Abstract
This study deals with the synthesis, pharmacological activity, and kinetic studies of mefenamic acid (MA) prodrugs of tyrosine and glycine. The synthesis involved a series of protection and deprotection reactions. The hydrolysis of these prodrugs in the intestine was confirmed by hydrolysis kinetics studies in simulated gastric fluid, simulated intestinal fluid, and 80% plasma. The prodrugs were also evaluated for analgesic, anti-inflammatory, and ulcerogenic activities. The glycine prodrug showed maximum analgesic activity of 86%, and both tyrosine and glycine prodrugs showed better anti-inflammatory activity of 74% and 81%, respectively, when compared to the 40% of MA. Further, the prodrugs showed fewer gastric ulcers compared to MA; tyrosine and glycine prodrugs had an average ulcer index of 9.1 and 4.5, respectively, while an average ulcer index of 24.2 was observed with MA. These findings suggest that both prodrugs are better in action as compared to MA, and are advantageous in having fewer gastrointestinal side effects.
Introduction
Mefenamic acid (MA), one of the widely used non-steroidal anti-inflammatory drugs (NSAIDs), has anti-inflammatory and analgesic activity. The main side effects of MA include gastrointestinal tract (GIT) disturbance, peptic ulceration, and gastric bleeding. A possible approach to solving these problems may be derivitization of the carboxylic function of the NSAIDs to prodrug forms, with adequate stability at the acidic pH of the stomach. Thus, such derivitization may, on the one hand, prevent local irritation of the stomach mucosa and, on the other hand, be capable of releasing the parent drug spontaneously or enzymatically in the blood following its absorption.
The literature reveals that many efforts have been made to synthesize prodrugs via masking the carboxylic acid group by forming ethyl ester, methyl ester, glycolamide ester, and amide prodrugs using various amino acidsCitation1–3. However, no attempts have been made to develop amide prodrugs of NSAIDs using amino acids, although this has been utilized as a major tool with other NSAIDs. The advantages of using amino acids for this purpose are associated with their characteristics such as normal dietary constituent, non-toxic in moderate doses, healing effect on gastric toxicity, marked anti-inflammatory activity, and site specificity. With this background, the present research aimed to synthesize the amide prodrugs of MA with methyl esters of tyrosine and glycine, and study their various physicochemical characters, hydrolysis kinetics, anti-inflammatory activity, analgesic activity, and ulcer index as prodrugs.
Materials and methods
Materials
The amino acids l-tyrosine and l-glycine were obtained from M/s Hi-Media Ltd., Mumbai, India, and the drug mefenamic acid was obtained as a gift sample from Alkem Laboratories, Mumbai, India. The other reagents and solvents used were of analytical grade. The reactions were monitored by thin layer chromatography (TLC) on pre-coated silica G plates using iodine vapor as detecting agent. The melting points were recorded using melting point determination apparatus by Sigma Instruments, Chennai, India, and are uncorrected. The elemental analysis was performed using a Carlo-Erba model 1108 analyzer (Italy), and the found values are very near to the theoretical values. 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were recorded in dimethylsulfoxide (DMSO) on a Bruker DRX 400 Fourier transform spectrometer with tetramethylsilane (TMS) as internal standard. Chemical shifts are expressed as δ (ppm) values. Mass spectra were recorded on a Thermo Finnigan trace DSQ GC-mass spectrometer. The hydrolysis data and drug content determination were performed by Elico UV spectrophotometer.
Synthesis of prodrugs
Mefenamic acid (MA) is 2-[(2,3-dimethylphenyl)amino]benzoic acid, and synthesis of the prodrugs was carried out by the Schotten–Baumann methodCitation4,Citation5.
Step 1: synthesis of methyl ester hydrochlorides of l-tyrosine and l-glycine
Freshly distilled thionyl chloride (0.05 M) was slowly added to methanol (100 mL) with cooling, and amino acid (0.1 M) was added to it. The mixture was refluxed for 6–8 h at 60–70°C with continuous stirring on a magnetic stirrer. Excess thionyl chloride and solvent were removed under reduced pressure giving crude amino acid methyl ester hydrochloride. It was titrated with a 20 mL portion of cold ether at 0°C until the excess of dimethyl sulfite was removed. The resulting solid product was collected and dried under vacuum. It was re-crystallized from hot methanol by the slow addition of 15–20 mL ether followed by cooling at 0°C. The crystals were collected the next day and washed twice with an ether:methanol mixture at a ratio 5:1 followed by pure ether, and dried under vacuum to give pure amino acid methyl ester hydrochloride.
Step 2: synthesis of mefenamic acid chloride
Mefenamic acid (0.05 M) was dissolved in the minimum amount of chloroform, and freshly distilled thionyl chloride (0.05 M) was added slowly to it. The mixture was refluxed for 15 h at 60–70°C with continuous stirring on a magnetic stirrer. The viscous liquid was immediately poured onto a Petri dish and was vacuum dried to give yellow-colored crude mefenamic acid chloride.
Step 3: synthesis of prodrugs of mefenamic acid with methyl esters of l-tyrosine (MAT) and l-glycine (MAG)
Ice cold, aqueous sodium hydroxide solution (5%) was taken in a 250 mL beaker and the methyl ester of l-tyrosine or l-glycine hydrochloride (0.05 M) was added to it. The reaction mixture was mechanically stirred for 30 min at room temperature, after which the beaker was transferred to an ice bath kept on a mechanical stirrer, maintaining the temperature at 10°C. Mefenamic acid chloride (0.01 M) was added in small portions with continuous stirring for 7–8 h. The solid that separated out was filtered off. The crude prodrug was re-crystallized from methanol.
Synthesis of the title prodrugs is illustrated in , and the structures were established by elemental analysis, 1H NMR, 13C NMR, and mass and Fourier transform infrared (FTIR) spectral methods.
Figure 1. Structures of newly synthesized compounds MAT and MAG.
Spectral analysis
The spectral data obtained for MAT (N-[α-amino-β-(p-hydroxyl phenyl propionate)]-2 [(2,3-dimethyl phenyl amino benzamide)]) and MAG (N-[methyl acetate]-2 [(2,3-dimethyl phenyl amino benzamide)]) are given as follows.
MAT: N-[α-amino β-(p-hydroxyl phenyl propionate)]-2 [(2,3-dimethyl phenyl amino benzamide)] UV spectra (λmax) in SGF 242 nm, in SIF 248 nm, and in phosphate buffer 282 nm; IR (KBr, cm−1) 3312 (NH str. of amide), 3203 (aromatic CH str.), 2921, 2861 (aliphatic CH str.), 1723 (C-O str. of ester), 1654 (amide I), 1456, 1378 (CH bend, aliphatic), 1245 (C-O str. of ester); 1H NMR (δ, ppm) (DMSO) 7.00–7.87 (m, 5H, aromatic ring), 6.59 (d, 1H, CH in ring), 5.69 (d, 1H, CH in ring), 3.79 (t, 2H, CH2 in ring), 2.99–3.24 (m, 2H, CH2 in ring), 9.83 (br, NH), 4.17 (q, 2H, OCH3) 1.28 (t, 3H, OCH3), 4.59–4.65 (m, 1H, CH3), 1.38 (d, 3H, CH3 saturated); 13C NMR (δ, ppm) (DMSO) 135.3, 139.6, 128.9, 126.2, 128.9, 17.3, 17.8, 139.6, 129, 128.5, 126.2, 119.2, 127.6, 128.2, 171.2, 63.2, 172.9, 62.1, 32.5, 128, 126, 138, 126, 135.6, 128.2; Mass (m/z) 418 (M+).
MAG: N-[methyl acetate]-2 [(2,3-dimethyl phenyl amino benzamide)] UV spectra (λmax) in SGF 255 nm, in SIF 263 nm, and in phosphate buffer 285 nm; IR (KBr, cm−1) 3424 (NH str. of amide), 3062 (aromatic CH str.), 2915, 2930 (aliphatic CH str.), 1635 (C-O str. of ester), 1616 (amide I), 1424, 1378 (CH bend, aliphatic), 1292 (C-O str. of ester); 1H NMR (δ, ppm) (DMSO) 6.98–7.01 (m, 5H, aromatic ring), 6.68 (d, 1H, CH in ring), 5.66 (d, 1H, CH in ring), 3.81 (t, 2H, CH2 in ring), 3.56–2.86 (m, 2H, CH2 in ring), 9.77 (br, NH), 4.37 (q, 2H, OCH3), 1.48 (t, 3H, CH3 saturated); 13C NMR (δ, ppm) (DMSO) 129.60, 126.5, 130.4, 138.4, 135.4, 139.6, 178.6, 32.5, 172.9, 65.2, 132, 132, 63.2, 20.10, 129, 120.1, 126.10, 128.10; Mass (m/z) 312 (M+).
Physicochemical characterization
The synthesized prodrugs of MA were subjected to solubility, physicochemical characterization, spectral analysis, and hydrolytic studies.
Solubility
Approximately 5 mg of prodrug was dissolved in 5 mL of each solvent at 37 ± 1°C in glass test tubes. The solvents used were 0.1 N NaOH, 0.1 N HCl, ethanol, methanol, ether, ethyl acetate, chloroform, acetone, dimethylformamide (DMF), and water. Test tubes were gently shaken and the solubility was observed. In the case of any observed insoluble fractions, a known amount of solvent was further added to ascertain the solubility of the compound.
Protein binding studiesCitation6
A solution of synthesized prodrug (10 mg/mL) was made in phosphate buffered saline (PBS, pH 7.4), and 100 mL of this solution was taken in a beaker. A cellophane membrane (molecular weight cut-off in the range of 10,000–12,000 Da obtained from Hi-Media, India) was first washed with distilled water and then with buffer solution (pH 7.4). It was tied at the opening end of a dialysis tube; the dialysis tube, containing (6%) egg albumin, was dipped into the drug solution and covered. The whole assembly was placed on a magnetic stirrer and switched to low revolutions per minute. The temperature was maintained at 37 ± 0.5°C. After every 1 h, 1 mL of the PBS containing drug solution was replaced with a fresh 1 mL of PBS. The withdrawn sample was diluted further with 1 mL phosphate buffer and the concentration of free MA was estimated using a spectrophotometer at 230 nm. indicates the physicochemical properties of the synthesized products, mefenamic acid tyrosine prodrug (MAT) and mefenamic acid glycine prodrug (MAT).
Table 1. Physicochemical properties of prodrugs.
In vitro hydrolysis studies
In vitro hydrolysis studies of the synthesized prodrugs were carried out in simulated gastric fluid (SGF), pH 1.2, simulated intestinal fluid (SIF), pH 7.4, and 80% human plasmaCitation7,Citation8. A solution of 10 mg of prodrug was prepared in 90 mL of SIF (pH 7.4) or SGF (pH 1.2). An aliquot of 15 mL of this solution was withdrawn repeatedly and kept in test tubes maintained at 37 ± 0.5°C. At a definite interval of time (0.5 h, 1–8 h) an aliquot was withdrawn from the different test tubes and was transferred to micro-centrifuge tubes followed by the addition of methanol to make up the volume. The tubes were placed in a freezing mixture in order to arrest further hydrolysis, followed by vortexing at high speed for 5 min. After vortexing, the tubes were centrifuged at high speed (3000 rpm) for 5 min. A 5 mL amount of clear supernatant obtained from each tube was measured on an ultraviolet (UV) spectrophotometer for the amount of free MA released after hydrolysis of prodrugs in SGF, SIF, and SIF + 80% human plasma at 230 nm. The kinetics of hydrolysis was monitored by the increase of free drug concentration with time, and the order of reaction and half life (t1/2) were also calculated. The rate of hydrolysis was calculated using the formula: K = (2.303/t) log(a/a – x), where K represents the hydrolysis constant, t is the time in min, a is the initial concentration of prodrug, x is the amount of prodrug hydrolyzed, and (a – x) is the amount of prodrug remaining.
Pharmacological studies
The synthesized compounds were evaluated for analgesic activity, anti-inflammatory activity, and ulcer index. Student’s t-test was performed to ascertain the significance of all the exhibited activities. The test compounds and the standard drugs were administered in the form of a suspension (1% carboxymethyl cellulose (CMC) as vehicle) by the oral route of administration for analgesic and anti-inflammatory activities. For ulcerogenicity studies, the drugs were administered intraperitoneally as a suspension in 10% (v/v) Tween 80. Wistar albino male rats, weighing 100–200 g, were used for the study. The selected animals were housed in acrylic cages under standard environmental conditions (20–25°C) in a well ventilated room maintained at a 12:12 h light:dark cycle, and fed with standard rodent diet and water ad libitum. Each group consisted of six animals. All the animals were acclimatized for a week before use.
All animal experiments were carried out according to the guidelines of the Committee for the Purpose of Control of Experiments on Animals (Reg. No. 930/a/06/CPCSEA) and approval of the Institutional Animal Ethics Committee was obtained.
Analgesic activity
The analgesic activity of the synthesized prodrugs was determined by thermal stimulus using the tail flick methodCitation9,Citation10. An analgesiometer was used for determination of the pain threshold of albino rats. Rats (100–200 g) were divided into four groups, each comprising six rats. A rat was placed in a holder through which the tail of the rat protruded. The reaction time was recorded at 1, 2, 3, and 4 h after treatment and the cut-off time was 9 s. The normal reaction time, i.e. the time taken to flick the tail, was noted. Animals showing delayed response were rejected. The prodrug (dose of each prodrug was equivalent to 20 mg/kg body weight) was administered orally in a 1% suspension of sodium CMC and compared with mefenamic acid as reference. The percent analgesic activity was calculated by the formula given as:
where T1 is the reaction time (s) before administration of the prodrug, T2 is the reaction time (s) after administration of the prodrug, and Tc is the cut-off time (s).
Anti-inflammatory activity
The anti-inflammatory activity was evaluated using carrageenan-induced edema of the rat pawCitation11. Albino rats (100–200 g) were divided into four groups of six animals each. Group I served as the control group, group II received mefenamic acid 20 mg/kg, and groups III and IV received prodrugs MAT and MAG, respectively, where the dose was molecularly equivalent to the free drug. The initial volume of the right hind paw of an albino rat was measured by plethysmometer without administration of drug. The drug was administered orally in a 1% suspension of sodium CMC. Thirty minutes after drug administration, carrageenan (0.1 mL, 1% (w/v) solution in normal saline) was injected into the plantar surface of the right hind paw of each animal as phlogistic agent. The volume of the right hind paw of albino rats was measured after 2, 4, and 6 h. The mean difference in volume of the right hind paw of the rats was compared with the control and standard. The percent inhibition of paw edema was calculated as:
where Vc is the mean relative change in paw edema volume in the control group and Vt is the mean relative change in paw edema volume in the test group.
Ulcerogenic activity
Gastrointestinal toxicity of the synthesized prodrugs was measured and compared with the drug by measuring the ulcer indexCitation12–14. For this purpose, male albino rats were selected, weighing between 100 and 200 g; the rats were divided into four groups each comprising six rats, including a control and a standard group. The prodrug was suspended in 10 mL of 2% (w/v) suspension of acacia. A measured volume of the suspension containing a dose equivalent to 20 mg/kg of body weight of MA was administered orally to the test group daily for 5 days. The rats were fasted after administration of the last dose; thereafter they were sacrificed by decapitation and the stomach was removed, opened, and washed with distilled water. Lesions on the gastric mucosa were counted by visual examination using a binocular magnifier. Ulcers greater than 0.5 mm were recorded. The ulcer index (UI) was calculated according to the severity of the gastric mucosal lesions, which were graded as: grade 1 = less than 1 mm erosions, grade 2 = 1–2 mm erosions, and grade 3 = more than 2 mm erosions. The UI was calculated as:
Histopathological studies
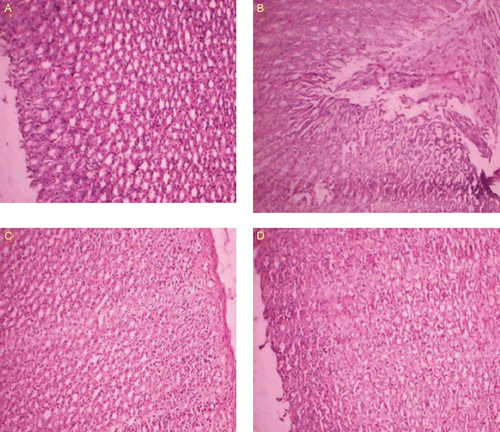
Histopathological studies of stomachs of ratsCitation15 were carried out using hematoxylin and eosin stain at the Pathology Department, Sri Venkateswara Veterinary University, Tirupati, India. The stomach tissues were removed from the rats and fixed in 10% normal saline for at least 48 h. These were then processed routinely and the tissues were embedded in paraffin wax. Histological sections were cut at 5–6 μm and stained with routine hematoxylin and eosin. These were then examined by a consultant histopathologist. The lesions observed were assessed for the following: mucosal atrophy, the presence of inflammatory cells in the wall, eosinophils, lymphocytes, and plasma cells. Photomicrographs of representative lesions at various magnifications were taken on a Zeiss optical microscope, Stemi 2000-C, with a resolution of 10 × to 45 ×, with a trinocular camera attached.
Statistical analysis
Statistical analysis of the pharmacological activity of the synthesized prodrugs in animals was evaluated using one-way analysis of variance (ANOVA). Student’s t-test was applied for expressing the significance, and a p value less than 0.05 was considered significant. Experimental data are expressed as mean ± SD (standard deviation).
Results and discussion
Synthesis and characterization
Synthesis of the prodrugs was carried out by the Schotten–Baumann method, and the structures were established by elemental analysis, 1H NMR, 13C NMR, mass, and FTIR spectral methods. The purity was determined by TLC. The results of elemental analysis of the synthesized prodrugs were very near to theoretical values and were in confirmation of the desired structure. Further, the molecular ions recorded in the mass spectra were also in agreement with the molecular weights of the compounds.
The synthesized prodrugs of MA were subjected to solubility and hydrolytic studies. The yields of the prodrugs were observed to be good. The greater solubility of the standard drug MA is mainly due to the presence of free carboxylic acid, which forms the sodium salt and makes the compound ionic. The prodrugs showed moderate to high solubility, compared to MA, in various solvents of chloroform, ethanol, and acetone, indicating their lipophilic behavior.
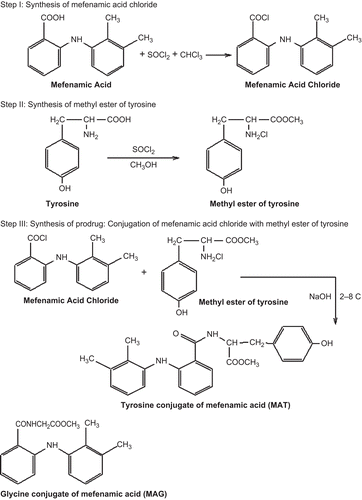
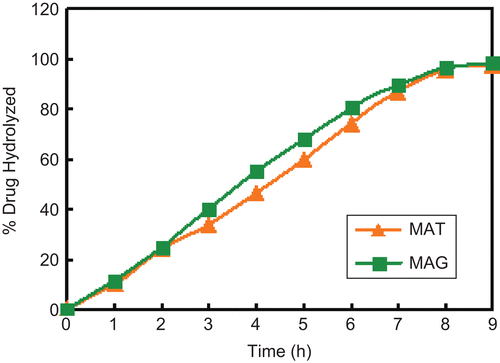
Hydrolytic studies of the prodrugs MAT and MAG were carried out in SGF (pH 1.2), SIF (pH 7.4), and SIF + 80% human plasma (pH 7.4). The comparative patterns of hydrolysis of these prodrugs in SIF and SIF + 80% human plasma (pH 7.4) are shown in and , respectively. The amount of MA regenerated on hydrolysis in SIF (pH 7.4) of MAT and MAG was found to be 78% and 89.2%, respectively, and that in SIF + 80% human plasma was found to be 97.8% and 98%, respectively. The prodrugs showed no hydrolysis in SGF, satisfactory hydrolysis in SIF, and an encouraging hydrolysis rate in SIF + 80% human plasma due to the presence of amidase in the plasma.
Figure 2. Comparative patterns of hydrolysis of MAT and MAG in simulated intestinal fluid (SIF).
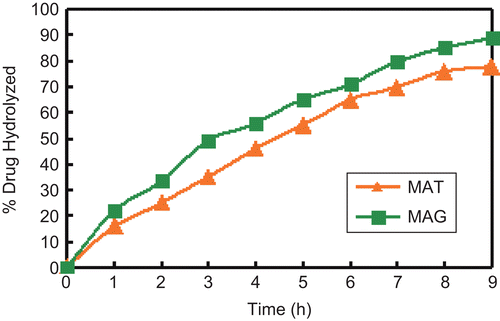
Figure 3. Comparative patterns of hydrolysis of MAT and MAG in SIF + 80% plasma.
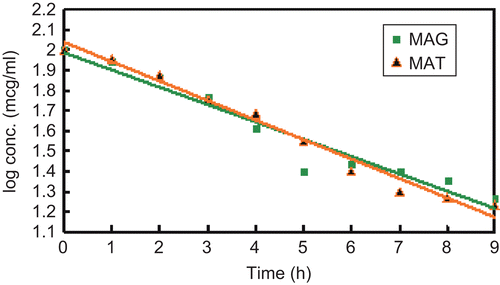
The results of the hydrolytic kinetics study revealed that both MAT and MAG followed first order kinetics as shown in . The half-life of MAT and MAG in SIF (pH 7.4) was calculated as 5.68 h and 4.62 h, suggesting that they are adequately stable to be absorbed intact from the intestine, compared to the half-life of 3 h of MA. The percentage protein binding of prodrugs MAT and MAG was found to be 62.80% and 54.40%, respectively, while that of standard drug was 89.10%. This increases the availability of the prodrugs for hydrolysis in plasma, and hence reduces the dose requirement.
Figure 4. First order hydrolysis plot of MAT and MAG.
Pharmacological studies
Analgesic activity
Evaluation of analgesic activity was performed by the tail-flick technique. The observations of analgesic testing as given in indicate that the test compounds exhibited moderate analgesic activity at 1 h of reaction time and an increase in activity which reached a peak level at 2 h. A decline in activity was observed at 3 and 4 h. The percent analgesia was calculated and compared with the reference drug. The analgesic activity calculated after 4 h of administration of the drug was 64% for MAT, 86% for MAG, and 74% for MA. The results thus confirm that MAG showed improved analgesic activity over mefenamic acid.
Table 2. Analgesic activity of MAT and MAG.
Anti-inflammatory activity
The anti-inflammatory activity developed by oral administration of the drugs is shown in . The anti-inflammatory activity was calculated as percentage inhibition of edema. Six hours after administration of drug, the prodrugs MAT and MAG showed good inhibition of edema at 74% and 81%, respectively, as compared to 40% by MA.
Table 3. Anti-inflammatory activity of MAT and MAG.
Ulcerogenic activity
The ulcerogenic liability of synthesized prodrugs MAT and MAG was tested in comparison to the parent drug mefenamic acid following single-dose oral administration in rats. The ulcer index was calculated and is shown in . Gross observation of the stomach revealed obvious widespread hemorrhagic spots in the mefenamic acid-treated animals compared to the prodrug-treated animals. The prodrug-treated groups showed intact mucosal layers, identical to the group receiving vehicle only. The ulcerogenic dose of prodrug was double that of the parent drug. These findings indicate that the prodrugs MAT and MAG are significantly less irritating to the gastric mucosa than is mefenamic acid.
Table 4. Mean ulcer index of MAT and MAG.
Histopathological studies
There was no death recorded in any of the experimental animal groups during the study period. The gastric tissues were investigated microscopically, and it was observed that tissue samples of the control group rats showed normal histological findings (). Microscopic investigation of the MA group revealed a focal erosive area in the gastric mucosa and a zone (clear zone) in the basal regions of the gastric glands that stained pale. This zone was parallel to the surface of the stomach lumen. In this zone, the structures of the gland were destroyed. They had disintegrated from the basal lamina and fallen into the lumen. The nuclei of these cells had become smaller and dense, and their cytoplasms were stained as dark eosinophilic bodies. Small hemorrhagic areas and patches of inflammatory cell infiltrations were present in the lumen of the glands and lamina propria. Normal histological findings were displayed for both MAT and MAG groups. This reveals that the prodrugs do not produce any ulceration in the gastric region.
Figure 5. Histopathological studies of prodrugs. (A) Healthy control. (B) Ulcer control showing mucosal injury induced by MA and massive mucosal infiltration of inflammatory cells. (C) Treated with MAT. (D) Treated with MAG.
Conclusions
In conclusion, the synthesis of amide prodrugs, MAT and MAG, containing mefenamic acid was carried out successfully and the structures were confirmed by elemental and spectral analysis. The prodrugs released the parent drug MA quantitatively at pH 7.4 but were resistant to hydrolysis at acidic pH, indicating that these prodrugs do not break down under acidic conditions. Both prodrugs showed good anti-inflammatory activity with fewer gastric ulcers when compared to the parent drug. The minimal ulcerogenic dose of the prodrugs was found to be twice as high as that of MA, and in equimolar quantities the prodrugs were consistently less ulcerogenic than the parent drug.
Acknowledgements
The authors express their thanks to M/s. Alkem Laboratories, Mumbai, for providing the gift sample of mefenamic acid. The authors are grateful to Padmashree Dr. M. Mohan Babu, Chairman, Sree Vidyanikethan Educational Trust, Tirupati, India, for providing the necessary facilities to carry out this work.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ravichandran V, Mishra A, Agrawal RK, Dixit VK. Synthesis, characterization and pharmacological evaluation of amide prodrugs of flubiprofen. J Braz Chem Soc 2008;19:89–100.
- Mishra A, Ravichandran V, Jain PK, Dixit VK, Agrawal RK. Synthesis, characterization and pharmacological evaluation of amide prodrugs of ketorolac. Eur J Med Chem 2008;43:2464–72.
- Khan MYS, Akheter M. Synthesis, pharmacological activity and hydrolytic behaviour of glyceride prodrugs of ibuprofen. Eur J Med Chem 2005;40:371–6.
- Streitweiser A Jr, Heathcock GH. Introduction to Organic Chemistry, 3rd ed. New York: Macmillan Publishing Company, 1989.
- Norman ROC, Coxon JM. Principles of Organic Synthesis, 3rd ed. London: ELBS and Chapman & Hall, 1993.
- Martin AN, Bustamante P, Chine AHC. Physical Pharmacy, 4th ed. New Delhi: BI Waverly Pvt. Ltd., 1993.
- Zhao X, Tao X, Wei D, Song Q. Pharmacological activity and hydrolysis behavior of novel ibuprofen glucopyranoside conjugates. Eur J Med Chem 2006;41:1352–8.
- Metwally KA, Yaseen SH, Lashine EM, El-Fayomi HM, El-Sadek ME. Non-carboxylic analogues of arylpropionic acids: synthesis, anti-inflammatory activity and ulcerogenic potential. Eur J Med Chem 2007;42:152–60.
- D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther 1941;72:74–9.
- Alagarsamy V, Shankar D, Solomon VR. Synthesis of some novel 2-mercapto-3-(substituted amino)-5,6,7,8-tetrahydro-3H-benzo[4,5] thieno[2,3d] pyrimidin-4-ones as analgesic and anti-inflammatory agents. ARKIVOC 2006;16:149–59.
- Neha G, Deepika N, Suneela SD, Dhaneshwar SR, Chaturvedi SC. Synthesis, hydrolysis, kinetics and pharmacodynamic profiles of novel prodrugs of flubiprofen. Indian J Pharm Sci 2005;67:369–73.
- Brodie DA, Cook PG, Bauer BJ, Dagle GE. Indomethacin-induced intestinal lesions in the rat. Toxicol Appl Pharmacol 1970;17:615–24.
- Main LH, Whittle BJR. Investigation of the vascodilator and antisecretory role of prostaglandin in the rat gastric mucosa by use of non-steroidal anti-inflammatory drugs. Br J Pharmacol 1975;53:217–24.
- Kulkarni SK. Handbook of Experimental Pharmacology, 3rd ed. New Delhi: Vallabh Prakash, 2001:128–30.
- Yagmurca M, Ucar M, Fadillioglu E, Erdogan H, Ozturk F. The effects of nitric oxide on rat stomach injury induced by acetylsalicylic acid. Turk J Med Sci 2009;39:13–19.