
Abstract
The main purpose of this study was to improve the oral bioavailability of sirolimus (SRL), a poorly water-soluble immunosuppressant, by encapsulating into lipids-based nanostructured lipid carriers (NLCs). SRL-loaded NLCs (SRL-NLCs) were prepared by a high-pressure homogenization method with glycerol distearates (PRECIROL ATO-5) as the solid lipid, oleic acid as the liquid lipids, and Tween 80 as the emulsifier. The SRL-NLCs prepared under optimum conditions was spherical in shape with a mean particle size of about 108.3 nm and an entrapment efficiency of 99.81%. In vitro release of SRL-NLCs was very slow, about 2.15% at 12 h, while in vitro lipolysis test showed fast digestion of the NLCs within 1 h. Relative oral bioavailability of SRL-NLCs in Beagle dogs was 1.81-folds that of the commercial nanocrystalline sirolimus tablets Rapamune®. In conclusion, the NLCs show potential to improve the oral bioavailability of SRL.
Introduction
Sirolimus (SRL) is a macrocyclic lactone immunosuppressant, which is widely used for anti-rejection therapy in organ transplantation (MacDonald et al., Citation2000; McEwan et al., Citation2005; Watson & Bradley, Citation2006; Kubiak et al., Citation2012). However, with a log P value of 4.3 at pH 7.0, SRL has extremely low aqueous solubility (2.6 μg/ml) (Simamora et al., Citation2001), which is generally accepted as the main rate-limiting factor for oral absorption of SRL (Solymosi et al., Citation2015). Currently, the leading commercial formulations of SRL are oral solution and tablets, which was launched under the product name Rapamune®. The oral bioavailability of the solution was about 14%, whereas the tablets, developed by the NanoCrystal® technology, show relatively higher bioavailability of about 17% (Hu et al., Citation2012). It is true that there is still much space left to improve the oral bioavailability of SRL.
For this purpose, various methods have been explored, such as inclusion complexes (Rouf et al., Citation2007; Zhang & Ma, Citation2013), liposomes (Haeri et al., Citation2013), supersaturable formulations (Kim et al., Citation2013), and self-microemulsifying drug delivery system (SMEDDS) (Sun et al., Citation2011; Hu et al., Citation2012). Among them, lipid-based drug delivery system seems to be more promising due to their unique absorption-enhancing effect taking advantage of the metabolic mechanisms of lipids (Mu & Høy, Citation2004; Pouton & Porter, Citation2008; Mu et al., Citation2013). Nanostructured lipid carriers (NLCs) composed of a solid lipid matrix with some amount of liquid lipids are new, improved generation of lipid nanoparticles based on solid lipid nanoparticles (SLN) (Müller et al., Citation2002). The solid nanoparticles have various advantages such as ability to immobilize drug in the solid matrix which makes the formulation more stable compared with SMEDDS (Wissing et al., Citation2004; Liu et al., Citation2014). Besides, they not only inherit the merits of SLN such as biodegradability, biocompatibility, and ease of scaling-up and manufacturing but also display more advantages than SLNs including high drug-loading capacity owing to the presence of liquid lipids which create more imperfections in the matrix to allow accommodation of more drug molecules (Das et al., Citation2012). Furthermore, drug expulsion is minimized during storage due to enhanced drug loading, and drug release patterns can be modulated by varying the structure of lipid matrices. Recently, nanostructured lipid carriers (NLCs) have been investigated to improve the oral bioavailability of many poorly water-soluble drugs (Zhuang et al., Citation2010; Tian et al., Citation2013; Shangguan et al., Citation2014).
In this study, we developed SRL-loaded NLCs (SRL-NLCs) using a high-pressure homogenization method, and optimized the preparative variables based on particle size, entrapment efficiency, drug loading capacity, and other parameters. Then, we studied the morphology, in vitro release and in vitro lipolysis of the formulations. Furthermore, oral bioavailability study in beagle dogs was carried out to evaluate the absorption of SRL-NLCs in comparison with commercial SRL tablets.
Materials and methods
Materials
Sirolimus was purchased from Fujian Kerui Pharmaceutical Co., Ltd. (Fuzhou, China). Ascomycin was purchased from Dalian Meilun Biotech Co., Ltd. (Dalian, China). Precirol ATO -5 was kindly gifted by Gattefosse Co. (Saint Priest, Cedex, France). Oleic acid and Tween-80 were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Commercial sirolimus tablet Rapamune® (1 mg) was purchased from Pfizer Ireland Pharmaceuticals (Kildare, Ireland). High-pressure liquid chromatographic grade methanol and acetonitrile were purchased from Sigma-Aldrich (Shanghai, China) Trading Co., Ltd. (Shanghai, China). Deionized water was prepared by a Milli-Q purification system (Millipore, Billerica, MA). All other reagents were of analytical grade.
Preparation of SRL-NLCs
SRL-NLCs were prepared by a high-pressure homogenization method with minor modifications (Tian et al., Citation2013). First, certain amount of precirol ATO-5 and oleic acid were mixed and melted at 80 °C. Then SRL was dissolved in ethanol and added into the lipid mixture under magnetic stirring at 500 rpm. Meanwhile, the aqueous phase was prepared by dispersing the surfactant, Tween 80, in deionized water and heated to the same temperature. After evaporation of ethanol, the hot aqueous phase was added into the oil phase under high shear at 5000 r/min for 1 min by a high-speed homogenizer (Ningbo scientz biotechnoligy Co., Ltd., Ningbo, China). Subsequently, the obtained coarse emulsion was further homogenized at a temperature of about 70 °C using a micro-jet homogenizer (Nano DeBEE, BEE, South Easton, MA) under 20 000 psi for four cycles. The resulting hot O/W nanoemulsions were cooled down to room temperature to solidify. The obtained NLCs formulations were stored at 4 °C.
Determination of SRL in vitro by HPLC-UV
SRL was determined by HPLC-UV on an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA) at a wavelength of 278 nm, a column temperature of 50 °C, and an injection volume of 20 μL. The mobile phase was acetonitrile/methanol/water in volume ratio of 45/34/21, and pumped by a quaternary pump at a flow rate of 1.0 mL/min (Hu et al., Citation2012). SRL was separated by a C18 column (Eclipse XDB-C18 column, 4.6 × 150 mm, 5 μm; Agilent, Santa Clara, CA). The linearity was ranged from 0.05 to 10 μg/mL, with a correlation coefficient of 0.9999. Accuracy of assay was about 100.8%, (n = 15). Intraday and interday precision were all below 2%.
Particle size and zeta potential analysis
Malvern Zetasizer Nano® Instruments (Malvern, UK) was used to measure the average particle size and zeta potential of SRL-NLCs. It was equipped with a 4 mW He–Ne laser (633 nm) at 25 °C based on photon correlation spectroscopy. Samples were diluted five times with deionized water before measurement. The results were analyzed by the Zetasizer Software provided by Malvern Instruments (Malvern, UK). Each sample was measured in triplicate.
Entrapment efficiency and loading capacity
The entrapment efficiency of SRL-NLCs was calculated by determining the amount of free drug using ultrafiltration technology (Araújo et al., Citation2010). Briefly, the NLCs dispersion was diluted 10 times with deionized water and added into the upper chamber of an ultrafiltration cell (Amicon ultra, Millipore Co., Billerica, MA, MWCO 100 kDa). After centrifugation for 10 min at 4000 rpm, the non-encapsulated drug was determined by HPLC. The total drug content in SRL-NLCs was determined after dissolving the vehicles using methanol under ultra-sonication. The drug entrapment efficiency (EE%) and loading capacity (LC%) could be calculated by the following equations:
where Wtotal is the amount of total drug in the preparation, Wfree is the amount of non-encapsulated drug and Wlipid is the amount of lipid added. Each sample was measured in triplicate.
Transmission electron microscopy (TEM)
The morphology of the NLCs was observed by transmission electron microscope (JEM-1230 Electron microscope, JEOL, Tokyo, Japan). The sample was negatively stained with 1% (w/v) uranyl acetate, placed onto copper grids and dried before TEM observation.
In vitro release
In vitro release of SRL-NLCs was evaluated by a dynamic dialysis method (Moreno-Bautista & Tam, Citation2011) in a dissolution tester (ZRS-8G, Tianjin, China) based on Chinese Pharmacopeia Appendix Method III (small beaker method). The dissolution medium was 250 mL of 0.4% sodium dodecylsulfate (SDS) with a pH of 7.4, which was confirmed sufficient to achieve sink conditions, thermostatically maintained at 37.0 ± 0.5 °C. The paddle rotation speed was set to 100 rpm. SRL-NLCs suspension (equivalent to 1 mg SRL) or the reference SRL formulation, Rapamune®, was sealed into a dialysis bag (MWCO 140 000, Spectrumlabs, Rancho Dominguez, CA) and immersed into the medium. Aliquots of medium were withdrawn at time intervals and the same volume of fresh medium of equal temperature was supplemented to keep constant volume. The withdrawn samples were filtered through 0.22 μm membrane filters (FINE Scientific Ltd., Beijing, China), and were assayed by HPLC-UV as described above.
In vitro lipolysis
In vitro lipolysis model has been well accepted as a useful tool to predict the gastrointestinal behavior of lipid-based drug delivery systems by mimicking in vivo digestive conditions (Ibrahim et al., Citation2012; Shangguan et al., Citation2015). In this study, the lipid digestion rate and extent of SRL-loaded NLCs were evaluated under both fasted and fed-state intestinal conditions. The simulated intestinal fluid for lipolysis was as follows: fed state: Tris-maleate 50 mM, NaCl 150 mM, CaCl2 20 mM, sodium taurocholate 20 mM, lecithin 5 mM, lipase 1000 TBU/mL; fasted state: Tris-maleate 50 mM, NaCl 150 mM, CaCl2 5 mM, sodium taurocholate 5 mM, lecithin 1.25 mM, lipase 1000 TBU/mL (Porter & Charman, Citation2001). Briefly, 3 mL NLC suspensions were added into 22 mL digestive medium. The final pH was adjusted to 7.50 ± 0.05. Then, 2 mL porcine pancreatic lipase dispersion was added to initiate the reaction under magnetic stirring, maintaining temperature at 37 °C. The pH of the medium was maintained at 7.5 by using a pH-stat titrator (TitraLab® 854, Radiometer Analytical, Spectrumlabs, Rancho Dominguez, CA) to neutralize the fatty acid produced by lipase digestion of ATO-5 and oleic acid with 0.1 M NaOH. When lipolysis completed, 4-bromophenylboronic acid (4-BPBA) was immediately added to inactivate the lipases. After that, the digestive product was separated into an aqueous phase and a precipitated phase by ultra-centrifuging (Hitachi ultracentrifuge CP100MX, Hitachi Inc., Tokyo, Japan) at 284 200 g for 90 min at room temperature. The drug content in each phase was analyzed by HPLC.
Bioavailability studies
The oral bioavailability of SRL-NLCs was evaluated in beagle dogs with commercial SRL tablets as a reference in a crossover design with a two-week washout period between dosing. Six healthy male beagle dogs (12.0 ± 2.0 kg) were fasted for 12 h before administration, but allowed free access to water. The formulations were administered orally in a dose of 2.0 mg SRL equivalent. Blood samples (2.0 mL) were withdrawn through the leg veins into heparinized tubes at time intervals, and kept frozen at −20 °C until analysis. All animal experiments were approved by Fudan University Institutional Animal Care and Use Committee.
Liquid–liquid extraction method was used to extract SRL from blood (Hu et al., Citation2012). Briefly, 10 μL internal standard solution (1.03 mg/L) and 500 μL sodium acetate buffer solution (pH 4.6) were added to 500 μL blood sample. After vortex-mixing for 1 min, 2 mL tert-butyl methyl ether was added, and extraction was preceded by mixing for 20 min in an oscillator. After centrifugation at 5000 rpm for 3 min, the organic layer was collected and evaporated under nitrogen flow at 40 °C. The residue was reconstituted with 100 μL mobile phase and analyzed by LC-MS/MS.
The main pharmacokinetic parameters such as the area under the concentration–time curve (AUC0–t) were obtained using a pharmacokinetic program Kinetica 4.4 (Thermo Fisher, Waltham, MA). The values of peak blood concentration (Cmax) and peak time (Tmax) were obtained directly from raw data. Statistical analysis was performed by one-way ANOVA. The relative bioavailability (Fr) of SRL-NLCs to commercial SRL tablets was calculated using the following equation:
Determination of SRL in dog blood by HPLC-MS/MS
The concentrations of SRL in dog blood were determined by HPLC-MS/MS (Agilent, Santa Clara, CA) with ascomycin (FK520) as internal standard (IS). SRL was separated by an Agilent ZOBAX C18 column (50 mm × 2.1 mm, 5 μm; Agilent, Santa Clara, CA) at 50 °C with acetonitrile/water in volumetric ratio of 9/1 (v/v) as the mobile phase at a flow rate of 0.3 mL/min (Hu et al., Citation2012). The detection was performed by an Agilent triple quadrupole detector (G6410, Agilent, Santa Clara, CA). The mass spectrometer was operated with electrospray ionization (ESI) interface in positive ionization mode and with multiple reactions monitoring mode. The selected reaction monitoring (SRM) of SRL and internal standard were m/z 936.7 → 409.3 and m/z 815.1 → 604.4, respectively. The linearity was observed within the range of 0.8440–54.00 ng/mL with r value of 0.9995. The limit of detection was 0.2 ng/mL, and the accuracy was 98.54 ± 2.20%. Extraction recovery of SRL and internal standard in dog plasma was 86.55 ± 2.37% and 77.43 ± 1.81%, respectively. At concentrations of 3.375, 13.50, and 54.00 ng/mL, intra-day precision was 7.53%, 7.55%, and 1.17%, respectively, while inter-day precision was 11.68%, 8.37%, and 1.08%, respectively.
Results
Preparation and characterization of SRL–NLCs
Precirol ATO-5 and oleic acid were chosen as the solid and liquid lipid, respectively, because they were reportedly used to improve oral bioavailability for poorly water-soluble drugs (Barnwell et al., Citation1992; O’Driscoll, Citation2002). SRL-NLCs were prepared successfully by the hot high-pressure homogenization method with good reproducibility. Since the in vitro and in vivo performance of nanoparticles are significantly influenced by particle size distribution, entrapment efficiency and drug loading capacity. Several preparative parameters that could significantly affect the physicochemical properties of NLCs were investigated. The effects of SRL/lipids (w/w), emulsifier/lipids (w/w), OA/ATO-5 (w/w), homogenization pressure and cycles are shown in . Entrapment efficiency of SRL was as high as 99.89% and hardly influenced by all of the preparative parameters (), which might be attributed to the high affinity of SRL to the lipid matrix (Simamora et al., Citation2001). With SRL/lipids increased from 2/100 to 10/100, the loading capacity increased from 1.95% to 8.25% substantially; however, the entrapment efficiency of NLCs decreased from 99.89% to 89.90% due to loss of SRL to the outer aqueous phase, and the particle size of NLCs increased slightly from 108 nm to 118 nm possibly due to increased drug loading (). A substantial decrease in particle size correlated with increased concentration of emulsifiers, i.e. a particle size of 165.87 nm at emulsifier/lipids ratio of 25/100 decreasing to 74.25 nm at 50/100 (). The increase in OA/ATO-5 ratio from 0 to 1 leads to increasing in particle size and slight fluctuation in entrapment efficiency (). The homogenization pressure had significant effect on particle size. At homogenization pressures of 10 000 and 20 000 psi, the mean diameters of NLCs were 117.93 and 111.97 nm, respectively, showing little difference, whereas at higher homogenization pressure of 30 000 psi the diameter was 159.53 nm, a significant increase (). But homogenization cycles showed no significant influence on particle size ().
Figure 1. Effects of SRL/lipids (A), emulsifier/lipids (B), OA/ATO 5 (C), homogenization pressure (D), and cycles (E) on entrapment efficiency (EE), drug loading (DL), and particle size of SRL-NLCs. Significance at p < 0.05 level. (A) EE: 10/100 versus each of other three groups. DL: between each two groups. Size: either 2/100 or 3/100 versus either 4/100 or 10/100. (B) Size: between each two groups. (C) Size: between each two groups. (D) Size: either 10 000 or 20 000 psi versus 30 000 psi. (E) Size: cycle 3 versus 4.
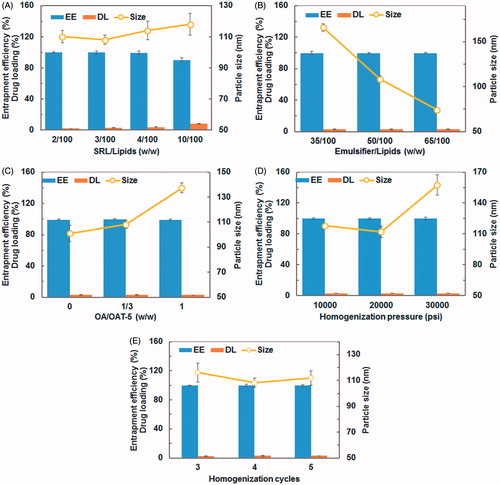
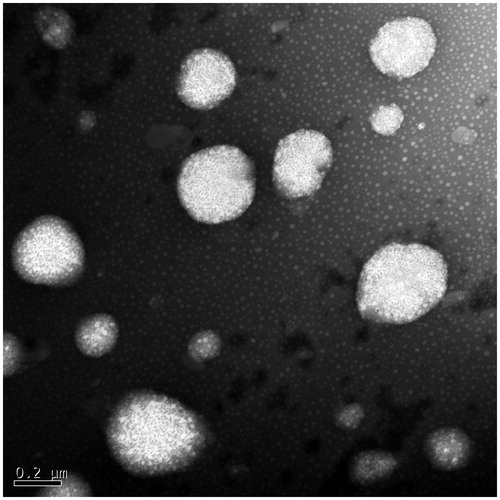
Taking together all the results of single-factor analysis, the optimal formulation was as follows: SRL/lipids ratio (w/w), 3/100; emulsifier/lipids ratio (w/w), 37.5/100; OA/ATO-5 ratio (w/w), 1/3; homogenization pressure, 20 000 psi; homogenization cycles, 4. The entrapment efficiency of SRL-NLCs prepared under optimum conditions was 99.81 ± 0.06%. The average particle size was 108.3 ± 3.47 nm (n = 3). The size distribution of SRL-NLCs is shown in . The morphology of SRL-NLCs determined by TEM is shown in . The TEM photograph showed that the nanoparticles were in spherical and uniform shape without aggregation. The mean diameter was in the range of 100–200 nm, which was in accord with that measured by Zetasizer Nano (Malvern Instruments, Malvern, UK).
Figure 2. Intensity Gaussian distribution of SRL-NLCs particle size.
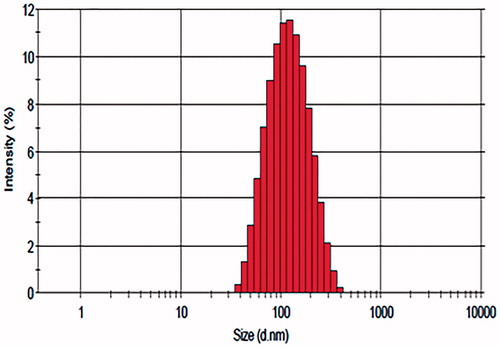
Figure 3. TEM photograph of SRL-NLCs.
In vitro release
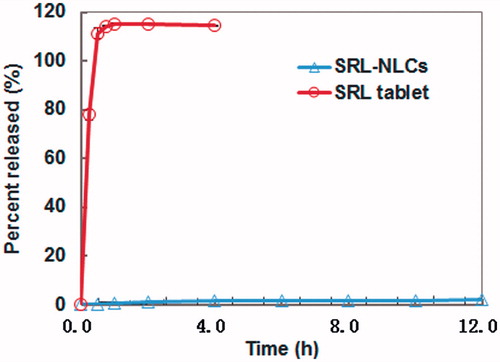
The in vitro release profiles of SRL-NLCs and commercial SRL tablets are shown in . Commercial SRL tablets showed rapid release with more than 78% released in 15 min, and almost complete release after 30 min. However, SRL was hardly released from NLCs, with less than 1% after 2 h and only 2.15% at 12 h.
Figure 4. In vitro release profile of SRL-NLCs and commercial SRL tablets.
In vitro lipolysis
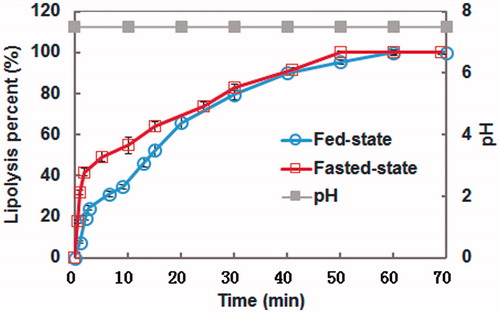
The in vitro lipolysis model was designed to simulate the digestive process of lipid-based drug delivery systems in the gastrointestinal tract. The cumulative percent digestion of SRL-NLCs versus time at constant pH 7.5 is shown in . It was observed that under both simulated fed and fasted-state, the lipolysis was very fast at the initial stage, and then slowed down, which completed at about 60 min. Within the first 20 min, the fed-state lipolysis was slower than the fasted-state lipolysis. To identify distribution of the drug in different phases, the lipolysate was separated through centrifugation into aqueous and precipitate phases. The proportions of SRL in the aqueous and precipitate phases under fasted conditions were 59.61% and 40.39%, in comparison with 70.41% and 29.59% under fed conditions.
Figure 5. Cumulative lipolysis percent–time profiles of SRL-NLCs titrated by NaOH at constant pH of 7.5. Data expressed as mean ± SD (n = 3).
Bioavailability studies
The blood concentration–time profiles and the main pharmacokinetic parameters are shown in and . High inter-individual variations were observed for both formulations. SRL-NLCs gave significantly higher levels of SRL than the reference SRL nanocrystal formulation. In contrary to previous reports (Sun et al., Citation2011; Hu et al., Citation2012), the blood SRL levels dropped to below the limit of quantification after 10 h, which might be contributed to variations in the physical state of the dogs used. In comparison with the relatively slow peak time of 3.0 h for the reference, the Tmax of SRL-NLCs was 1.5 h, indicating very quick absorption. The Cmax of SRL-NLCs was 18.29 ng/mL, more than twice of the reference Cmax. The relative bioavailability of SRL-NLCs to commercial nanocrystal tablets was 181%.
Figure 6. Blood concentration–time profiles of SRL after oral administration of optimal SRL-NLCs and commercial SRL tablets to beagle dogs (mean ± SD, n = 6).
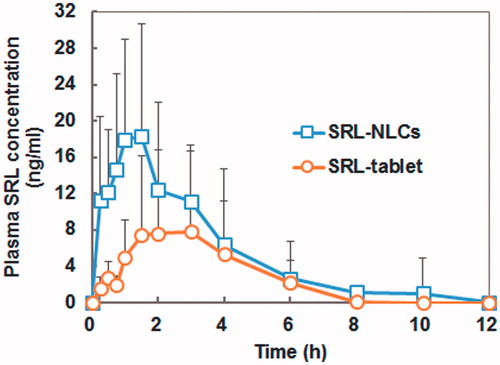
Table 1. Pharmacokinetic parameters after oral administration of SRL-NLCs and commercial SRL tablets to beagle dogs (mean ± SD, n = 6).
Discussion
Due to high affinity of SRL with the lipid phase, the entrapment efficiency of SRL in NLCs is very high. Except at extreme SRL loadings with a SRL/lipid ratio of 10/100, the entrapment efficiencies are almost 100%, indicating negligible leakage of SRL. The extremely limited in vitro release also confirms that the outwards diffusion of SRL as well as the erosion of the lipid matrix in the presence of surfactants is negligible. Based on the above observations, it is deduced that the in vivo release of SRL from NLCs due to factors other than lipolysis can be neglected, and has little effect on absorption of the drug. It has long been established that lipolysis is the principle mechanism governing in vivo release (Porter & Charman, Citation2001; Shangguan et al., Citation2015). Therefore, lipolysis is studied by mimicking the gastrointestinal physiology. It is the rate of lipolysis and following transformation into mixed micelles, rather than inherent release, that play the dominating role in the whole absorption process of NLCs.
The most outstanding feature of this study is the finding that the oral bioavailability of SRL is greatly enhanced to as high as 181% that of the commercial nanocrystal tablet, which is nevertheless higher than the result of 136% obtained for a self-nanoemulsifying SRL delivery system (Hu et al., Citation2012). The use of a nanocrystal-based SRL commercial tablet, instead of crude SRL microcrystals, as the reference provokes interesting discussion on the mechanisms of oral absorption of NLCs. Obviously, the enhanced oral bioavailability by NLCs should not be ascribed to improved dissolution because NLCs showed extremely limited in vitro release in sharp contrast with the fast release of SRL nanocrystal tablet. In another aspect, this result also proves that the currently used in vitro release test protocols may not be applicable to lipid-based delivery systems. To obtain good in vitro–in vivo correlation, the in vitro release test should take into account of the contribution of lipolysis, which, however, awaits future investigation. As indicated by in vitro lipolysis study, NLCs could be digested by lipases very quickly within about 20 min, and be degraded completely at 60 min. There is no doubt that lipolysis is the main mechanism governing the in vivo digestion and absorption of NLCs (Shangguan et al., Citation2015).
Another distinct finding of this study is the faster absorption rate of NLCs, as observed from the Tmax values, in accordance with our previous results (Tian et al., Citation2013; Shangguan et al., Citation2014). There are a few interpretations, albeit speculative, to this finding. First of all, NLCs may experience fast digestion once was transported to small intestine and transformed into mixed micelles that are ready for absorption, whereas SRL nanocrystals may not dissolve as fast in vivo and, therefore, exhibits prolonged absorption profile with slow peak time. Second, SRL nanocrystals may be released very quickly, but precipitates quickly as well, whereas SRL may be transferred to mixed micelles without significant precipitation upon digestion of NLCs by lipases and co-lipases with the help of intrinsic surfactants. Our study on the in vivo fate of solid lipid nanoparticles confirmed the very quick digestion of the lipid-based nanocarriers after oral administration (Hu et al., Citation2015), which in part highlights the advantages of lipid-based nanoparticles over nanocrystals.
One may argue that NLCs can also be absorbed through the M cell pathway. However, the very fast digesting rate and the very fast peak time of NLCs apparently do not comply with this assumption because M cell-based absorption means absorption as intact vehicles via lymphatic route which may take longer time. Since there are a series of barriers to the absorption of intact lipid-based nanocarriers, the contribution of the M cell pathway to overall absorption may be trivial, if there is.
Conclusions
SRL-NLCs were successfully prepared by modified high-pressure homogenization method with high entrapment efficiency. In vitro release of SRL-NLCs showed very limited release indicating high affinity of the drug to the lipid matrix. In vitro lipolysis test demonstrated fast digestion of NLCs in simulated intestinal fluid by lipases. The oral bioavailability of SRL-NLCs was enhanced about 1.82-fold that of commercial SRL tablets in beagle dogs. SRL-NLCs show potential for the enhancement of oral delivery of SRL.
Declaration of interest
The authors report that they have no conflicts of interest. This work was financially supported by Shanghai Commission of Science and Technology (14JC1490300), and National Key Basic Research Program (2015CB931800).
Related Research Data
References
- Araújo JE, Gonzalez-Mira MA, Egea ML, et al. (2010). Optimization and physicochemical characterization of a triamcinolone acetonide-loaded NLC for ocular antiangiogenic applications. Int J Pharm 393:168–76
- Barnwell SG, Laudanski T, Story MJ, et al. (1992). Improved oral bioavailability of propranolol in healthy human volunteers using a liver bypass drug delivery system containing oleic acid. Int J Pharm 88:423–32
- Das S, Ng WK, Tan RH. (2012). Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci 47:139–51
- Haeri A, Sadeghian S, Rabbani S, et al. (2013). Sirolimus-loaded stealth colloidal systems attenuate neointimal hyperplasia after balloon injury: a comparison of phospholipid micelles and liposomes. Int J Pharm 455:320–30
- Hu X, Lin C, Chen D, et al. (2012). Sirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluation. Int J Pharm 438:123–33
- Hu X, Zhang J, Yu Z, et al. (2015). Environment-responsive aza-BODIPY dyes quenching in water as potential probes to visualize the in vivo fate of lipid-based nanocarriers. Nanomedicine 11:1939–48
- Ibrahim F, Gershkovich P, Sivak O, et al. (2012). Assessment of novel oral lipid-based formulations of amphotericin B using an in vitro lipolysis model. Eur J Pharm Sci 46:323–8
- Kim MS, Kim JS, Cho W, et al. (2013). Supersaturatable formulations for the enhanced oral absorption of sirolimus. Int J Pharm 445:108–16
- Kubiak DW, Koo S, Hammond SP, et al. (2012). Safety of posaconazole and sirolimus coadministration in allogeneic hematopoietic stem cell transplants. Biol Blood Marrow Transplant 18:1462–5
- Liu Y, Wang L, Zhao Y, et al. (2014). Nanostructured lipid carriers versus microemulsions for delivery of the poorly water-soluble drug luteolin. Int J Pharm 476:169–77
- Müller RH, Radtke M, Wissing SA. (2002). Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm 242:121–8
- MacDonald A, Scarola J, Burke JT, et al. (2000). Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus. Clin Ther 22:B101–21
- McEwan P, Baboolal K, Conway P, et al. (2005). Evaluation of the cost-effectiveness of Sirolimus versus cyclosporin for immunosuppression after renal transplantation in the United Kingdom. Clin Ther 27:1834–46
- Moreno-Bautista G, Tam KC. (2011). Evaluation of dialysis membrane process for quantifying the in vitro drug-release from colloidal drug carriers. Colloid Surf A: Physicochem Eng Asp 389:299–303
- Mu H, Holm R, Müllertz A. (2013). Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm 453:215–24
- Mu H, Høy CE. (2004). The digestion of dietary triacylglycerols. Prog Lipid Res 43:105–33
- O’Driscoll CM. (2002). Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci 15:405–15
- Porter CJH, Charman WN. (2001). In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev 50:S127–47
- Pouton CW, Porter CJH. (2008). Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev S 60:625–37
- Rouf MA, Bilensoy E, Vural İ, et al. (2007). Inclusion complexation of rapamycin with beta-cyclodextrin to improve solubility and stability of the drug. Eur J Pharm Sci 32:S46–7
- Shangguan M, Lu Y, Qi J, et al. (2014). Binary lipids-based nanostructured lipid carriers for improved oral bioavailability of silymarin. J Biomater Appl 28:887–96
- Shangguan M, Qi J, Lu Y, et al. (2015). Comparison of the oral bioavailability of silymarin-loaded lipid nanoparticles with their artificial lipolysate counterparts: implications on the contribution of integral structure. Int J Pharm 489:195–202
- Simamora P, Alvarez JM, Yalkowsky SH. (2001). Solubilization of rapamycin. Int J Pharm 213:25–9
- Solymosi T, Angi R, Basa Dénes O, et al. (2015). Sirolimus formulation with improved pharmacokinetic properties produced by a continuous flow method. Eur J Pharm Biopharm 94:135–40
- Sun M, Zhai X, Xue K, et al. (2011). Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci 43:132–40
- Tian Z, Yi Y, Yuan H, et al. (2013). Solidification of nanostructured lipid carriers (NLCs) onto pellets by fluid-bed coating: preparation, in vitro characterization and bioavailability in dogs. Powder Technol 247:120–7
- Watson CJE, Bradley JA. (2006). Sirolimus and everolimus: inhibitors of mammalian target of rapamycin in liver transplantation. Transplant Rev 20:104–14
- Wissing SA, Kayser O, Müller RH. (2004). Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev 56:1257–72
- Zhang J, Ma PX. (2013). Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv Drug Deliv Rev 65:1215–33
- Zhuang CY, Li N, Wang M, et al. (2010). Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm 394:179–85