Abstract
Fourteen 5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamides (3a–n) were synthesized and evaluated for their in vitro activity against Mycobacterium tuberculosis H37Rv (MTB), multidrug-resistant Mycobacterium tuberculosis (MDR-TB), and Mycobacterium smegmatis (MC2), as well as their cytotoxicity and MTB isocitrate lyase (ICL) inhibition activity. 1-Cyclopropyl-6-fluoro-8-methoxy-7-(3-methyl)-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid (3n) was found to be the most active compound in vitro with MICs of < 0.17 and 0.17 μM against log-phase MTB and MDR-TB, respectively. Some compounds showed 20–45% inhibition against MTB ICL at 10 μM.
Introduction
Although effective chemotherapeutic agents have been developed, Mycobacterium tuberculosis (MTB), the causative agent of tuberculosis (TB), continues to be the greatest single infectious cause of mortality worldwide, killing roughly two million people annuallyCitation1. The World Health Organization has estimated that one-third of the world’s population is infected with MTB. The TB bacilli may remain in the body for decades without causing the symptoms of TB. TB is a leading cause of death among people who are human immunodeficiency virus (HIV)-positive (13% of AIDS deaths worldwide)Citation1. The synergy between TB and the acquired immunodeficiency syndrome (AIDS) epidemic and the surge of multidrug-resistant isolates of M. tuberculosis (MDR-TB) have reaffirmed tuberculosis as a primary public-health threat. It has been predicted that by 2020, one billion people will be newly infected if new anti-TB treatments are not developed. It is therefore necessary to discover new, safe, and more efficient antibiotics against this disease. In an effort to discover new and effective chemotherapeutic agents for the treatment of TB, we recently reported the antimycobacterial activity of various thiazolyl thiourea/thiosemicarbazonesCitation2,Citation3, spiro-piperidin-4-onesCitation4, oxobenzothiazolo[3,2-a]quinoline-6-carboxylic acidsCitation5, thiazeto[3,2-a]quinoline-3-carboxylic acidsCitation6, 8-naphthyridine-3-carboxylic acidCitation7, 5H-thiazolo[3,2-a]quinoline-4-carboxylic acidCitation8, and pyrano[3,2-c]pyridine-3-carbonitrilesCitation9. In the course of screening to discover new antimycobacterial compounds, we identified 5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamides which inhibited MTB and MDR-TB. We present herein the results concerning the synthesis, in vitro antimycobacterial activity (log- and starved-phase cultures), cytotoxicity, and MTB isocitrate lyase (ICL) inhibition studies.
Materials and methods
Chemistry
Melting points were taken on an electrothermal melting point apparatus (Buchi BM530) in open capillary tubes and are uncorrected. Infrared (IR) spectra (KBr disk) were run on a Jasco IR Report 100 spectrometer. 1H-nuclear magnetic resonance (NMR) spectra were scanned on a Jeol Fx 400 MHz NMR spectrometer using CDCl3, and dimethyl-sulfoxide (DMSO)-d6 as solvent. Chemical shifts are expressed in δ (ppm) relative to tetramethylsilane. Elemental analyses (C, H, and N) were performed on a PerkinElmer model 240C analyzer and the data were within ± 0.4% of the theoretical values.
General procedure for the preparation of amides (3a–n)
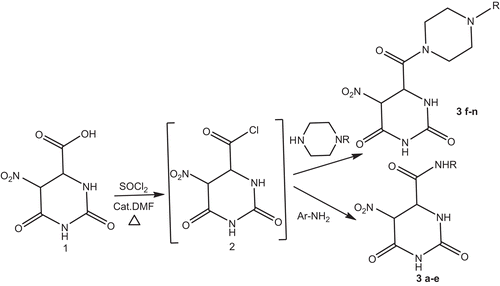
5-Nitro-2,6-dioxohexahydro-4-pyrimidinecarboxylic acid (1) (1.5 mol) was converted to the acid chloride (2) by refluxing in thionyl chloride with a catalytic amount of dimethylformamide (DMF). After 2–4 h, the reaction mixture was concentrated and suspended in pyridine. The mixture was cooled to 0°C under N2, and the corresponding amine (1 mol) was added. After stirring for 24 h at room temperature, the reaction mixture was concentrated, neutralized with 1 N HCl, and extracted with EtOAc (×3). The organics were washed with brine, dried over anhydrous MgSO4, and concentrated in vacuo to give the title compounds (3a–n).
5-Nitro-2,6-dioxo-N-phenylhexahydro-4-pyrimidinecarboxamide 3a Yield: 59.2 %; m.p.: 167–168°C; IR (KBr): 3520, 3000, 1720, 1560, 1355, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.90–7.66 (m, 5H, Ar-H), 8.12 (s, 1H, CONH, D2O exchangeable), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 172.9, 153.9, 138.6, 129.5, 124.4, 121.4, 94.2, 47.3; Anal (C11H10N4O5) C, H, N.
N-(4-Chlorophenyl)-5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamide 3b Yield: 61.3%; m.p.: > 250°C; IR (KBr): 3524, 3010, 1720, 1560, 1360, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 7.22–7.60 (m, 4H, Ar-H), 8.12 (s, 1H, CONH, D2O exchangeable), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 172.9, 153.9, 136.6, 129.5, 123.4, 94.2, 47.3; Anal (C11H9ClN4O5) C, H, N.
N-(4-Bromophenyl)-5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamide 3c Yield: 58.0%; m.p.: > 250°C; IR (KBr): 3524, 3000, 1720, 1560, 1350, 1320, 750, 600 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 7.35–7.55 (m, 4H, Ar-H), 8.12 (s, 1H, CONH, D2O exchangeable), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 172.9, 153.9, 137.6, 131.8, 123.4, 118.6, 94.2, 47.3; Anal (C11H9BrN4O5) C, H, N.
N-(2,4-Dinitrophenyl)-5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamide 3d Yield: 85.1%; m.p.: 169–170°C; IR (KBr): 3524, 3010, 1720, 1540, 1350, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 8.05–9.10 (m, 3H, Ar-H), 8.12 (s, 1H, CONH, D2O exchangeable), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 172.9, 153.9, 145.2, 142.1, 141.3, 127.4, 123.4, 118.6, 94.2, 47.3; Anal (C11H8N6O9) C, H, N.
N-(5-Bromo-2-pyridinyl)-5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamide 3e Yield: 62.4%; m.p.: 171–172°C; IR (KBr): 3524, 3000, 1740, 1720, 1560, 1350, 1320, 760, 600 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 7.60–8.62 (m, 3H, Ar-H), 8.12 (s, 1H, CONH, D2O exchangeable), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C NMR (DMSO-d6) δ (ppm): 175.1, 172.9, 153.9, 151.6, 142.4, 140.9, 123.4, 115.6, 94.2, 47.3; Anal (C10H8BrN5O5) C, H, N.
5-Nitro-6-[(4-phenylpiperazino)carbonyl]dihydro-2,4(1H,3H)-pyrimidinedione 3f Yield: 71.0%; m.p.: 152–153°C; IR (KBr): 3524, 3000, 1720, 1560, 1350, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 3.45 (t, 4H, -2CH2), 3.68 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.50–7.15 (m, 5H, Ar-H), 8.12 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 170.1, 153.9, 149.6, 130.1, 118.2, 114.5, 94.2, 49.8, 47.3, 46.3; Anal (C15H17N5O5) C, H, N.
6-[4-(4-Methoxyphenyl)piperazino]carbonyl-5-nitrodihydro-2,4(1H,3H)-pyrimidinedione 3g Yield: 58.4%; m.p.: 155–157°C; IR (KBr): 3524, 3000, 1720, 1560, 1350, 1320, 1270 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 3.45 (t, 4H, -2CH2), 3.58 (s, 3H, -OCH3), 3.68 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.50–6.77 (m, 4H, Ar-H), 8.12 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 170.1, 153.9, 150.5, 142.2, 115.5, 94.2, 56.3, 49.8, 47.3, 46.3; Anal (C16H19N5O6) C, H, N.
6-[4-(4-Chlorophenyl)piperazino]carbonyl-5-nitrodihydro-2,4(1H,3H)-pyrimidinedione 3h Yield: 53.9%; m.p.: 106–108°C; IR (KBr): 3524, 3000, 1720, 1560, 1350, 1320, 750, 600 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 3.45 (t, 4H, -2CH2), 3.68 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.50–7.11 (m, 4H, Ar-H), 8.12 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 170.1, 153.9, 147.8, 130.1, 123.9, 115.5, 94.2, 49.8, 47.3, 46.3; Anal (C15H16ClN5O5) C, H, N.
6-[4-(4-Fluorophenyl)piperazino]carbonyl-5-nitrodihydro-2,4(1H,3H)-pyrimidinedione 3i Yield: 80.2%; m.p.: > 250°C; IR (KBr): 3524, 3000, 1720, 1560, 1350, 1320, 1100 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 3.45 (t, 4H, -2CH2), 3.68 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.50-6.84 (m, 4H, Ar-H), 8.12 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 170.1, 153.9, 152.1, 145.3, 116.6, 115.5, 94.2, 49.8, 47.3, 46.3; Anal (C15H16FN5O5) C, H, N.
5-Nitro-6-(4-[3-(trifluoromethyl)phenyl]piperazino-carbonyl)dihydro-2,4(1H,3H)-pyrimidinedione 3j Yield: 52.5%; m.p.: 178–180°C; IR (KBr): 3524, 3000, 1720, 1560, 1400, 1350, 1320, cm−1; 1H-NMR (DMSO-d6) δ (ppm): 3.45 (t, 4H, -2CH2), 3.68 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.70-7.05 (m, 4H, Ar-H), 8.12 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 175.1, 170.1, 153.9, 150.5, 131.8, 130.2, 124.3, 117.4, 114.9, 110.2, 94.2, 49.8, 47.3, 46.3; Anal (C16H16F3N5O5) C, H, N.
1-Ethyl-6-fluoro-7-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid 3k Yield: 50.2%; m.p.: 150–152°C; IR (KBr): 3520, 3300, 3000, 1700, 1720, 1695, 1560, 1530, 1420, 1355, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 1.15 (t, 3H, -CH3), 3.21 (q, 2H, -CH2), 3.44 (t, 4H, -2CH2), 3.62 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 5.92 (s, 1H, C8-H), 7.15 (s, 1H, C5-H), 7.95 (s, 1H, C2-H), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 12.00 (s, 1H, COOH), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 178.1, 175.1, 169.9, 166.5, 153.9, 148.3, 144.6, 143.7, 140.9, 118.6, 116.4, 109.6, 100.2, 94.2, 49.2, 47.3, 46.1, 13.2; Anal (C21H21FN6O8) C, H, N.
1-Cyclopropyl-6-fluoro-7-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid 3l Yield: 70.9%; m.p.: > 250°C; IR (KBr): 3520, 3300, 3000, 1710, 1720, 1695, 1540, 1530, 1420, 1355, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.28–0.46 (m, 4H, cyclopropyl), 1.32 (m, 1H, cyclopropyl), 3.44 (t, 4H, -2CH2), 3.62 (t, 4H, -2CH2), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 5.92 (s, 1H, C8-H), 7.15 (s, 1H, C5-H), 7.95 (s, 1H, C2-H), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 12.00 (s, 1H, COOH), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 178.1, 175.1, 169.9, 166.5, 153.9, 148.3, 144.6, 143.7, 140.9, 118.6, 116.4, 109.6, 100.2, 94.2, 49.2, 47.3, 46.1, 36.3, 5.7; Anal (C22H21FN6O8) C, H, N.
7-(3,5-Dimethyl)-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid 3m Yield: 51.4%; m.p.: > 250°C; IR (KBr): 3520, 3300, 3010, 1700, 1720, 1695, 1560, 1530, 1420, 1355, 1300 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 1.15 (t, 3H, -CH3), 1.62 (d, 3H, -CH3), 3.21 (q, 2H, -CH2), 3.44 (d, 2H, -CH2), 3.62 (t, 2H, -CH2), 3.51 (t, 2H, -CH2), 4.02 (m, 1H, -CH), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.95 (s, 1H, C5-H), 7.95 (s, 1H, C2-H), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 12.00 (s, 1H, COOH), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 178.1, 175.1, 169.9, 166.5, 153.9, 148.3, 146.6, 136.1, 118.6, 112.4, 109.6, 94.2, 50.6, 49.2, 47.3, 46.1, 44.5, 16.2, 13.2; Anal (C22H22F2N6O8) C, H, N.
1-Cyclopropyl-6-fluoro-8-methoxy-7-(3-methyl)-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid 3n Yield: 65.3%; m.p.: 162–164°C; IR (KBr): 3520, 3300, 3000, 1700, 1720, 1695, 1560, 1530, 1420, 1355, 1320 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.28–0.46 (m, 4H, cyclopropyl), 1.32 (m, 1H, cyclopropyl), 1.62 (d, 3H, -CH3), 3.44 (d, 2H, -CH2), 3.51 (t, 2H, -CH2), 3.53 (s, 3H, -OCH3), 3.62 (t, 2H, -CH2), 4.02 (m, 1H, -CH), 4.84 (dd, 1H, -CH of pyrimidinyl ring), 5.62 (d, 1H, -CH of pyrimidinyl ring), 6.71 (s, 1H, C5-H), 7.95 (s, 1H, C2-H), 9.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable), 12.00 (s, 1H, COOH), 14.00 (s, 1H, NH of pyrimidinyl ring, D2O exchangeable); 13C-NMR (DMSO-d6) δ (ppm): 178.1, 175.1, 169.9, 166.5, 153.9, 148.3, 146.2, 144.6, 132.6, 129.5, 118.6, 109.6, 108.9, 94.2, 56.4, 49.2, 47.3, 46.1, 44.8, 36.3, 16.2, 5.7; Anal (C24H25FN6O9) C, H, N.
Antimycobacterial activity in log-phase cultures
All compounds were screened for their in vitro antimycobacterial activity against MTB, MDR-TB, and Mycobacterium smegmatis ATCC 14468 (MC2) in Middlebrook 7H11 agar medium supplemented with OADC (oleic acid–albumin–dextrose–catalase) by an agar dilution method similar to that recommended by the National Committee for Clinical Laboratory Standards for the determination of minimum inhibitory concentration (MIC) in triplicateCitation10. The MDR-TB clinical isolate was obtained from the Tuberculosis Research Center, Chennai, India, and was resistant to isoniazid, rifampicin, ethambutol, and ofloxacin. The MIC is defined as the minimum concentration of compound required to give complete inhibition of bacterial growth.
Antimycobacterial activity in 6-week-starved cultures
For starvation experimentsCitation11, MTB cells were grown in Middlebrook 7H9 medium supplemented with 0.2% (v/v) glycerol, 10% (v/v) Middlebrook OADC enrichment, and 0.025% (v/v) Tween 80 at 37°C with constant rolling at 2 rpm until they reached an optical density at 600 nm of ∼0.6. The cells were then washed twice and re-suspended in phosphate-buffered saline (PBS) at the same cell density. Cells (50 mL of culture) were incubated at 37°C for an additional 6 weeks in 1 L roller bottles. Compounds, dissolved in DMSO, were added to 1 mL PBS containing ∼1 × 107 starved MTB cells at various concentrations. Cultures were incubated in 15 mL conical tubes at 37°C with constant shaking for 7 days and then washed twice in PBS before dilutions were plated on Middlebrook 7H11 plates supplemented with 0.2% (v/v) glycerol, 10% (v/v) Middlebrook OADC enrichment, and 0.025% (v/v) Tween 80, containing no antibiotics. Bacterial growth was determined after incubation for 4 weeks at 37°C. The MIC is defined as the minimum concentration of compound required to give complete inhibition of bacterial growth. All values were determined in triplicate.
ICL enzyme assay
Isocitrate lyase activity was determined at 37°C by measuring the formation of glyoxylate phenylhydrazone at 324 nmCitation12. The reaction mixture contained 100 µL of 0.5 mM potassium phosphate buffer, 1.2 µL of 1 mM magnesium chloride, 24 µL of 100 mM 2-mercaptoethanol, 7 µL of 4 mM phenylhydrazine hydrochloride, 6 µL of 50 mM trisodium isocitric acid, and ICL enzyme (usually 3–6 µL). This mixture was made up to 200 µL with MilliQ water (water that has been purified and deionized to a high degree by a water purification system manufactured by Millipore Corporation). At the end of the 10th minute this reaction mixture was made up to 1 mL and ultraviolet (UV) absorbance was measured at 324 nM, which served as a control. For the test compounds, 3 µL of 100 mM 3-nitropropionic acid (3-NPA) was used, and in the case of the candidate molecules, 10 µL of 10 mM concentration was added to the abovementioned reaction mixture. At the end of the 10th minute this reaction mixture was made up to 1 mL and UV absorbance was measured at 324 nM, which served as the test. The percentage inhibition was calculated by the formula: (control absorbance) – (test absorbance)/(control absorbance) × 100.
Cytotoxicity
Some compounds were further examined for toxicity (IC50) in a mammalian Vero cell line at concentrations of 62.5 μg/mLCitation13. After 72 h of exposure, viability was assessed on the basis of cellular conversion of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) into a formazan product using the Promega CellTiter 96 non-radioactive cell proliferation assay.
Docking studies
The crystal structure of MTB ICL with its bound inhibitor nitropropionate taken from the Protein Data Bank (PDB entry 1F8I) was used for docking. Using the “protein preparation wizard” in Schrödinger Maestro software, the bond order was assigned for all atoms of the receptor, hydrogen atoms were added, and all the water molecules were removed. The H-bond was optimized by the exhaustive sampling method. The protein was minimized using the OPLS (optimized potentials for liquid simulations) force field to 0.3 Å root-mean-square deviation (rmsd) by keeping the protein backbone constrained. All of the amino acids within a 10 Å radius from the center of the bound ligand were considered to constitute the active site. Docking was carried out using GOLDCitation14, which uses the genetic algorithm (GA). For each of the 50 independent GA runs, a maximum number of 100,000 GA operations were performed, whereby all variables for the GA were set to their default values. Default cutoff values of 2.9 Å (dH-X) for hydrogen bonds and 6.0 Å for van der Waals were employed. When the top three solutions attained rmsd values within 1.5 Å, GA docking was terminated. All the ligand structures were built in Schrödinger Maestro and geometry minimized by applying the OPLS force field.
Results and discussion
Synthesis
The synthesis of 5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamides was achieved in two steps (). 5-Nitro-2,6-dioxohexahydro-4-pyrimidinecarboxylic acid (1) was activated by refluxing with thionyl chloride in the presence of a catalytic amount of DMF to form the acid chloride (2), which in turn was reacted with various primary and secondary amines to afford the title compounds (3a–n) with 50–80% yields. The purity of the compounds was checked by thin layer chromatography (TLC) and elemental analysis, and the compounds of this study were identified by spectral data.
Scheme 1. Synthesis protocol of the compounds.
Antimycobacterial activity
In vitro studies
The compounds initially were screened for their in vitro antimycobacterial activity against log-phase MTB, MDR-TB, and MC2 by the agar dilution method for the determination of minimum inhibitory concentration (MIC) in triplicate. The MDR-TB clinical isolate was resistant to isoniazid, rifampicin, ethambutol, and ofloxacin. The MIC is defined as the minimum concentration of the compound required to give complete inhibition of bacterial growth, and MICs of the synthesized compounds along with those of the standard drugs for comparison are presented in .
Table 1. Antimycobacterial activities of 5-nitro-pyrimidinecarboxylic acid derivatives.
In the first phase of screening against log-phase MTB, all the compounds showed excellent in vitro activity against MTB, with MIC values less than 25 μM. Four compounds (3k–n) inhibited MTB with MICs of less than 1 μM, and were more potent than the standard ciprofloxacin (MIC: 4.71 μM). When compared to isoniazid (MIC: 0.66 μM), three compounds (3l–n) were found to be more active against MTB, and two compounds (3m, n) were found to be more active than rifampicin (MIC: 0.23 μM). Compound 1-cyclopropyl-6-fluoro-8-methoxy-7-3-methyl-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid (3n) was found to be the most active compound in vitro, with an MIC of < 0.17 μM against MTB, and was 3.8 and 1.3 times more potent than isoniazid and rifampicin, respectively. Subsequently, some of the compounds were evaluated against MDR-TB, and among the six compounds screened, all compounds inhibited MDR-TB with MICs ranging from 0.17 to 4.98 μM, and were found to be more active than isoniazid (MIC: 45.57 μM) and ciprofloxacin (MIC: 37.68 μM). Four compounds (3k–n) inhibited MDR-TB with MIC values of less than 1 μM. Compound 3n was found to be most active in vitro with an MIC of 0.17 μM against MDR-TB, and was 22 and 268 times more potent than rifampicin and isoniazid, respectively. The compounds were also evaluated against MC2, in which all the compounds inhibited MC2 with MIC values ranging from 1.39 to 89.86 μM; 10 compounds were found to be more active than isoniazid (MIC: 45.57 μM), and four compounds were more active than rifampicin (MIC: 1.89 μM).
With respect to the structure–MTB activity relationship, the results demonstrated that the antimycobacterial activity was enhanced to varying degrees (up to 720-fold) by conversion of the starting material 5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxylic acid (1) to the amides (3a–n). Among the phenyl and pyridine substituted amides, pyridine substituted compounds (3a–e) showed better activity. Introduction of the fluoroquinolone moiety enhanced the activity many-fold. Introducing bulky groups at the acidic function of 1 enhanced the antimycobacterial activity, which might be due to more penetration of these compounds into mycobacterial cells.
Activity against dormant mycobacteria
The compounds that showed good activity against the log-phase culture of MTB were further screened against 6-week-starved cells of MTB according to the literature procedure. All five compounds (3e, 3k–n) inhibited the starved culture of MTB with MIC values ranging from 3.96 to 52.08 μM (). Isoniazid (INH) had poor activity against starved cells, with an MIC of 729.1 μM. As previously observedCitation11, rifampicin (RIF) retained activity, although it was considerably less active against non-growing than against log-phase cells. All five tested compounds were more potent than INH, and four compounds (3k–n) were found to be more potent than RIF (MIC: 15.2 μM). The presence of persistent and dormant MTB is thought to be the cause for the lengthy TB chemotherapy, since the current TB drugs are not effective in eliminating persistent or dormant bacilli. This study revealed that these molecules, active against slowly growing or non-growing persistent bacilli, are thought to be important to achieve a shortened therapy. Compound 7-3,5-dimethyl-4-[(5-nitro-2,6-dioxohexahydro-4-pyrimidinyl)carbonyl]piperazino-1-ethyl-6,8-difluoro-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid (3m) was found to be the most active compound, with an MIC of 3.96 μM.
Table 2. Inhibitory activities of selected compounds against log-phase and 6-week-starved MTB.
Isocitrate lyase inhibition studies
As these synthesized compounds showed activity against dormant Mycobacterium, we decided to explore the possible mechanism by screening some compounds against the ICL enzyme of MTB. ICL is an important enzyme in the glyoxylate cycle during carbohydrate starvation; in MTB it catalyzes the cleavage of isocitrate to glyoxylate and succinate, allowing the organism to survive on acetate or fatty acidsCitation15. The glyoxylate cycle is not present in higher animals, and due to its necessity for survival for the persistent phase of the infection, ICL is considered an ideal drug target for persistent MTB. Several small-molecule inhibitors have been describedCitation16 as MTB ICL inhibitors; however, none has been developed as a drug for MTB. Isocitrate lyase activity was determined at 37°C by measuring the formation of glyoxylate phenylhydrazone in the presence of phenylhydrazine and isocitrate lyase at 324 nm, based on the method describedCitation12. The compounds were screened at a single concentration of 10 µM, and percentage inhibitions of the screened compounds along with that of the standard MTB ICL inhibitor 3-NPA (at 100 µM) for comparison are reported (). All five compounds inhibited MTB ICL with percentage inhibitions ranging from 20.11 to 45.70% at 10 µM. Compound 3m was found to be the most active compound in the enzyme inhibition studies. This is the first report of the screening of newer synthetic compounds that have an inhibition to MTB ICL. Further investigation could provide lead compounds for drug development against persistent TB. No correlation between ICL enzyme inhibition studies and MIC was found.
Table 3. Inhibitory activities of selected compounds and 3-nitropropionic acid (3-NPA) against MTB ICL.
Cytotoxicity activity
Some compounds were further examined for toxicity (IC50) in a mammalian Vero cell line at 62.5 μg/mL concentrations. After 72 h of exposure, viability was assessed on the basis of cellular conversion of MTT into a formazan product, and the results are reported in . Six compounds, when tested, showed IC50 values ranging from 111.5 to < 199.8 μM. These results are important, as these compounds with their increased cytoliability are much less attractive in the development of TB drugs. This is primarily due to the fact that the eradication of TB requires a lengthy course of treatment, and the need for an agent with a high margin of safety becomes a primary concern. The IC50 value of compound 3n was found to be > 111.5 μM, and it showed a selectivity index (IC50/MIC) of 655.8.
Docking studies on 3m for MTB ICL binding
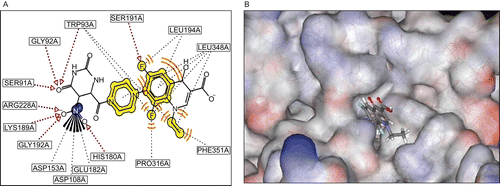
The main objective of the docking study was to assess the binding pattern and the interaction pattern of the ligand 3m with the ICL enzyme. The docking calculations run in GOLD showed very good polar and hydrophobic interaction for the ligand, indicating a high affinity to the enzyme (). Polar interactions such as H-bond and electrostatic interactions were predominant at the interior part of the active site, whereas hydrophobic/van der Waals interactions were overwhelming at the exterior part of the active site. It can be seen that binding forces between ICL and the ligand 3m manifested in the multicenter (bifurcated) H-bond between the NO of the nitro group and Arg228 (NH…ON, 2.89 Å; NH…ON, 2.64 Å), Gly192 (NH…ON, 2.9 Å), His180 (NH…ON, 3.00 Å), and the multicenter H-bond between 4-oxo-pyrimidine and Ser91 (OH…O=C, 2.9 Å), Trp93 (NH…O=C, 2.6 Å), Gly92 (NH…O=C, 2.7 Å). An H-bond between the fluorine (6th position) of the quinoline moiety and the OH group of Ser191 (2.68 Å) and a weak dipole-induced dipole interaction with Leu194 (3.56 Å) were also found. Van der Waals interactions were seen between the pyrazine moiety and Leu348 (3.23 Å), and a hydrophobic interaction between 8-fluoroquinoline and Leu348 (2.23 Å), Trp93 (3.62 Å), Pro316 (3.81 Å), the quinoline ring and Leu194 (3.12 Å), Leu348 (3.20 Å), and 1-ethylquinoline and Phe351 (3.33 Å), Leu348 (3.01 Å), Trp93 (3.14 Å). A strong electrostatic interaction between the electropositive N of the nitro group and Asp153 (2.9 Å), Asp108 (3.06 Å), Asp182 (3.44 Å) was also indicated. This result suggests that the ligand structure has good affinity for the enzyme. This also supports the wet laboratory experiment results on the inhibition of ICL by 3m.
Figure 1. (A) Schematic representation of interactions of the ligand 3m isocitrate lyase (ICL) complex in the active site. H-bonds are shown as dark colored dashed lines, hydrophobic/van der Waals interactions are shown as light dashed lines, electrostatic interactions are shown as dashed lines with “flower” at base of origin. (B) Ligand 3m docked inside the active site pocket of the MTB ICL.
Screening of the antimycobacterial activity of these 5-nitro-2,6-dioxohexahydro-4-pyrimidinecarboxamides has identified some new leads endowed with high activity toward log- and starved-phase MTB. The present study reveals the importance of these compounds effective for the treatment of TB, particularly persistent infection. In conclusion, it has been shown that the potency, selectivity, and low cytotoxicity of these compounds make them valid leads for synthesizing new compounds that possess better activity with low cytotoxicity. Further structure–activity and animal studies should prove fruitful.
Declaration of interest
The authors are thankful to the Department of Bio-technology (BT/01/COE/05/06/01), Government of India, for their financial assistance.
References
- World Health Organization. Global tuberculosis control: surveillance, planning, financing: WHO report 2008. Geneva: WHO, 2008.
- Sriram D, Yogeeswari P, Dinakaran M, Thirumurugan R. Antimyco-bacterial activity of novel 1-(5-cyclobutyl-1,3-oxazol-2-yl)-3-(sub)phenyl/pyridylthiourea endowed with high activity toward multi-drug resistant tuberculosis. J Antimicrob Chemother 2007;59:1194–6.
- Sriram D, Senthilkumar P, Dinakaran M, Yogeeswari P, China A, Nagaraja V. Antimycobacterial activities of novel 1-(cyclopropyl/tert-butyl/4-fluorophenyl)-1,4-dihydro-6-nitro-4-oxo-7-(substituted secondary amino)-1,8-naphthyridine-3-carboxylic acid. J Med Chem 2007;50:6232–9.
- Kumar RR, Perumal S, Senthilkumar P, Yogeeswari P, Sriram D. Discovery of antimycobacterial spiro-piperidin-4-ones: an atom economic, stereoselective synthesis, and biological intervention. J Med Chem 2008; 51:5731–5.
- Dinakaran M, Senthilkumar P, Yogeeswari P, China A, Nagaraja V, Sriram D. Antimycobacterial and phototoxic evaluation of novel 6-fluoro/nitro-4-oxo-7-(sub)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid. Int J Antimicrob Agents 2008;31:337–44.
- Dinakaran M, Senthilkumar P, Yogeeswari P, China A, Nagaraja V, Sriram D. Novel ofloxacin derivatives: synthesis, antimycobacterial and toxicological evaluation. Bioorg Med Chem 2008;16:3408–18.
- Sriram D, Yogeeswari P, Thirumurugan R, Pavana RK. Discovery of newer antitubercular oxazolyl thiosemicarbazones. J Med Chem 2006; 49:3448–50.
- Dinakaran M, Senthilkumar P, Yogeeswari P, China A, Nagaraja V, Sriram D. Synthesis, antimycobacterial activities and phototoxic evaluation of 5H-thiazolo[3,2-a]quinoline-4-carboxylic acid derivatives. Med Chem 2008;4:482–91.
- Kumar RR, Perumal S, Senthilkumar P, Yogeeswari P, Sriram D. An atom efficient, solvent-free, green synthesis and antimycobacterial evaluation of 2-amino-6-methyl-4-aryl-8-[(E)-arylmethylidene]-5,6,7,8-tetrahydro-4H-pyrano[3,2-c]pyridine-3-carbonitriles. Bioorg Med Chem Lett 2007;17:6459–62.
- National Committee for Clinical Laboratory Standards. Antimyco-bacterial susceptibility testing for Mycobacterium tuberculosis. Proposed standard M24-T. Villanova, PA: National Committee for Clinical Laboratory Standards. 1995.
- Xie Z, Siddiqi N, Rubin ER. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Anitimicrob Agents Chemother 2005;49:4778–80.
- Bai B, Xie JP, Yan JF, Wang H, Hu C. A high throughput screening approach to identify isocitrate lyase inhibitors from traditional Chinese medicine sources. Drug Dev Res 2006;67:818–23.
- Gundersen LL, Meyer NJ, Spilsberg B. Synthesis and antimycobacterial activity of 6-arylpurines: the requirements for the N-9 substituent in active antimycobacterial purines. J Med Chem 2002;45:1383–6.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267:727–48.
- Mc Kinney JD. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000;406:735–8.
- Bentrup KHZ, Miczak A, Swenson DL, Russell DG. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J Bacteriol 1999;181:7161–7.