centrifuged at 8000g for 15 min. The supernatant was decanted into a china dish and evaporated to dryness at room temperature. This was further reconstituted with 100 ml of mobile phase and vortexed for 30 s and 20 ml was injected into an HPLC system. ATV was detected at a wavelength of 249 nm. The adeptness of nanoparticulate formulations was appraised by administering pure drug orally and measuring the blood levels at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16, 20 and 24 h.
In-vitro/in-vivo correlation
Attempts were made to correlate the in vivo plasma level data obtained for OPT formulation and pure drug with the corresponding in vitro drug-release data and establish IVIVC. For establishing Level A IVIVC, percent drug absorbed data at various time points were obtained using modified Wagner-Nelson method and correlated with percent drug release data (Lu et al., Citation2011).
In situ SPIP studies
In situ SPIP studies were performed in male Wistar rats. The animals were before made to abstain from solid food at least 24 h prior to the study. The SPIP studies were carried out on the OPT formulation and pure drug, each in hexaplicate, employing an in-house fabricated assembly. The animals were anesthetized using thiopental sodium (50 mg/kg) intraperitoneally. Following mid-line incision of the abdomen, the proximal part of the jejunum, 2–5 cm below the Ligament of Trietz, was cannulated with a glass cannula and connected to a reservoir (Ho et al., Citation2008). The second incision was made 10–15 cm below the first incision and connected with an outflow glass cannula. The intestinal segments were perfused with Krebs-Ringer's solution until the perfusate was clear. The intestinal segments were consequently perfused with OPT formulation and pure drug maintained at 37 ± 1 °C at a perfusion rate of 0.25 ml/min. Steady state was achieved within 30 min, after which aliquots of samples (1 ml each) were periodically withdrawn at regular intervals of 10 min each. The volume of sample for each time interval was 2 ml. Samples were stored at -20 °C until analysis. Samples were filtered and directly injected onto HPLC column and required no sample preparation prior to analysis.
Stability studies
The OPT formulation were subjected to stability studies carried out at 25 ± 2 °C /60% ± 5% RH, as per the ICH guidelines for the climatic zone IV. The formulation was kept in air-tight glass vials and assayed periodically, at the time points of 0, 1, 3 and 6 months, for Dnm, DEE, %Rel24h and ζ potential.
Statistical analysis
For pharmacokinetic analysis, the plasma concentrations of each rat were analyzed with the Kinetica 5.0.11 version software (Thermo Fisher Scientific Inc., Waltham, MA), using a non-compartmental model. All results were expressed as means ± SD.
Results
Response surface analyses
The coefficients of the polynomial equation (Equation (1)) for Dnm, DEE and %Rel24h formed excellent fits to the data, with the values of r2 ranging between 0.9956 and 0.9985 (p < 0.001 in all the cases).
(1)
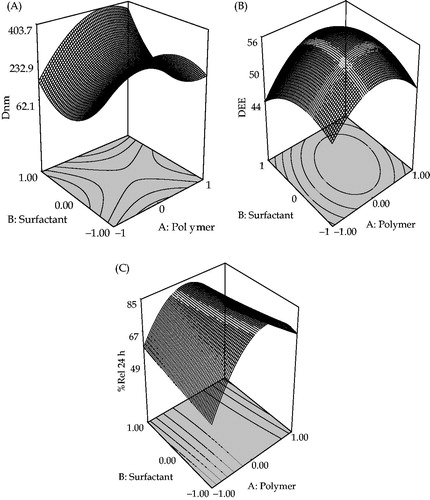
As observed in , a somewhat linear increase in Dnm was observed from lower to intermediate levels of polymer following which there was decrease in Dnm with higher levels of polymer. With increase in surfactant concentrations, however, a relatively little curvilinear descending trend was observed on Dnm. shows a linear increase in the values of DEE with rise in the levels of polymer. Analogous observation was noticeable in case of surfactant. shows a “canopy type” of response surface, characterizing initial non-linear increase, followed by a decline in the values of %Rel24h with further rise in the levels of polymer. Analogous observation, though of less magnitude, was noticeable in case of surfactant.
Figure 1. Response surface plot showing the influence of TTAB (surfactant) and Eudragit RL 100 (polymer) on (A) particle size (Dnm), (B) drug encapsulation efficiency (DEE) and (C) mean percent drug release in 24 h (%Rel24h) for NPs formulations of ATV.
Particle size (Dnm) and Zeta (ζ) potential
Dnm of all the 13 NPs formulations, prepared as per the experimental design, ranged between 390 and 487 nm. Remarkably small Dnm was observed at the higher levels of the TTAB and intermediate levels of the Eudragit RL 100. The ζ-potential was found to be positive of all the 13 NPs formulations ranged between +21.63 and +26.47.
Drug encapsulation efficiency
DEE is drug-loading capacity of any formulations. In the current study, the centrifugation method was used to determine DEE. The Eudragit RL 100 concentration has a marked influence on drug loading capacity of NPs. The DEE of all the 13 NPs formulations ranged between 41.3% and 56.9% as per experimental design.
In vitro drug release studies
All the formulations tended to release ∼85.14% of drug within 24 h, with relatively miniscule amount of drug release after 24 h.
Optimization data analysis and validation
The optimum formulation was selected by “trading off” various response variables while adopting the following criteria:
The OPT formulation (containing polymer 278 mg and emulsifier 249 mg) was found to fulfill maximal criteria for optimal performance.
The said formulation exhibited Dnm of 465.59 nm, polydispersity index (PDI) of 0.372, ζ potential of +24.53, DEE of 59.28%, and %Rel24h of 89.33%. Linear correlation plots drawn between the predicted and observed responses after forcing the line through the origin, also demonstrated high values of r, ranging between 0.9984 and 0.9991, indicating excellent goodness of fit (***p < 0.001). The corresponding residual plots also showed nearly uniform and random scatter around the zero-axis.
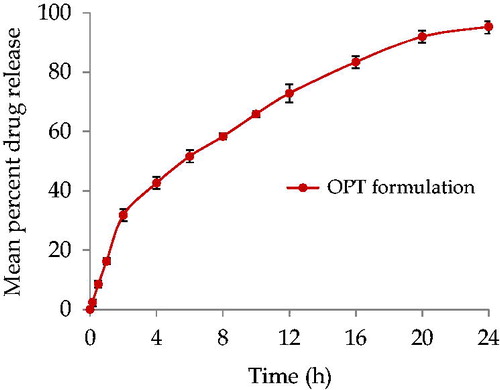
Drug release studies of OPT formulation
Marked improvement was observed in the release profile of OPT formulation (). Drug dissolution was nearly completed within 24 h (drug release 95.33%).
Figure 2. In vitro release profile of ATV loaded Eudragit RL 100 NPs with TTAB as surfactant in pH 7.4 phosphate buffer. Data points shown are mean ± standard deviation (SD) (n = 3).
Transmission electron microscopy
The TEM image () unambiguously reveals that most of the emulsion particles of OPT formulation were below 500 nm in size and were spherical in shape. Dnm distribution distinctly reveals the particles size of the OPT formulation was 465.59 nm.
Figure 3. TEM image shows particle size of OPT formulation.

In vivo pharmacokinetic studies in rats
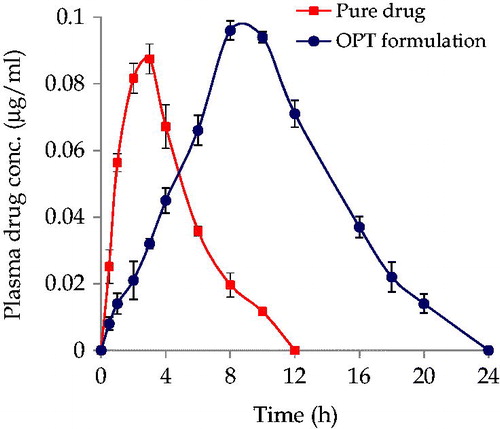
The mean plasma concentration–time profile () depicts significant higher plasma levels from OPT formulation with respect to pure drug (***p < 0.001). Further, the lower magnitude of standard error means (SEM) of OPT formulation indicated that the pharmacokinetic profile was more reproducible than that of pure drug. Plasma drug profile of OPT formulation exhibited more sustained release vis-à-vis pure drug. Maximal change in the pharmacokinetic parameter was observed in case of absorption rate constant (Ka), which is known to reflect the first-order rate of drug absorption. Nearly 2.35-fold augmentation in the magnitude of Ka was perceptible with the OPT formulation. On the whole, the values of Cmax and AUC0-24 rose considerably vis-à-vis pure drug to 1.10 and 2.91-fold, respectively, in case of the OPT formulation ratifying distinct improvement in extent of bioavailability too (***p < 0.001). Subsequent one-way ANOVA carried out on various pharmacokinetic parameters viz. AUC0-24, Ka and Tmax connoted highly statistically significant variation (p < 0.001) in the drug absorption potential of ATV from OPT formulation vis-à-vis pure drug.
Figure 4. Plasma drug level profiles of pure drug and OPT formulation. Each point represents mean of six replicates and each cross bar indicates 1 SEM.
In-vitro/in-vivo correlation
Both linear and non-linear Level A correlations were investigated between the two variables. Overall, the linear relationship was found to be better fitted than the linear and cubic non-linear relationship, as is observed from the smaller magnitude of p values.
In situ SPIP studies
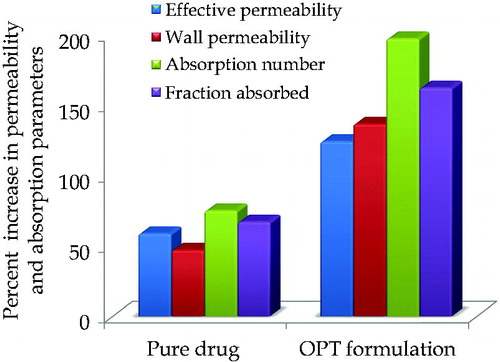
In the current study (), OPT formulation significantly improved the magnitude of absorption number by 2.65-fold, with reference to the pure drug. Nearly identical values of absorption number for the pure drug suggest analogous absorption mechanisms. Another dimensionless absorption parameter, fraction absorbed, provides an estimate of the extent of drug absorption (Fagerholm et al., Citation1996). As depicted in , the marked increase in the magnitude of fraction absorbed in case of OPT formulation (2.42-fold) vis-à-vis pure drug indicates high bioavalaiblity improvement potential of the OPT formulation. The effective permeability provides a direct measurement of absorption rate across the intestinal epithelium. As shown in , nearly 2.11-fold enhancement in the values of effective permeability of ATV was noticeable upon formulating it as NPs.
Figure 5. Percent increase in permeability parameters and absorption parameters in OPT formulation vis-à-vis pure drug using in situ single pass intestinal perfusion technique.
Stability studies
shows miniscule variation in the formulation parameters during 6 months of storage at the stability conditions of 25 °C ± 2 °C/60% RH ± 5% RH.
Table 2. Various parameters of the OPT formulation analyzed at different time points during stability studies.
Discussion
This is the first pharmacokinetic, in vitro/iv vivo correlation and in situ SPIP studies of nanoparticulate drug-delivery system of ATV to assess the potential for a NPs stabilized Eudragit RL 100-based formulation to improve the absorption of a poorly water soluble drug for ratifying its biopharmaceutical superiority. The OPT formulation provided enhanced bioavailability when compared to the pure drug.
The CCD is documented as the most efficient design. All the formulations, prepared as per the experimental design, exhibited nano Dnm, ranging between 64 nm and 403 nm. As we all know that the Dnm plays an important role in the alternation of pharmacokinetics through adjusting the in vivo distribution and clearance. The polydispersity of all NPs formulations was less than 0.31, demonstrating a unimodal size distribution.
ζ-potential is also of interest for the characterizations of these NPs since it reflects the surface charge of NPs and has been usually used to predict and control stability. Positive surface charge noted for the NPs were due to the polymer. It is a cationic polymer having quaternary ammonium groups.
DEE is also an important characteristic, which can directly influence the amount of the drug-loaded NPs at a given dose level. The DEE of all the 13 NPs formulations ranged between 41.3% and 56.9% as per experimental design. A high DEE of 59.28 ± 2.5% for OPT formulation was obtained. Eudragit RL 100 NPs have been reported to give high DEE for water-insoluble drugs. Present investigation also reveals that nanoprecipitation method can lead to NPs with high entrapment with water insoluble drug such as ATV.
We next investigated the in vitro release profiles of all the 13 NPs formulations in pH 7.4 PBS at 37 °C. A sustained release pattern is a key issue in the development of colloidal drug-delivery systems used in the field of nanomedicine. As the drug content in the formulation was higher, the release rate also increased. This may be due to the saturation of quaternary ammonium group present in Eudragit RL 100 polymer by drug molecules, occurred at a high drug content, which results in increased drug release from the formulations. None of the formulations showed burst release pattern. This indicates that the drug is homogeneously dispersed in polymer matrix. Other factors including the polymer composition, percentage of drug-loading, Dnm, as well as the characteristics of the encapsulated drug also affect the drug release from Eudragit RL 100 NPs.
Significant augmentation in the values of dissolution time for OPT formulation is quite indicative of higher bioavailability potential of ATV, a BCS class II drug exhibiting dissolution-limited absorption. Analogous in vitro dissolution profiles from OPT formulation construe that the Eudragit RL 100 seem to sustain the drug release from the NPs.
ATV NPs were designed to improve the oral bioavailability, reducing the side effects and dosing frequency of the drug. To our knowledge, there was no comprehensive study reported in the literature demonstrating the pharmacokinetic and in situ SPIP studies of ATV loaded Eudragit RL 100 NPs.
We believe that no single parameter can alone influence the fate of the formulations in vivo; rather it is a collective influence of many parameters. Blood levels after oral administration of pure drug were compared with oral administration of OPT formulation are shown in . From the graphs obtained by plotting plasma drug concentration versus time, it was observed that OPT formulation showed the release for 24 h with Cmax of 0.096 ± 1.52 µg/ml and AUC0-24 of 1.407 ± 2.18 µg/ml at Tmax of 8.01 h. Oral administration of pure drug exhibited only 12 h plasma profile, where oral pure drug suspension showed a Cmax of 0.87 ± 1.55 µg/ml at Tmax of 3 h. Low values of AUC0-24 and Cmax after oral administration of adequate dose of pure drug (7.2 mg/kg) also point toward incomplete absorption of drug from rats. The higher Cmax levels coupled with higher values of AUC0-24 observed in that OPT formulation indisputably vouch distinct improvement in rate and extent of drug bioavailability.
IVIVC allows prediction of the in vivo performance of a drug based on the in vitro drug release profiles. Excellent Level A correlation was observed with OPT formulation. Since ATV is a poorly water-soluble drug exhibiting nearly complete and dissolution limited absorption, it is anticipated to establish good point-to-point correlation between in vitro and in vivo performance (i.e. Level A). For Class 2 compounds dissolution is the rate- limiting step in absorption, therefore the establishment of IVIVC is expected vis-à-vis other BCS classes. The IVIVC established in the current studies has a definitive novel nuance, as hardly any study on fruitful establishment of level A IVIVC has been reported for a BCS class II drug ATV encapsulated in Eudragit RL 100 nanoparticulate drug delivery system.
Significant improvement in the magnitudes of fraction absorbed and absorption number exhibited by the OPT formulation () indicated distinct improvement in the amount of drug transported across the intestine. This corroborated their high bioavailability improvement potential vis-à-vis pure drug. Much higher values of the absorptivity parameters from the Eudragit RL 100 NPs can be ascribed to the transport of drug via M cells and the Peyer's patces, in accordance with an earlier literature report (Ensign et al., Citation2012). Size is also an important characteristic for efficient uptake. Numerous studies have been conducted for investigating the effect of size in various animal models and experimental systems, with the common consensus being that particles less than 1 μm in size can be transcytosed by M cells (Norris et al., Citation1998). Also, the improvement in permeability of OPT formulation may be attributed to the high concentration of emulsifier present in the formulation. Analogous results of increased intestinal permeability on increasing the surfactant concentration have been reported in literature (Gao et al., Citation2011). The close analogy of the in vivo results with that of the SPIP studies ratifies the usefulness of this in situ technique in the estimation of bioavailability and/or permeation parameters. shows miniscule variation in the values of Dnm, DEE, %Rel24h and ζ potential during six months of their storage.
Conclusion
The novelty of this work was to develop ATV NPs using Eudragit RL 100, and optimizing their composition using systematic “DoE” methodology. In vitro release test showed that ATV released from Eudragit RL100 NPs in a sustained manner for about 24 h. Considerable enhancement in the rate and extent of oral drug absorption ratified the superior performance of the nanoparticulate drug delivery systems in enhancing the bioavailability of ATV. Besides drug release enhancement and nano Dnm, significant improvement in oral bioavailability of ATV from nanoparticulate drug delivery system may be attributed ostensibly to reduced metabolism.
Acknowledgements
The authors would like to thank Prof. B.G Shivananda for his advice and support to carry out this research work. The authors are also grateful to Hetero Drugs, Hyderabad, India, for providing the gift sample of Atazanavir.
Declaration of interest
The authors report no declarations of interest. The authors gratefully acknowledge financial support and granting research fellowship (45/38/2011/Nan-BMS) from ICMR (Indian Council of Medical Research, Govt of India, New Delhi).
References
- Al-Qadi S, Grenha A, Carrión-Recio D, et al. (2012). Microencapsulated chitosan nanoparticles for pulmonary protein delivery: in vivo evaluation of insulin-loaded formulations. J Control Release 157:383–90
- Baba K, Tanaka Y, Kubota A, et al. (2011). A method for enhancing the ocular penetration of eye drops using nanoparticles of hydrolysable dye. J Control Release 153:278–87
- Balkundi S, Nowacek AS, Veerubhotla RS, et al. (2011). Comparative manufacture and cell based delivery of antiretroviral nano formulations. Int J Nanomedicine 6:3393–404
- Beloqui A, Solinís MA, Gascón AR, et al. (2013). Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release 166:115–23
- Bender AR, Von Briesen H, Kreuter J, Duncan IB, Rubsamen-Waigmann H. (1996). Efficiency of nanoparticles as a carrier system for antiviral agents in human immunodeficiency virus-infected human monocytes/macrophages in vitro. Antimicrob Agents Chemother 4:1467–71
- Benet LZ, Broccatelli F, Oprea TI. (2011). BDDCS applied to over 900 drugs. AAPS J 13:519–47
- Dahan A, Hoffman A. (2007). The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm 67:96–105
- Dodiya SS, Chavhan SS, Sawant KK, Korde AG. (2011). Solid lipid nanoparticles and nanosuspension formulation of Saquinavir: preparation, characterization, pharmacokinetics and biodistribution studies. J Microencapsul 28:515–27
- Devi KV, Pai RS. (2006). Antiretrovirals: need for an effective drug delivery. Indian J Pharm Sci 68:1–6
- Ensign LM, Cone R, Hanes J. (2012). Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 64:557–70
- Fagerholm U, Johansson M, Lennernas H. (1996). Comparison between permeability coefficients in rat and human jejunum. Pharm Res 13:1336–42
- Fukushima K, Terasaka S, Haraya K, et al. (2007). Pharmaceutical approach to HIV protease inhibitor atazanavir for bioavailability enhancement based on solid dispersion system. Biol Pharm Bull 30:733–8
- Gao F, Zhang Z, Bu H, et al. (2011). Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J Control Release 149:168–74
- Ho YF, Lai MY, Yu HY, et al. (2008). Application of rat in situ single-pass intestinal perfusion in the evaluation of presystemic extraction of indinavir under different perfusion rates. J Formos Med Assoc 107:37–45
- Kawabata Y, Wada K, Nakatani M, et al. (2011). Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm 420:1–10
- Lu Y, Kim S, Park K. (2011). In vitro–in vivo correlation: Perspectives on model development. Int J Pharm 418:142–148
- Mastan SK, Kumar K (2010). Influence of atazanavir on the pharmacodynamics and pharmacokinetics of gliclazide in animal models. Int J Diabetes Mellit 2:56–60
- Merisko-Liversidge EM, Liversidge GG. (2008). Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol 36:43–8
- Merisko-Liversidge EM, Liversidge GG. (2011). Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv Rev 63:427–40
- Mora-Huertas CE, Fessi H, Elaissari A. (2010). Polymer-based nanocapsules for drug delivery. Int J Pharm 385:113–42
- Norris DA, Puri N, Sinko PJ. (1998). The effect of physical barriers and properties on the oral absorption of particulates. Adv Drug Deliv Rev 34:135–54
- Pongpaibul Y, Price JC, Whitworth CW. (1984). Preparation and evaluation of controlled release indomethacin microspheres. Drug Dev Ind Pharm 10:1597–616
- Quevedo MA, Nieto LE, Brinon MC. (2011). P-glycoprotein limits the absorption of the anti-HIV drug zidovudine through rat intestinal segments. Eur J Pharm Sci 43:151–9
- Shi J, Votruba AR, Farokhzad OC, Langer R. (2010). Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett 10:3223–30
- Simpson KN, Jones WJ, Rajagopalan R, Dietz B. (2007). Cost effectiveness of lopinavir/ritonavir tablets compared with atazanavir plus ritonavir in antiretroviral-experienced patients in the UK, France, Italy and Spain. Clin Drug Investig 27:807–17
- Singh G, Pai RS, Devi VK. (2012a). Optimization of pellets containing solid dispersion prepared by extrusion/spheronization using central composite design and desirability function. J Young Pharmacists 4:146–56
- Singh G, Pai RS, Devi VK. (2012b). Response surface methodology and process optimization of sustained release pellets using Taguchi orthogonal array design and central composite design. J Adv Pharm Tech Res 3:30–40
- Singh G, Pai RS. (2013). Pharmacokinetics and in vivo biodistribution of optimized PLGA nanoparticulate drug delivery system for controlled release of emtricitabine. Drug Deliv DOI: 10.3109/10717544.2013.867382.
- Singh G, Pai RS (2014a). Optimized PLGA nanoparticle platform for orally dosed trans-resveratrol with enhanced bioavailability potential. Expert Opin Drug Deliv 11:647–59
- Singh G, Pai RS (2014b). In-vitro/in-vivo characterization of trans-resveratrol-loaded nanoparticulate drug delivery system for oral administration. J Pharm Pharmacol Doi: 10.1111/jphp.12232.
- Singh G, Pai RS (2014c). Optimized self-nanoemulsifying drug delivery system of atazanavir with enhanced oral bioavailability: in vitro/in vivo characterization. Expert Opin Drug Deliv Doi: 10.1517/17425247.2014.913566.