
Abstract
Background. Myocardial ischemia-reperfusion injury (IRI) is associated with activation of the innate immune system and the resultant inflammatory response. Myocardial ischemic preconditioning (IPC) is the most powerful endogenous protective mechanism against myocardial IRI, probably via the role of anti-inflammation. Regulatory T cells (Tregs), which are characterized by the expression of the forkhead/winged-helix transcription factor FoxP3, play an important role in the negative modulation of immune responses. We tested the hypothesis that Tregs may contribute to the protective effect of myocardial IPC through anti-inflammatory mechanisms. Methods and results. The left anterior descending coronary arteries of rats were occluded for a 30-min ischemia, followed by a 48-h reperfusion. Myocardial IPC was induced by 4 cycles of 5-min ischemia and 5-min reperfusion. Ischemia was achieved by ligation of the left anterior descending coronary artery (LAD), and reperfusion was initiated by releasing the ligature. Rats were injected with a Treg cell-depleting antibody or normal rat immunoglobulin (IgG), after IPC. The accumulation of Tregs was observed at indexed time points following IPC. The protein expression of FoxP3 significantly increased in the myocardium after IPC, and peaked at day-2. Treatment of preconditioned rats with the Treg cell-depleting antibody demonstrated less protein expression of FoxP3 (p < 0.001), more infiltration of inflammatory cells in the myocardium (p < 0.01), and larger myocardial infarct size (p < 0.001), compared with the IgG injection group. Conclusion. Cardioprotection by IPC is associated with Tregs.
Introduction
Prompt reperfusion is the most effective therapy to salvage an ischemic myocardium from irreversible necrosis, but also induces a side-effect, the so-called ischemia-reperfusion (IR) injury (IRI), which accounts for up to 50% of the final infarct size of a myocardial infarct (Citation1). The prevention of myocardial IRI, albeit of great clinical importance, remains a technical challenge. Although the pathogenesis of IRI is multifactorial, the activation of an inflammation cascade following reperfusion of an ischemic heart has been proven as a key and detrimental process for triggering or mediating myocardial IRI (Citation2,Citation3). Myocardial IPC is the most powerful endogenous myocardial protection against IRI, via multiple mechanisms including inhibition of the innate inflammatory reaction (Citation4–6). Accordingly, the inflammatory response could be a key link between IPC-afforded heart protection and IR-induced myocardial injury.
Tregs are anti-inflammatory lymphocytes. Expression of the interleukin-2 (IL-2) receptor (CD25) and the transcription factor FoxP3 identify a highly suppressive subset of CD4 + Tregs, which possess multiple mechanisms to inhibit the function and proliferation of pro-inflammatory leukocytes (Citation7–10). Tregs, therefore, might be the bridge linking IPC and IRI.
We hypothesized that Tregs might contribute to the heart protection against IRI, afforded by IPC. To test this hypothesis, we investigated the temporal accumulation of Tregs in a rat myocardial IPC model, and then targeted the deletion of the Tregs’ function with a Treg cell-depleting antibody, to observe the alterations of the IPC-induced cardioprotection.
Methods
Animals
Male syngeneic inbred Wistar rats, weighing 250– 300 g, were obtained from Shanghai SLAC Laboratory Animal Co. Ltd., China, and were housed in a specific pathogen-free facility. Our investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996). All animal experiments were conducted under protocols approved by the Experimental Animal Care Committee of the Affiliated Union Hospital, Fujian Medical University.
Surgical procedures
Ischemic preconditioning
All rats were anesthetized using pentobarbital sodium (40 mg/kg, i.p.) with supplemental doses as needed, and ventilated via a tracheostomy on a rodent respirator. A midline sternotomy was performed, and a reversible coronary artery snare occluder was placed around the left anterior descending coronary artery (LAD). Ischemia was achieved by ligating the proximal LAD using a 5-0 silk suture with a section of PE-10 tube placed over the LAD. Reperfusion was initiated by releasing the ligature and removing the PE-10 tube. A run of myocardial IPC was conducted by 4 cycles of 5-min ischemia followed by 5-min reperfusion. At the end of the fourth reperfusion, the snare was removed and the chest wall was closed. The above surgical procedures were also performed in sham groups with placement of a snare around the proximal LAD, but with no occlusion.
Ischemia-reperfusion
A run of myocardial IR was achieved by ligating the LAD for 30 min and then releasing by using the snare occluder. The loosened snare was left in place for the purpose of evaluating the ischemic area later, and then the chest wall was closed and the animals were recovered for continuous reperfusion. When reperfused for 48 h, the animals were sacrificed and their hearts were harvested. Myocardial samples were cut down from the IR region, for later analysis.
Antibody-mediated Treg cell depletion
Rats were injected via the tail vein with 2.0 mg of the anti-rat CD25 monoclonal antibody (clone NDS61) or normal rat immunoglobulin (IgG), 24 h before the 30-min ischemia or the second sham operation. Depletion of Tregs was verified by flow cytometry and Western blot analysis.
Experimental protocols
In the first experimental stage, 96 rats were randomly divided into 2 groups (n = 48 per group) (): the control group (sham), receiving sham operation without LAD occlusion; and the ischemic preconditioning group (IPC), given a run of IPC. Each group was then divided into 6 subgroups (n = 8 per subgroup), and the rats from each subgroup were sacrificed respectively at successive time points (days- 0, 1, 2, 3, 5 and 7) after myocardial IPC or sham operation.
Figure 1. Experimental protocols and animal groups. (A) 96 rats were divided into 2 groups (n = 48 per group): the control group (sham) and the ischemic preconditioning group (IPC). In each group, the hearts were harvested at successive time points (days 0, 1, 2, 3, 5 and 7) after myocardial IPC or sham operation (n = 8 per time point). Sham hearts were operated without ligating the coronary artery. (B) Rats were divided into 4 groups (n = 8 per group) and injected with the CD25 monoclonal antibody, NDS61, or control IgG, 24 h before 30-min ischemia (30 min I) or the second sham surgery. The hearts were harvested after a 48-h reperfusion (48 h R).
The protocols of the second experimental stage are schematically depicted in . The rats were randomly divided into 4 groups (n = 8 per group): the Sham/Sham-IgG group, which underwent two sham operations at an interval of 48 h, and was injected with placebo IgG at 24 h before the second sham operation; the Sham/IR-IgG group, which underwent a sham operation and a run of 30 min IR (30 min myocardial ischemia, then a 48-h reperfusion) at an interval of 48 h, was injected with placebo IgG at 24 h before 30-min ischemia, and then reperfused for 48 h; the IPC/IR-IgG group, which underwent a run of IPC and a run of 30-min IR at an interval of 48 h, was injected with placebo IgG at 24 h before 30-min ischemia, and then reperfused for 48 h; the IPC/IR-NDS61 group, which underwent a run of IPC and a run of 30-min IR at an interval of 48 h, was injected with NDS61 at 24 h before 30-min ischemia, and then reperfused for 48 h. All rats were sacrificed after 48-h reperfusion.
Flow cytomety
Flow cytometry was used to determine the number of Tregs before and after depletion of Tregs. Single-cell suspensions were prepared from rat spleens. Erythrocytes were lysed with RBC lysis buffer (150 mM NH4Cl, 1.0 mM KHCO3, and 0.1 mM Na2EDTA, pH 7.2), and the resulting leukocytes were resuspended at 106 cells/ml, for flow cytometric analyses. The antibodies used for the surface staining were: fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (0.25 μg per 106 cells; eBioscience), and phycoerythrin (PE)-conjugated anti-CD25 (0.125 μg per 106 cells; eBioscience). The cells were analyzed by flow cytometry using the FACSCalibur system.
Histological studies
Transverse midsections of hearts or spleens were fixed in formalin, embedded in paraffin, sectioned at 4 μm, and processed for histologic study. The number of Treg cells at successive time points (days- 0, 1, 2, 3, 5 and 7) after myocardial IPC was detected by immunohistochemical staining. The antibody used for immunohistochemical staining was anti-FoxP3. The total inflammatory cell infiltration after myocardial IR was assessed by hematoxylin and eosin staining (H&E). The quantification of positive cells was performed under 200 × magnification (field area = 0.12 mm2) or 400 × magnification (field area = 0.24 mm2). Positive cells from 3 sections were counted on 30 randomly chosen visual fields per section, and averaged.
Western blot analysis
The myocardial extracts were prepared as described previously, and Western blotting was used to determine the expression of FoxP3 (Citation11). After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the proteins were electro-transferred to nitrocellulose membranes, and the membranes were incubated overnight at 4°C with one of the following primary antibodies: anti-FoxP3, anti- myeloperoxidase, anti-macrophage marker, or anti-CD4. The membranes were then incubated with a secondary antibody, donkey anti-rabbit IgG, or goat anti-mouse IgG, for 1 h at room temperature. Target signals were normalized relative to the GAPDH expression, and assessed by ImageJ 1.36 software (NIH).
Infarct size determination
At the end of the 48-h reperfusion, the left coronary artery was reoccluded using the snare left in place. To assess the ischemic area at risk, 1% Evans blue was infused into the left atrial appendage. The excised hearts were cut transversely into five 1.5-mm-thick slices. The slices were incubated with 1% triphenyltetrazolium chloride solution (Sigma, USA) at 37°C for 15 min. Viable myocardium stained red, and infarcted tissue appeared white. The infarct size (IS, white), the area at risk (AAR, red and white), and the total left ventricle (LV) area from each section were measured using ImageJ 1.36 software (NIH). Ratios of AAR/LV and of IS/AAR were calculated and expressed as a percentage.
Statistical analysis
All data were analyzed with SPSS Version 11.5 (SPSS Inc., Chicago, Illinois, USA). All data were presented as mean ± SD. For comparison between multiple groups, the data were analyzed first by ANOVA, following the least significant difference (LSD) procedure if statistically different. A value of p < 0.05 was considered to be statistically significant.
Results
IPC promotes Treg cell accumulation in the heart
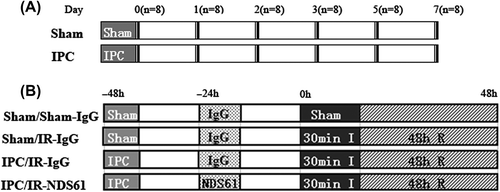
On the basis of the previously demonstrated ability of Treg cells to traffic to areas of inflammation (Citation12,Citation13), we hypothesized that myocardial IPC would cause accumulation of Treg cells in the heart. To test this, we measured the protein expression of FoxP3 in rat heart and spleen at successive time points (0, 1, 2, 3, 5 and 7 days), after either sham surgery or myocardial IPC, by Western blotting analyses. As compared to each sham subgroup at the corresponding time point, Western blotting showed that each IPC subgroup had a significant increase in the protein expression of FoxP3 in the heart at days-1, 2, and 3 after IPC, with a peak in protein expression at day-2 and a return to baseline level at day-5 ().
Figure 2. Myocardial ischemic preconditioning (IPC) caused the protein expression of FoxP3. At 0, 1, 2, 3, 5, and 7 days after myocardial IPC or sham (non-preconditioned) surgery, rat hearts were harvested and quantitatively analyzed for FoxP3 expression, using the Western blotting method. The FoxP3 protein levels are represented as a percentage of their respective sham control. Data are presented as the mean ± S.D.; ***p < 0.001, compared to nonpreconditioned rats, n = 8 per group.
There were no significant differences in the protein expression of FoxP3 observed in the spleen after IPC.
CD25 depletion significantly decreases CD4+CD25+FoxP3+ Tregs
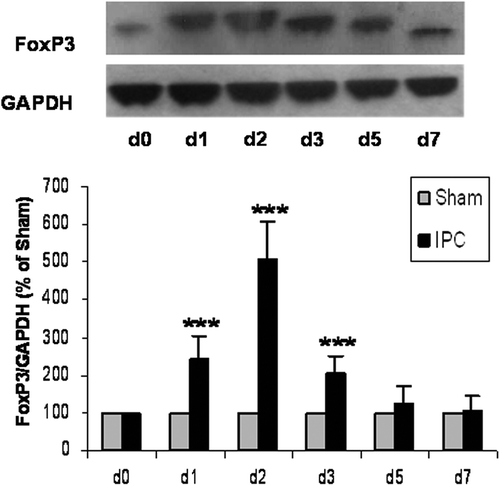
To test the function of Tregs in heart protection against IRI afforded by myocardial IPC, we used NDS61, a monoclonal antibody specific to CD25, which is highly expressed on Tregs, to selectively delete the functions of CD4+CD25+FoxP3+Tregs in rats. The depletion of Tregs was verified in the Tregs-enriched organ, the spleen, by Western blotting (). As compared to the placebo IgG-treated preconditioned rats (IPC/IR-IgG), a significant (≈50%) decrease in the protein expression of FoxP3 in the spleen was detected in the NDS61-treated preconditioned rats (IPC/IR-NDS61) (0.26 ± 0.06 vs. 0.14 ± 0.04, p < 0.001). Meanwhile, the protein expression of FoxP3 was also significantly reduced by approximately 50% in the heart (0.16 ± 0.05 vs. 0.09 ± 0.03, p < 0.001) ().
Figure 3. NDS61 administration significantly reduced Tregs in preconditioned rats. Preconditioned rats were injected with NDS61 or control IgG on day-1, before 30-min myocardial ischemia. Just before 30-min myocardial ischemia, FoxP3 expression in the Spleen (A) and heart (B) were measured by Western blotting analysis. Data are presented as the mean ± S.D.; ***p < 0.001 compared to IPC/IR-IgG. IPC, ischemic preconditioning; IR, ischemia-reperfusion injury; Treg, regulatory T.
Treg cell depletion inhibits the protective effect of IPC
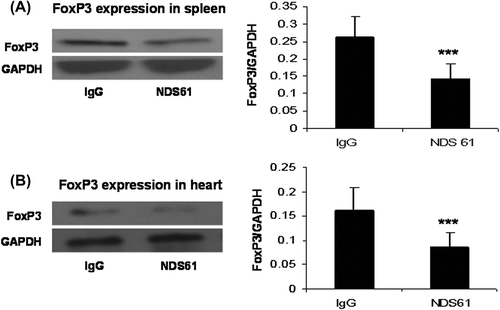
The histologic and functional changes in each experimental group after Treg cell depletion are shown in and .
Figure 4. Treg cell depletion inhibits anti-inflammatory effect of IPC. On day-1, before 30-min myocardial ischemia, preconditioned rats were injected with NDS61 or control IgG. At the end of 48-h reperfusion, some heart sections were stained with hematoxylin and eosin to assess total inflammatory cell infiltration in the different treatment groups. Data are presented as the mean ± S.D.; ***p < 0.001 compared to Sham/IR-IgG; †p < 0.05 compared to IPC/IR-IgG. IPC, ischemic preconditioning; IR, ischemia-reperfusion (original magnification 200×).
Figure 5. Treg cell depletion increases myocardial infarct size. On day-1, before 30-min myocardial ischemia, preconditioned rats were injected with NDS61 or control IgG. At the end of the 48-h reperfusion, the infarct size was determined by TTC staining (n = 8 per group). Values are expressed as mean ± S.D. ***p < 0.001 compared to Sham/IR-IgG; †††p < 0.001 compared to IPC/IR-IgG. LV, left ventricle; AAR, area at risk; IS, infarct size; IPC, ischemic preconditioning; IR, ischemia-reperfusion injury.
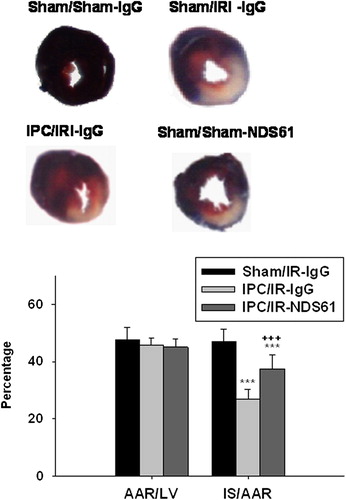
As compared to the non-preconditioned IRI rats (Sham/IR-IgG), the preconditioned rats treated with placebo (IPC/IR-IgG) had significant decrease in the number of inflammatory cells [(6.43 ± 2.64) × 103/mm2 vs (16.03 ± 5.30) × 103/mm2, p < 0.001] and in infarct size [(27.00% ± 3.27%) vs (47.5% ± 4.3%), p < 0.001]
As compared to the preconditioned rats treated with placebo (IPC/IR-IgG), the preconditioned rats treated with NDS61 (IPC/IR-NDS61) had more accumulation of inflammatory cells [(11.44 ± 4.56) × 103/mm2 vs (6.43 ± 2.64) × 103/mm2, p < 0.01] and larger infarct size [(37.50% ± 4.75%) vs (27.00% ± 3.27%), p < 0.001)], but these differences significantly reduced as compared to the non-preconditioned IRI rats (Sham/IR-IgG) [inflammatory cells :(11.44 ± 4.56) × 103/mm2 vs (16.03 ± 5.30) × 103/mm2, p < 0.05; infarct size: (37.50% ± 4.75)% vs (47.5% ± 4.31)%, p < 0.001].
Discussion
Inflammation plays a critical role in the pathogenesis of IRI, and this has been confirmed by abundant experimental and clinical studies (Citation2,Citation3). Recently, several studies have suggested that immune cells such as Tregs could suppress the activation of inflammation, thus protecting the kidney or liver from IRI (Citation14,Citation15). Our study firstly demonstrated that, in a rat heart model, IPC-mediated Tregs accumulation in the myocardium in a time-dependent manner is accompanied by less infiltration of pro-inflammatory cells following myocardial IR; in contrast, the targeted deletion of Tregs’ function enhanced the inflammatory response by increased recruitment of pro-inflammatory cells following myocardial IR, and attenuated the cardioprotective effects against IRI afforded by IPC, highlighting that IPC-induced heart protection is associated with Tregs.
Many studies regarding myocardial IR have suggested that inflammatory cells, particularly monocytes, macrophages, and T-cells, are involved in myocardial IRI, more exactly by initiating or exacerbating reperfused injury, and that depletion of inflammatory cells such as neutrophils, macrophages or CD4+ T cells in animal models is sufficient to reduce myocardial IRI (Citation16–19).
Tregs are anti-inflammatory lymphocytes that have been identified in various disease models (Citation20,Citation21). Such lymphocytes are multi-functional in inhibiting or regulating inflammatory reaction (Citation7–10). Another important property of Tregs is their propensity to home in to areas of ongoing inflammation (Citation12,Citation13).
IPC is a powerful intrinsic cardiac protection against IRI. Many complex mechanisms or signaling molecular pathways, such as PI3K/Akt, PKC/NF-κB, JAK/STAT and glycogen synthase kinase-3β (GSK-3β), are involved in the cardiac protection provided by IPC (Citation22–25), among which inhibition of the innate inflammation activation may be of importance (Citation4–6). However, there are no data regarding the roles of Tregs in cardiac protection against IRI, and their relation to IPC is unclear, even though several recent studies have suggested that Tregs could protect the kidney or liver from IRI via their suppression of innate immune cells (Citation14,Citation15).
In a rat heart model, we found that IPC increased the protein expression of FoxP3 in the myocardium in a time-dependent manner, which increased at days-1, 2 and 3 after IPC, peaking at day-2 and returning to baseline level at day-5. This is consistent with the finding that the delayed protective effect of myocardial IPC begins 24 h after IPC and continues for approximately 72 h (Citation26,Citation27). The results indicated that IPC promoted the accumulation of Tregs.
To determine the function of Tregs in myocardial IPC, we administered the Treg-deleting antibody to preconditioned rats, resulting in an approximately 50% reduction in the protein expression of FoxP3, compared to placebo IgG-treated preconditioned rats. Decreasing Treg cells in preconditioned rats caused inhibition of the anti-inflammatory effect of IPC and partially reversed the infarct-sparing protection, indicating that the Tregs’ negative modulation of inflammatory response does play a crucial role in the mediation of heart IPC.
Despite evidence that preconditioning works both in animal models and humans, the concept has not been well accepted as routine clinical therapy by clinicians, since in most clinical settings, the conventional mechanical IPC cannot possibly be performed prior to the onset of myocardial ischemia due to unpredictable ischemic events. Over the last 30 years, there have been intensive investigations into the biochemical pathways that lead to myocardial preconditioning. A number of pharmacologic agents are thought to act as triggers or effectors of ischemic preconditioning (Citation28–30), including both the early phase as well as the late phase of IPC. Some of these agents, such as adenosine, adenosine agonists, and KATP channel/openers that simulate ischemic preconditioning without inducing ischemia, have been studied in humans. In general, however, there have been no definitive and satisfactory outcomes found in the clinical realm.
Our study revealed that myocardial IPC, via the Tregs’ negative modulation of immune cells, inhibits the recruitment and activation of inflammation cells that are the major mediators or effectors for myocardial IRI; and that the deletion of Tregs’ functions attenuated such anti-inflammatory effects, thus abolishing IPC-afforded cardioprotection. These findings would provide a novel and feasible therapeutic target, i.e. anti-inflammation or immune modulation, as preconditioning mimetics. For this point, the present study might afford a broad range of potential clinical applications, not only for the prevention of myocardial IRI but also for more widespread systemic protection to other organs which are susceptible to acute IRI, such as the brain, liver, intestine, kidney, and so on, and for the donor protection to all solid organ transplantations as well.
In conclusion, the findings of our study suggest that cardioprotection afforded by IPC is associated with the anti-inflammatory property of Tregs.
Limitations
First, since the recently activated effector T cells and some dendritic cells may also express CD25 transiently, it is difficult to exclude the possibility that the anti-CD25 antibody exerts its effect via other cell subsets, therefore the conclusion in our study that the CD25-specific antibody exerted its effect via Tregs depletion may not be very precise. Second, because polyclonal IgG may have multiple biological effects, it is best to use the monoclonal IgG from the same species (mouse) as a negative control. Third, although the present study confirmed that Tregs play a crucial role in mediation of heart IPC via their negative modulation of inflammatory responses, answers to how and by what mechanisms Tregs negatively regulate innate immune system and inhibit inflammatory responses warrant investigation.
Acknowledgments
We would like to thank Bo Gao and Wei Xu from Shanghai Immunity Institute, and Lin Fan, Yunling Lin, Wei Cai and Ziwen Zhao from Fujian Provincial Institute of Coronary Disease, for their kindly help and excellent technical support.
Sources of Funding
This work was supported mainly by the National Natural Science Foundation of China (Grant No. 81170196), and partially by the National Natural Science Foundation of China (Grant No. 81100151), the Science Foundation for Distinguished Young Scholars of Fujian Province, China (Grant No.2013J06015), and the Science and Technology Project of Education Department of Fujian Province, China (Grant No.JA12132).
Declaration of interest: We declare that none of the authors have financial or other conflicts of interest in connection with the submitted article.
References
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1074–135.
- Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–97.
- Hansen PR. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995;91:1872–85.
- Wang NP, Bufkin BL, Nakamura M, Zhao ZQ, Wilcox JN, Hewan-Lowe KO, et al. Ischemic preconditioning reduces neutrophil accumulation and myocardial apoptosis. Ann Thorac Surg. 1999;67:1689–95.
- Nakamura M, Wang NP, Zhao ZQ, Wilcox JN, Thourani V, Guyton RA, et al. Preconditioning decreases Bax expression, PMN accumulation and apoptosis in reperfused rat heart. Cardiovasc Res. 2000;45:661–70.
- Huang SS, Wei FC, Hung LM. Ischemic preconditioning attenuates postischemic leukocyte–endothelial cell interactions: role of nitric oxide and protein kinase C. Circ J. 2006; 70:1070–5.
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by FoxP3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 2007;110: 1225–32.
- Buckner JH, Ziegler SF. Regulating the immune system: the induction of regulatory T cells in the periphery. Arthritis Res Ther. 2004;6:215–22.
- Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6.
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008; 28:546–58.
- Ke D, Fang J, Fan L, Chen Z, Chen L. Regulatory T cells contribute to
- Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632–6.
- Wei S, Kryczek I, Zou W. Regulatory T-cell compartmentalization and trafficking. Blood 2006;108:426–31.
- Burne-Taney MJ, Liu M, Baldwin WM, Racusen L, Rabb H. Decreased capacity of immune cells to cause tissue injury mediates kidney ischemic preconditioning. J Immunol. 2006; 176:7015–20.
- Arias-Diaz J, Ildefonso JA, Muñoz JJ, Zapata A, Jiménez E. Both tacrolimus and sirolimus decrease Th1/Th2 ratio, and increase regulatory T lymphocytes in the liver after ischemia/reperfusion. Lab Invest. 2009;89:433–45.
- Zuidema MY, Zhang C. Ischemia/reperfusion injury: The role of immune cells. World J Cardiol. 2010;2: 325–32.
- Svendsen JH, Hansen PR, Ali S, Baandrup U, Haunsø S. Leucocyte depletion attenuates the early increase in myocardial capillary permeability to small hydrophilic solutes following ischaemia and reperfusion. Cardiovasc Res. 1993; 27:1288–94.
- Gao XM, Liu Y, White D, Su Y, Drew BG, Bruce CR, et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: A predominant role of anti-inflammation. J Mol Cell Cardiol. 2011;50:991–9.
- Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4 + T lymphocytes. Circulation. 2006;114:2056–64.
- Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M, et al. Regulatory T cells protect mice against coxsackievirus- induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. 2010;121:2624–34.
- Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20: 1744–53.
- Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006; 70:240–53.
- Speechly-Dick ME, Mocanu MM, Yellon DM. Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res. 1994;75:586–90.
- Gross ER, Hsu AK, Gross GJ. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3β. Am J Physiol Heart Circ Physiol. 2006;291:H827–34.
- Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, et al. Cardioprotection by ischemic postconditioning is lost in aged and STAT3- deficient mice. Circ Res. 2008;102:131–5.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36.
- Baxter GF, Goma FM, Yellon DM. Characterisation of the infarct-limiting effect of delayed preconditioning: time course and dose-dependency studies in rabbit myocardium. Basic Res Cardiol. 1997;92:159–67.
- Okubo S, Xi L, Bernardo NL, Yoshida K, Kukreja RC. Myocardial preconditioning: basic concepts and potential mechanisms. Mol Cell Biochem. 1999;196:3–12.
- Nakano A, Cohen MV, Downey JM. Ischemic preconditioning. From basic mechanisms to clinical applications. Pharmacol Ther. 2000;86:263–75.
- Rubino A, Yellon DM. Ischemic preconditioning of vasculature: an overlooked phenomenon for protecting the heart? Trends Pharmacol Sci. 2000;21:225–30.