Abstract
The CHCl3-soluble fraction of the whole plant of Duranta repens showed anti-plasmodial activity against the chloroquine-sensitive (D6) and chloroquine-resistant (W2) strains of Plasmodium falciparum, with IC50 values of 8.5 ± 0.9 and 10.2 ± 1.5 μg/mL, respectively. From this fraction, two new flavonoid glycosides, 7-O-α-d-glucopyranosyl-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (1) and 7-O-α-d-glucopyranosyl(6′′′-p-hydroxcinnamoyl)-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (2), along with five known flavonoids, 3,7,4′-trihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (3), 3,7-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6,4′-trimethoxyflavone (4), 5,7-dihydroxy-3′-(2-hydroxy-3-methyl-3-butenyl)-3,6,4′-trimethoxyflavone (5), 3,7-dihydroxy-3′-(2-hydroxy-3-methyl-3-buten-yl)-5,6,4′-trimethoxyflavone (6), and 7-O-α-d-glucopyranosyl-3,5-dihydroxy-3′-(4′′-acetoxy-3′′-methylbutyl)-6,4′-dimethoxyflavone (7), have been isolated as anti-plasmodial principles. Their structures were deduced by spectroscopic analysis including 1D and 2D NMR techniques. The compounds (1–7) showed potent anti-plasmodial activities against D6 and W2 strains of Plasmodium falciparum, with IC50 values in the range of 5.2–13.5 μM and 5.9–13.1 μM, respectively.
Introduction
The genus Duranta (Verbenaceae) comprises about 35 species which mainly occur in the West Indies and Tropical and South America. It is represented in Pakistan by two species, namely Duranta repens and Duranta stenostachyaCitation1.
Duranta repens Linn. (syn. D. erecta, D. microphylla, D. plumieri, common name Sky Flower, Golden Dew Drop, Pigeon Berry) is a large, subtropical, shrub to small tree up to 18 feet in height. It is commonly grown as a hedge plant, and when trimmed, forms a strong compact hedge almost impenetrable to cattleCitation2. Duranta repens is widely distributed in the northern area of Pakistan. Medicinally, the fruit of this plant is used for the treatment of malariaCitation3. The MeOH extract of the plant also shows insecticidal and antifeedant properties against Aedes aegypti and Attagenus piceus, respectivelyCitation4.
Previous studies on the genus Duranta have resulted in the isolation of various compounds, including coumarinolignoidsCitation5, (E)-cinnamic acid, (E)-p-methoxycinnamic acidCitation6, diterpenoidsCitation7,Citation8, flavonoidsCitation7,Citation8, steroidsCitation9, glycosides of phenylpropanoidsCitation10,Citation11, triterpenesCitation12, and iridoidsCitation6,Citation13,Citation14.
Malaria is one of the most common infectious diseases in tropical and subtropical countries, including parts of the Americas, Asia, and Africa. Each year, it affects about 400–900 million people, and causes approximately one to three million deathsCitation15. Human malaria is caused by Plasmodium falciparum, P. malariae, P. ovale, and P. vivax; however, P. falciparum is the most prevalent for the disease, and is responsible for about 80% of infections and 90% of deathsCitation16. The first effective treatment (17th century) against the P. falciparum parasite was the bark of the cinchona tree, which contains quinine, a quinoline alkaloid. A number of medicines have been developed to treat malaria, with chloroquine and its derivativesCitation17 as the mainstay therapy. In recent years, P. falciparum has become increasingly resistant to conventional antimalarial drugs, and the search for new antimalarial compounds by combining natural sources and synthetic approaches is still under wayCitation18,Citation19. The spread of resistance of Plasmodium falciparum to commonly used antimalarial drugs has created an urgent need to develop new antimalarial treatments, preferably drugs that are affordable to developing countries where malaria is prevalent. Although powerful new technologies such as high-throughput screening and combinatorial chemistry are revolutionizing drug discovery, natural products still offer structural diversity, which makes them a valuable source of novel lead compounds.
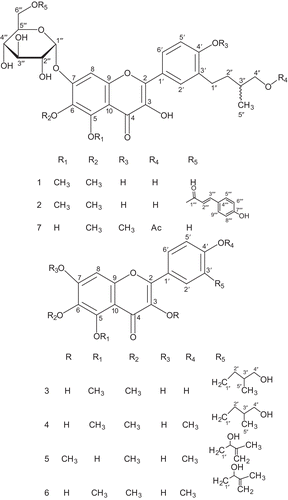
Herein we report the isolation and characterization of two new flavonoid glycosides, 7-O-α-d-glucopyranosyl-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (1) and 7-O-α-d-glucopyranosyl(6′′′-p-hydroxcinnamoyl)-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (2), along with five known flavonoids, 3,7,4′-trihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (3)7, 3,7-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6,4′-trimethoxyflavone (4)7, 5,7-dihydroxy-3′-(2-hydroxy-3-methyl-3-butenyl)-3,6,4′-trimethoxyflavone (5)8, 3,7-dihydroxy-3′-(2-hydroxy-3-methyl-3-buten-yl)-5,6,4′-trimethoxyflavone (6)8, and 7-O-α-d-glucopyranosyl-3,5-dihydroxy-3′-(4′′-acetoxy-3′′-methylbutyl)-6,4′-dimethoxyflavone (7)20 (). The compounds (1–7) showed potent anti-plasmodial activities against D6 and W2 strains of Plasmodium falciparum, with IC50 values in the range 5.2–13.5 μM and 5.9–13.1 μM, respectively.
Figure 1. Structures of flavonoids 1–7.
Materials and methods
General experimental procedure
Optical rotations were measured on a Jasco DIP-360 digital polarimeter. Infrared (IR) spectra were recorded on a 460 Shimadzu spectrometer. Ultraviolet (UV) spectra were obtained on a Hitachi UV-3200 spectrophotometer. The 1H and 13C nuclear magnetic resonance (NMR), heteronuclear multiple quantum coherence (HMQC), and heteronuclear multiple bond coherence (HMBC) spectra were recorded on Bruker NMR spectrometers operating at 400 MHz for 1H and 100 MHz for 13C NMR, respectively. The chemical shift values are reported in ppm (δ) units relative to SiMe4 as an internal standard and the coupling constants (J) are in Hz. Electron ionization (EI), fast atom bombardment (FAB), and high resolution-electron ionization mass spectra (HR-EIMS) were recorded on Jeol JMS-HX-110 and JMS-DA-500 mass spectrometers, m/z (rel. int). Aluminum sheets precoated with silica gel 60 F254 (20 × 20 cm, 0.2 mm thick; E. Merck) were used for thin layer chromatography (TLC), and silica gel (230–400 mesh; E. Merck) was used for column chromatography. For recycling high-performance liquid chromatography (HPLC) (LC 908 W), a semi-preparative column (ODS-M80) was used.
Plant material
The whole plant of Duranta repens Linn. (Verbenaceae) was collected from Swat (Pakistan) in March 2007 and identified by Nisar Ahmad, Department of Plant Sciences, Kohat University of Science and Technology, Kohat. A voucher specimen (PSK-61) was deposited in the herbarium of Kohat University.
Extraction and isolation
The shade-dried whole plant (8 kg) was chopped and soaked in MeOH for 10 days, extracted three times at room temperature in the same solvent, and filtered. The filtrate was evaporated in vacuo to give a dark-greenish residue (300 g), which was suspended in water and partitioned successively with n-hexane, CHCl3, and n-BuOH to obtain n-hexane-soluble (60 g), CHCl3-soluble (40 g), and n-BuOH-soluble (120 g) fractions, respectively. The CHCl3-soluble fraction (40 g) was subjected to column chromatography (CC) using hexane–CHCl3 in increasing order of polarity to give five fractions. The fraction which eluted with n-hexane–CHCl3 (7:3) was rechromatographed over flash silica gel using n-hexane–CHCl3 (9:1–1:1) as a solvent system to give three successive fractions. The third fraction was further subjected to CC (silica gel) using n-hexane–CHCl3 (7:3 and 6:4) as eluent to afford compounds 3 (18 mg), 4 (11 mg), 5 (14 mg), and 6 (15 mg) by repeated column chromatography. The fraction obtained from n-hexane–CHCl3 (3:7) was subjected to CC using MeOH:CHCl3 (1:9, 2:8) followed by CC over Sephadex LH-20 with pure water to get semi-pure compounds 1, 2, and 7, which were finally purified on recycling HPLC (LC 908 W) (ODS-M80 semi-preparative column, MeOH:H2O (1:1), flow rate 3 mL/min); UV and refractive index (RI) detectors; retention time (tR) 13 min (1, 21 mg), 20 min (2, 19 mg), and 26 min (7, 15 mg).
7-O-α-d-Glucopyranosyl-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (1) Yellowish gummy solid. [α]D25 = +91.3° (c = 0.01, MeOH). UV λmax (MeOH) nm (log ϵ): 273 (4.76), 340 (4.53). IR υmax (KBr) cm−1: 3396, 2925, 1656, 1597, 1190. HR-FAB-MS (pos.): 579.2072 [M + H]+. EI-MS: 578 (2), 416 (100), 401 (37), 398 (12.3), 373 (7), 343 (9), 342 (5.1), 329 (7.5), 207 (2.3), 152 (3.1). For 1H and 13C NMR data see .
Table 1. 1H (400 MHz) and 13C NMR (100 MHz) assignments (δ ppm) of flavonoids (1 and 2) in CD3OD.
7-O-α-d-Glucopyranosyl(6′′′-p-hydroxcinnamoyl)-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (2) Yellowish gummy solid. [α]D25 = +82.9° (c = 0.01, MeOH). UV λmax (MeOH) nm (log ϵ): 271 (4.70), 342 (4.55). IR υmax (KBr) cm−1: 3387, 2920, 1732, 1655, 1590, 1250, 1193. HR-FAB-MS (pos.): 725.2439 [M + H]+. EI-MS: 724 (1), 578 (1), 577 (6), 561 (9), 416 (100), 401 (35.2), 398 (10.9), 373 (5), 343 (10), 342 (6.4), 329 (5.6), 207 (2.6), 152 (3). For 1H and 13C NMR data see .
3,7,4′-Trihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone (3) Gummy solid. [α]D25 = +110° (c = 0.01, MeOH). UV λmax (MeOH) nm (log ϵ): 273 (8.06), 343 (8.02). IR υmax (KBr) cm−1: 3385, 2900, 1655, 1595, 1193. For EI-MS, 1H and 13C NMR data see reference 7.
3,7-Dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6,4′-trimethoxyflavone (4) Gummy solid. [α]D25 = +73.6° (c = 0.02, MeOH). UV λmax (MeOH) nm (log ϵ): 271 (8.25), 340 (8.33). IR υmax (KBr) cm−1: 3380, 2905, 1650, 1590, 1190. For EI-MS, 1H and 13C NMR data see reference 7.
5,7-Dihydroxy-3′-(2-hydroxy-3-methyl-3-butenyl)-3,6,4′-trimethoxyflavone (5) Yellow gummy solid. [α]D25 = +18.5° (c = 0.01, MeOH). UV λmax (MeOH) nm (log ϵ): 272 (4.41), 338 (4.48). IR υmax (CHCl3) cm−1: 3430, 2927, 1680, 1655, 1600, 1193. For EI-MS, 1H and 13C NMR data see reference 8.
3,7-Dihydroxy-3′-(2-hydroxy-3-methyl-3-buten-yl)-5,6,4′-trimethoxyflavone (6) Yellow gummy solid. [α]D25 = +33.3° (c = 0.03, MeOH). UV λmax (MeOH) nm (log ϵ): 272 (4.62), 344 (4.70). IR υmax (CHCl3) cm−1: 3425, 2900, 1684, 1650, 1590, 1190. For EI-MS, 1H and 13C NMR data see reference 8.
7-O-α-d-Glucopyranosyl-3,5-dihydroxy-3′-(4′′-acetoxy-3′′-methylbutyl)-6,4′-dimethoxyflavone (7) Yellowish gummy solid. [α]D25 = +27° (c = 0.01, MeOH). UV λmax (MeOH) nm (log ϵ): 273 (4.63), 343 (4.69). IR υmax (KBr) cm−1: 3396, 2925, 1656, 1597, 1190. For EI-MS, 1H and 13C NMR data see reference 20.
In vitro anti-plasmodial activity
The CHCl3-soluble fraction and pure compounds were tested for anti-plasmodial activities based on the inhibition of [3H]hypoxanthine uptake as described in the previously established protocolCitation21. Briefly, the CHCl3-soluble fraction and pure compounds were assayed using an automated microdilution technique to determine 50% growth inhibition of cultured parasitesCitation22,Citation23. Two different strains, chloroquine-sensitive Sierra Leone I (D6) and chloroquine-resistant Indochina I (W2), of Plasmodium falciparum were grown as described in the literatureCitation22,Citation23. The samples were serially diluted across the plate to provide a range of concentrations, used to determined IC50 values. Plates were incubated in a mixed gas incubator for 24 h. [3H]Hypoxanthine was then added and parasites allowed to grow for an additional 18 h. Cells were processed with a plate harvester (TomTec) onto filter paper and washed to eliminate unincorporated isotope. Filters were measured for activity in a microtiter plate scintillation counter (Wallac). Data from the counter were imported into a Microsoft Excel spreadsheet, which was then imported into an Oracle Database program to determine IC50 values. IC50 values were calculated from triplicate experiments.
Acid hydrolysis of compound 1
A solution of 1 (8 mg) in MeOH (5 mL) containing 1 N HCl (4 mL) was refluxed for 4 h, concentrated under reduced pressure, and diluted with H2O (8 mL). It was extracted with EtOAc and the residue recovered from the organic phase was found to be an inseparable mixture of products. The aqueous phase was concentrated, and d-glucose was identified by co-TLC using the solvent system EtOAc:MeOH:H2O:HOAc (11:2:2:2) and the sign of its optical rotation ([α]D20 +52.6°, c 0.02, H2O). It was also confirmed based on the retention time of its tetramethylsilane (TMS) ether (α-anomer 4.1 min, β-anomer 7.8 min) compared with a standard in gas chromatography (GC).
Results and discussion
Compound 1 was isolated as a yellowish gummy solid and its molecular formula was established as C28H34O13 by a [M + H]+ peak at m/z 579.2072 (calcd. for C28H35O13: 579.2077) in HR-FAB-MS. It gave a red color in the Shinoda test, indicating it to be a flavonoidCitation24. Negative results in the Quastel test indicated the absence of an ortho dihydroxyl moietyCitation25. The UV absorption maxima in MeOH at 273 and 340 nm suggested that it was a flavonoidCitation26. The IR spectrum showed the aromatic ring at 1597 cm−1, α,β-unsaturated carbonyl group at 1656 cm−1, methoxyl groups at 2925 and 1190 cm−1, and hydroxyl group at 3396 cm−1.
The 1H NMR spectrum of 1 exhibited an ABX system in ring B of the flavonoid, resulting in signals at δ 6.89 (1H, d, J = 8.6 Hz, H-5′), 7.80 (1H, dd, J = 8.6, 1.8 Hz, H-6′), and 7.83 (1H, d, J = 1.8 Hz, H-2′). A singlet at δ 6.51 could be assigned to the aromatic proton of ring ACitation22. The signals of two methoxyl groups appeared at δ 3.86 (3H, s, MeO-5) and 3.78 (3H, s, MeO-6), while the benzylic methylene protons of ring B resonated at δ 2.63 (2H, t, J = 7.1 Hz, H-1′′). The signals of the side chain were observed at δ 0.99 (3H, d, J = 6.8 Hz, H-5′′), 1.40 (2H, m, H-2′′), 1.65 (1H, m, H-3′′), and 3.50 (2H, d, J = 6.5 Hz, H-4′′), which identified the side chain as 3′′-methylbutyl. The above spectral data showed striking resemblance to those of 37, except differing in the presence of the additional sugar moiety, which was revealed by a doublet of the anomeric proton at δ 5.31 (1H, d, J = 3.6 Hz, H-1′′′). The signals of four protons geminal to the hydroxyl groups of the sugar appeared at δ 3.56 (1H, t, J = 9.6 Hz, H-4′′′), 3.68 (1H, m, H-5′′′), 3.85 (1H, t, J = 9.6 Hz, H-3′′′), and 3.98 (1H, dd, J = 9.6, 3.6 Hz, H-2′′′) and methylene protons at δ 3.66 (1H, dd, J = 11.1, 4.6 Hz, H-6′′′a) and 3.77 (1H, dd, J = 11.1, 5.2 Hz, H-6′′′b).
The 13C NMR (broadband (BB) and distortionless enhancement by polarization transfer (DEPT)) spectra of 1 showed a total of 28 carbons, with signals of the anomeric carbon at δ 99.9 and carbonyl carbon at δ 180.2. The assignments of all carbons were achieved by HMQC experiment.
Both the 1H and 13C NMR spectra indicated the presence of a carbohydrate moiety by the appearance of characteristic resonances at δH 5.31 and δC 99.9 (due to anomeric proton and carbon, respectively). This was further supported by resonances due to other oxygen-bearing carbons at δ 71.1, δ 73.0, δ 73.7, δ 74.2, and δ 62.9 in the 13C NMR spectrum. The identity of sugar as α-D-glucose was confirmed through comparison of the chemical shifts of its carbons with literature valuesCitation27, and was further confirmed through acid hydrolysis, which provided glycone identified as D-glucose through the sign of its optical rotation ([α]27 + 52.5°) and comparison of the retention time of its TMS ether with that of the standard in gas–liquid chromatography (GLC). The α-configuration of the glucose moiety was assigned on the basis of a small coupling constant of the anomeric proton H-1′′′ (J = 3.6 Hz).
The EI-MS spectrum showed a strong fragment peak at m/z 416 due to loss of the sugar moiety from the molecule. The other characteristic fragments originated from this peak at m/z (343 [416 – C4H9O]+, 342 [416 – C4H8O]+, 329 [416 – C4H8O–CH3]+, 207 [416 – C10H9O5]+, and 152 [416 – C14H16O5]+) were similar to those of 37. These fragments confirmed the presence of two methoxyl groups and one hydroxyl group on ring A of 1, one hydroxyl and the side chain on ring B, and the remaining hydroxyl at the C-3 positionCitation28. The occurrence of benzylic cleavage explained the loss of 73 amu, while the loss of 72 amu was due to β-cleavage of the side chain with hydrogen transfer to the aromatic nucleus via a 1,6 rearrangement. Since this requires at least one free ortho positionCitation29, the side chain therefore had to be located at either C-3′ or C-4′.
In the HMBC experiment of 1 (), the anomeric proton at δ 5.31 showed 3J correlations to C-7 (δ 156.0), C-3′′′ (δ 73.7), and C-5′′′ (δ 74.2). This confirmed attachment of the sugar moiety at the C-7 position. The 2J and 3J connectivities of H-1′′ at δ 2.63 with C-3′ (δ 131.2), C-2′′ (δ 34.4), C-2′ (δ 130.7), and C-4′ (δ 158.3) confirmed the position of the side chain at C-3′. The methyl protons of the side chain at δ 0.99 showed 2J and 3J correlations with C-3′′ (δ 35.2), C-4′′ (δ 68.6), and C-2′′ (δ 34.4). The protons at δ 3.50 (H-4′′) correlated with C-5′′ (δ 17.3), C-3′′ (δ 35.2), and C-2′′ (δ 34.4). The methoxyl group at δ 3.78 exhibited 3J correlation with C-6 (δ 131.2), and the other methoxyl group at δ 3.86 showed 3J interaction with C-5 (δ 159.8), thereby confirming the position of the two methoxyl groups at C-6 and C-5, respectively.
Figure 2. Important HMBC correlations of 1.
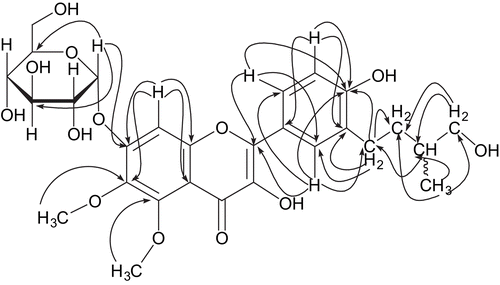
The carbons at positions C-5, C-7, and C-9 were assigned through HMQC and HMBC experiments. The H-8 proton at δ 6.51 showed 2J correlations with C-7 (δ 156.0) and C-9 (δ 153.7) and 3J correlations with C-6 (δ 131.2) and C-10 (δ 106.3). The other aromatic protons of ring B also showed HMBC interactions: the proton at δ 6.89 (H-5′) coupled with C-4′ (δ 158.3), C-3′ (δ 131.2), and C-1′ (δ 122.3), the proton at δ 7.83 (H-2′) coupled with C-4′ (δ 158.3), C-2 (δ 154.7), C-1′′ (δ 28.1), and C-6′ (δ 128.5), and the proton at δ 7.80 (H-6′) showing correlations with C-4′ (δ 158.3), C-2 (δ 154.7), and C-2′ (δ 130.7). All the ongoing data confirmed the structure of compound 1 as 7-O-α-d-glucopyranosyl-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone.
Compound 2 was obtained as a yellowish gummy solid, molecular formula C37H40O15 by [M + H]+ peak at m/z 725.2439 in HR-FAB-MS spectrometry (calcd. for C37H41O15: 725.2445). Its IR spectrum showed similar absorptions to 1, plus additional resonances due to an ester group (1732 and 1250 cm−1).
The 1H and 13C NMR spectra of 2 () were almost identical to those of 1, except differing in having additional characteristic signals for the p-hydroxycinnamoyl moiety (aromatic protons showing AA′, BB′ pattern with δ 7.40 (2H, d, J = 8.0 Hz) and δ 6.89 (2H, d, J = 8.0 Hz); trans olefinic protons at δ 7.51 (1H, d, J = 15.9 Hz) and δ 6.41 (1H, d, J = 15.9 Hz)). The 13C NMR spectrum of 2 showed additional signals due to the ester carbonyl group (δ 165.7), olefinic C-atoms (δ 144.7, 117.0), oxygenated aromatic C-atom (δ 159.3), and aromatic C-atoms (δ 125.9, 130.2 × 2, 116 × 2).
The EI mass spectrum of 2 showed prominent peaks at m/z 577 and 561, arising from the loss of p-hydroxycinnamoyl and p-hydroxycinnamate moieties, respectively. The remaining fragmentation pattern was similar to that of 1.
Comparison of the NMR spectroscopic data of 1 and 2 revealed a downfield shift for the sugar-OCH2 1H and 13C NMR signals of 2, indicating that the p-hydroxycinnamoyl moiety was attached at the 6′′′-O-atom of the glucosyl moiety. This was further confirmed by 3J HMBC correlations of CH2 (6′′′) to the ester carbonyl moiety at δ (C) 165.7. Thus, the structure of 2 was identified as 7-O-α-d-glucopyranosyl (6′′′-p-hydroxcinnamoyl)-3,4′-dihydroxy-3′-(4-hydroxy-3-methylbutyl)-5,6-dimethoxyflavone.
The structures of the known compounds (3–7) were established by comparison of their spectral data with literature valuesCitation7,Citation8,Citation20.
The two new flavonoid glycosides (1, 2) and the known flavonoids (3–7) described in this study were tested for anti-plasmodial activities against the chloroquine-sensitive (D6) and chloroquine-resistant (W2) strains of Plasmodium falciparum. Activities were observed for all of the tested compounds (1–7) (), with IC50 values in the range 5.2–13.5 μM and 5.9–13.1 μM for D6 and W2 strains of Plasmodium falciparum, respectively. These flavonoids appeared to be responsible for the observed anti-plasmodial activities of the crude CHCl3-soluble fraction of the whole plant of Duranta repens.
Table 2. In vitro IC50 values of flavonoids (1–7) against W2 and D6 strains of P. falciparum.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Nasir E, Ali SI. Flora of West Pakistan. Karachi: Department of Botany, University of Karachi, 1974;77:18–19.
- Sastri BN. The Wealth of India New Delhi, Publication and Information Directorate CSIR, 1952;III:117–19.
- Perry LM, Metzger J. Medicinal Plants of East and Southeast Asia. Cambridge, MA: MIT Press, 1980:432.
- Grainge M, Ahmad S. Handbook of Plants with Pest-Control Properties. New York, John Wiley & Sons, 1988:118.
- Iqbal K, Anis I, Muhktar N, Malik A. Phosphodiesterase inhibitory coumarinolignoids from Duranta repens. Heterocycles 2003;60:151–7.
- Kuo YH, Chen ZS, Lin YL. Chemical components of the leaves of Duranta repens Linn. Chem Pharm Bull 1996;44:429–36.
- Anis I, Anis E, Ahmed S, Mustafa G, Malik A, Amtul Z, et al. Thrombin inhibitory constituents from Duranta repens. Helv Chim Acta 2001;84:649–55.
- Anis I, Ahmed S, Malik A, Yasin A, Choudhary MI. Enzyme inhibitory constituents from Duranta repens. Chem Pharm Bull 2002;50:515–18.
- Ahmed S, Nizami TA, Nawaz HR, Malik A. A new steroid from Duranta repens. Fitoterapia 1998;LXIX:448–50.
- Lin YL, Kuo YH. A new glycoside, brachynoside, isolated from Clerodendron brachyanthum Schauer. Chem Pharm Bull 1992;40:1928–9.
- Yoshio T, Youko M, Takashi M, Choei O, Eiji H, Anki T, et al. Iridoid glucosides from the leaves and stems of Duranta erecta. Phytochemistry 1995;39:829–33.
- Hiradate S, Yada H, Ishii T, Nakajima N, Ohnishi-Kameyama M, Sugie H, et al. Three plant growth inhibiting saponins from Duranta repens. Phytochemistry 1999;52:1223–8.
- Rimpler H, Timm H. Iridoids and ecdysones from verbenaceae. V. Iridoids from Duranta repens L. Z Naturforsch 1974;29c:111–15.
- Rao CB, Rao TN, Kumar VEK, Vijay EKS. Chemical examination of the fruits of Duranta plumeri Jacq. Indian J Chem 1978;16B: 844–5.
- Breman JG. The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am J Trop Med Hyg 2001;64:1–11.
- Mendis K, Sina B, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 2001;64:97–106.
- Wataya Y, Kim HS. Development of antimalarial drugs: past, present and future. Jikken Igaku 2005;23:2741–7.
- Gessler MC, Nkunya MHH, Mwasumbi LB, Heinrick M, Tanner M. Screening Tanzanian medicinal plants for antimalarial activity. Acta Trop 1994;56:65–77.
- Tran QL, Tezuka Y, Ueda JY, Nguyen NT, MaruyamaY Begum, K, et al. In vitro antiplasmodial activity of antimalarial medicinal plants used in Vietnamese traditional medicine. Ethnopharmacology 2003;86:249–52.
- Iqbal K, Malik A, Muhktar N, Anis I, Khan SN, Choudhary MI. α-Glucosidase inhibitory constituents from Duranta repens. Chem Pharm Bull 2004;52:785–9.
- Yenesew A, Derese S, Irungu B, Midiwo JO, Waters NC, Liyala P, et al. Flavonoids and isoflavonoids with anti-plasmodial activities from the roots of Erythrina abyssinica. Planta Med 2003;69:658–61.
- Chulay JD, Hayne DJ, Diggs CL. Plasmodium falciparum: assessment of in vitro growth by (3H)hypoxanthine incorporation. Exp Parasitol 1983;55:138–46.
- Desjardins RE, Canfield CJ, Hayne DJ, Chulay JD. Quantative assessment of antimalarial activity in vitro by semiautomatic microdilution technique. Antimicrob Agents Chemother 1979;16:710–18.
- Sachdev K, Kulshrestha DK. Aliarin, a new flavonoid from Dodonaea viscose Linn. Indian J Chem B 1982;21:798–99.
- Quastel JH. Color test for o-dihydroxyphenols. Analyst (Lond) 1951;56:311.
- Voirin B. UV spectral differentiation of 5-hydroxy- and 5-hydroxy-3-methoxyflavones with mono (4′), di (3′,4′) or tri (3′,4′,5′)-substituted B rings. Phytochemistry 1983;22:2107–45.
- Malik A, Riaz M, Akbar E, Rafiq M, Afza N. Alkylated coumarin and coumarin glucoside from Daphne oleoides. Heterocycles 2003;60:947–51.
- Mabry TJ, Markham KR. In:Harborn JB, Mabry TJ, Mabry H, eds. The Flavonoids. London, Chapman and Hall, 1975:866.
- Budzikiewicz H, Djerassi C, Williams DH. Mass Spectrometry of Organic Compounds. San Francisco, CA: Holden-Day, 1967:82.