Abstract
Structural modifications around 8-HETE (8-hydroxyeicosatetraenoic acid), a natural agonist of the PPAR (peroxisome proliferator-activated receptor) nuclear receptors have led previously to the identification of a promising analog, the quinoline S 70655. Series of novel quinoline or benzoquinoline derivatives were designed through the modification of this lead. Variations of the nature of the aromatic core and of the side chains were carried out. The SAR studies indicated the high sensitivity of the upper acid chain to modifications as well as the strong effect of the length and size of the lipophilic side chain. They afforded several new promising PPARα/γ dual agonists with a high PPARα activity in vitro.
Introduction
The main features of metabolic syndrome (MS) include insulin resistance (IR), central or abdominal obesity, abnormal lipidemia (hypertriglyceridemia and low high-density lipoprotein (HDL) cholesterol), elevated blood pressure, and impaired glucose toleranceCitation1. MS is one of the factors that increases the risk of developing type 2 diabetes (T2D), which is defined by peripheral IR, insulin-production defect, and, as a consequence, hyperglycemiaCitation2.
Cardiovascular events are the primary cause of mortality among T2D and MS patients, and during recent decades the incidence of these diseases has dramatically increasedCitation3. As a result, efficient treatments of both lipid and glucose disorders are required.
Discovery of the peroxisome proliferator-activated receptors (PPARs) and their central role in lipid and glucose metabolisms has created a new approach for the treatment of T2D and MS. PPARs are members of the nuclear receptor superfamily, comprising steroid, thyroid, retinoic acid, and vitamin D receptors. Three subtypes of PPAR have already been identified to date: PPARα, PPARβ/δ, and PPARγ. PPARα promotes lipid uptake and oxidation in high-metabolism tissuesCitation4. PPARβ is expressed broadly, and seems to be involved in the regulation of lipid and lipoprotein metabolism. PPARγ is implied in lipid storage, adipocyte differentiation, and regulation of IR factorsCitation5. All subtypes of PPAR are activated by saturated and unsaturated fatty acids and their metabolites, even though the affinities are weak, and this retro-control is one of the mechanisms that maintain the physiological equilibrium level of fatty acids. Synthetic ligands have also been identified, such as the antidyslipidemic fibrates for PPARαCitation6 and the antidiabetic thiazolidinediones (TZDs) for PPARγCitation7.
Classical structure–activity relationship (SAR) studies have been carried out on the fibrates and TZD structures, and have provided a breakthrough in the preparation of dual PPARα/γ () with a full-agonist profile on PPARγ. The clinical development of this class of compounds clearly demonstrates their efficacy for the treatment of T2D and MS, by improving both lipid and glucose homeostasisCitation8,Citation9.
Figure 1. PPARα/γ dual agonists.
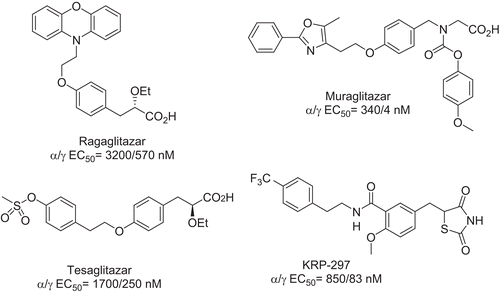
However, identification of adverse effects has stopped the development of several promising candidatesCitation10,Citation11. Even though the exact toxic mechanisms are not yet established, they seem to be clearly related to PPARγ activity. These results give good support to our strategy involving the preparation of dual PPARα/γ agonists with a full-agonist profile on PPARα and a partial-agonist profile on PPARγ. As we have previously reportedCitation12, several dual agonists were prepared by structural modifications of a natural ligand, 8(S)-HETE (8-hydroxyeicosatetraenoic acid), that presented a submicromolar activity on PPARα and a micromolar activity on PPARγ. One of these PPARα/γ dual agonists, the quinoline S 70655 (), exhibited the desired profile in vitro but was not active in vivo.
Figure 2. From 8(S)-HETE to quinoline S 70655.
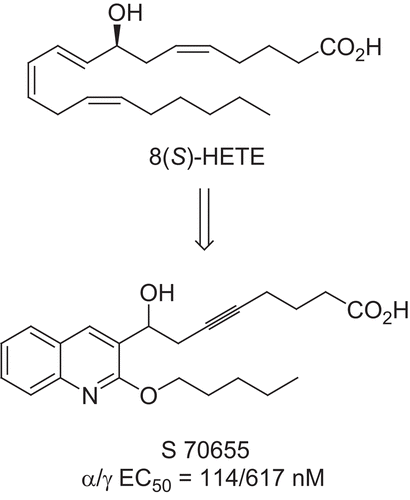
In the earlier SAR studies carried out on S 70655, we established the central role of the quinoline core, the free hydroxyl, and the triple bond for biological activityCitation13. In order to increase the activity and the pharmacokinetic parameters of S 70655, we have considered three new points: the nature of the lipophilic chain, the substitution of the acid moiety, and the substitution on the quinoline core. In this article, we report the synthesis and biological evaluation of the new derivatives corresponding to these three major modulations.
Materials and methods
Chemistry
Nuclear magnetic resonance (NMR) data were recorded in CDCl3 on a Bruker ARX 400 (400 MHz) spectrometer, using tetramethylsilane (TMS) (1H and 13C) or CCl3F (19F) as the internal standard, or on a Bruker Avance 300 (300 MHz) or a Bruker ARX 200 (200 MHz) spectrometer. Chemical shifts are expressed as parts per millions (ppm) in δ units. High-resolution mass spectra (HRMS) were recorded with a Varian MAT 311 spectrometer under electron impact at 70 eV. Microanalyses were carried out with a Flash E812 CHNS/O Thermo Electron analyzer. Chemicals were from commercial suppliers and were used without any further purification. Freshly distilled solvents under anhydrous conditions were used, unless otherwise mentioned.
Strategy of synthesis
The preparation of these new analogs followed the same strategy as previously described for S 70655: nucleophilic substitution of the 2-chloroquinoline moiety (introduction of the lipophilic chain) followed by introduction of the homopropargylic alcohol in position 3 (elaboration of the acid moiety)Citation13,Citation14. All these derivatives were prepared in racemic form only, since previous studies demonstrated, on a similar series of molecules, that racemic analogs exhibited a better bioactivity than individual enantiomersCitation13.
Synthesis of compounds 9a–9e
For the first series of modulations, we studied the role of the lipophilic chain (chain length, steric parameters, and prevention of metabolism) on the activity. Following the same strategy, we introduced diversity during the nucleophilic substitution step on the 2-chloroquinoline ().
Scheme 1. Synthesis of esters 8a–8g and sodium salts 9a–9g. Reagents and conditions: (a) CH(OMe)3, NH4NO3, MeOH, reflux, 4 h, 96%; (b) appropriate ROH, NaH, NMP, 0°C to rt, 12 h, 70–98%; (c) PTSA, THF/H2O, reflux, 4 h, 84–99%; (d) propargyl bromide, Mg, HgCl2, Et2O, −78°C to rt, 2 h, 72–99%; (e) TBDMSCl, Im., DMF, 0°C to rt, 12 h, 70–94%; (f) n-BuLi, THF, −78°C, 30 min, Br(CH2)3C(OCH3)3, HMPA, −60°C to rt, 12 h, then aq. NH4Cl 38–89%; (g) TBAF, THF, 45°C, 2 h, 47–82%; (h) LiOH·H2O, MeOH/H2O, rt, 48 h, (CO2H)2, 44–99% then NaOH, 87–95%.
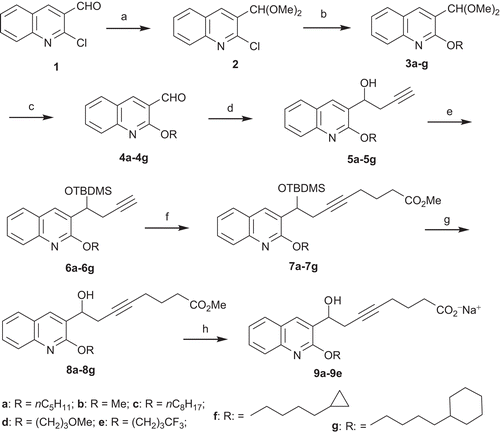
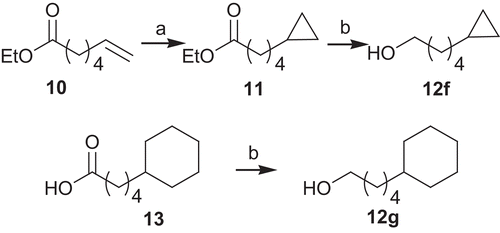
The commercially available quinoline 1 was submitted, after protection to acetal 2, to a nucleophilic substitution by various alcohols to afford ethers 3a–3g. Most of the required alcohols were commercially available except for 12f and 12g. These latter derivatives were prepared by standard procedures as indicated in . The cyclopropanation of ethyl hept-6-enoate 10 gave in excellent yield the ester 11, which, after reduction, afforded the desired alcohol 12f. On the other hand, 5-cyclohexylpentan-1-ol 12g was obtained in 99% yield by reduction of the corresponding acid 13.
Scheme 2. Synthesis of alcohols 12f and 12g: (a) CF3CO2H, Et2Zn, 1,2-diiodoethane, DCM, 0°C, 99%; (b) LiAlH4, Et2O, 92–99%.
After ketal deprotection to 4a–4g and the Grignard reaction with propargyl magnesium bromide, the homopropargylic alcohols 5a–5g were protected as silyl ethers 6a–6g. These key intermediates were alkylated by trimethyl 4-bromoorthobutyrate to give the derivatives 7a–7g in moderate to good yields. After silyl deprotection, the desired methyl esters 8a–8g were obtained and then the corresponding sodium salts 9a–9e.
Synthesis of compounds 14a, 18a, 18b
The second series of modulations performed on the quinoline S 70655 dealt with the acid moiety (), mainly in order to reduce the metabolism on this chain.
Scheme 3. Synthesis of compounds 14a and 18a, 18b: (a) LiOH·H2O, MeOH/H2O, rt, 48 h, (CO2H)2, 92%; (b) HO(CH2)2NH2, Et3N, BOPCl, CH2Cl2, 0°C to rt, 1 h, 50%; (c) n-BuLi, (HCHO)n, THF, −78°C to rt, 4 h, 72–78%; (d) BrCH2CO2t-Bu, n-Bu4NBr, toluene, NaOH aq., rt, 4 h, 68–90%; (e) TBAF, THF, 45°C, 2 h, 64–72%; (f) NaOH, MeOH/H2O, rt, 48 h, (CO2H)2, 54–84% then NaOH, 99%.
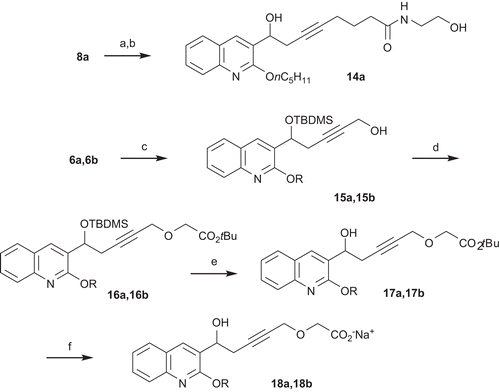
The first target amide 14a was obtained from the methyl ester 8a by saponification followed by coupling with ethanolamine. The second target compound presented an oxygen β to the carboxylic acid to avoid the metabolization of this chain. For that purpose, the propargyl derivatives 6a, 6b were reacted with n-butyllithium (BuLi) and paraformaldehyde to give the desired propargylic alcohols 15a, 15b. These derivatives were reacted with t-butyl bromoacetate to afford the intermediates 16a, 16b. After silyl deprotection, the esters 17a, 17b were obtained and the corresponding sodium salts 18a, 18b were prepared as previously describedCitation12.
Synthesis of compounds 26a, 26b, 27a, 27b
We finally explored the structure–activity relationships of the quinoline core, and two examples were selected (). The first compound presented a methoxy group in position 6 and the corresponding starting material 19a was commercially available. The second derivative presented a more hindered aromatic core, a benzoquinoline. The corresponding starting material, 19b, was prepared with moderate yield by Vilsmeier–Haack cyclization starting from the N-naphthalenacetamideCitation14.
Scheme 4. Synthesis of esters 26a, 26b and sodium salts 27a, 27b: (a) POCl3, DMF, reflux, 6 h, 40%; (b) HC(OMe)3, NH4NO3, MeOH, reflux, 4 h, 80–98%; (c) n-C5H11OH, NaH, NMP, 0°C to rt, 12 h, 68–80%; (d) PTSA, THF/H2O, reflux, 4 h, 85–99%; (e) propargyl bromide, Mg, HgCl2, Et2O, −78°C to rt, 2 h, 90–99%; (f) TBDMSCl, Im., DMF, 0°C to rt, 12 h, 97–98%; (g) n-BuLi, THF, −78°C, 30 min, Br(CH2)3C(OCH3)3, HMPA, −60°C to rt, 12 h, then aq. NH4Cl 50–65%; (h) TBAF, THF, 45°C, 2 h, 22–62%; (i) LiOH,·H2O, MeOH/H2O, rt, 48 h, (CO2H)2, 44–65% then NaOH, 99–100%.
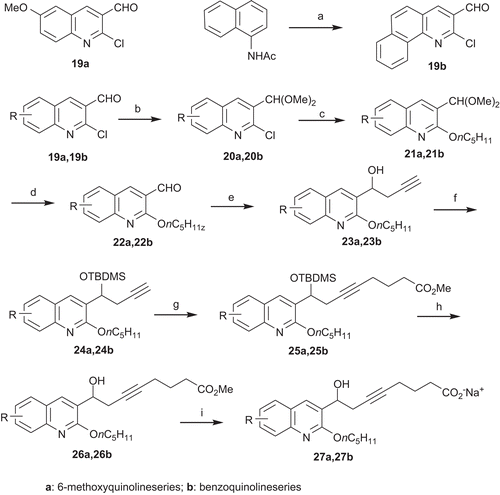
Starting from the aldehydes 19a and 19b, the desired compounds were prepared following the previously described synthesis route. In addition, these new analogs 26a, 27a and 26b, 27b were prepared with the same lipophilic C5 alkyl chain as S 70655.
Procedures and spectroscopic data
General procedure for the preparation of 2
To a suspension of the carbaldehyde in MeOH was added trimethyl orthoformate followed by NH4NO3. The resulting suspension was refluxed during 4 h, and after cooling to room temperature, the reaction was quenched with a saturated solution of Na2CO3 and Et2O was added. The organic layer was separated and the aqueous phase was extracted with Et2O. The collected organic phases were washed with brine, dried over MgSO4, and evaporated. The crude product was purified by flash chromatography.
2-Chloro-3-dimethoxymethyl-quinoline (2)
Compound was obtained with 2-chloroquinoline-3-carbaldehyde (6.0 g, 31.3 mmol), trimethylorthoformate (4.12 mL, 37.6 mmol), NH4NO3 (126 mg, 1.56 mmol) and MeOH (30 mL). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded a white solid (7.14 g, 96% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.33 (s, 1H, H-Ar), 7.94 (d, J = 8.4 Hz, 1H, H-Ar), 7.77 (d, J = 8.1 Hz, 1H, H-Ar), 7.73 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.48 (ddd, J = 8.1, 7.0, 1.0 Hz, 1H, H-Ar), 5.64 (s, 1H, CH(OCH3)2), 3.35 (s, 6H, CH(OCH3)2); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 149.30 (C), 147.46 (C), 137.26 (CAr), 130.86 (CH), 129.26 (C), 128.23 (CH), 128.07 (CH), 127.25 (CH), 126.73 (C), 100.40 (CH), 53.90 (2C, CH3).
General procedure for the preparation of 3a–3g
The alcohol was added dropwise to a suspension of NaH (60% in mineral oil, first washed with petroleum ether) in N-methyl-2-pyrrolidone (NMP) at 0°C. After 30 min, acetal 2 (or 20a, 20b) was added, the cooling bath was removed, and the mixture was stirred overnight. The reaction was quenched by adding water, the organic layer was separated, and the aqueous phase was extracted with Et2O. The collected organic phases were washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was purified by column chromatography on silica gel.
3-Dimethoxymethyl-2-pentyloxy-quinoline (3a)
Compound was obtained with NaH (60% in mineral oil) (364 mg, 9.10 mmol), 1-pentanol (555 µL, 9.10 mmol), 2 (1.08 g, 4.55 mmol), and NMP (4.5 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded an off-white solid (1.26 g, 96% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.22 (s, 1H, H-Ar), 7.94 (dd, J = 8.4, 1.0 Hz, 1H, H-Ar), 7.76 (dd, J = 8.1, 1.5 Hz, 1H, H-Ar), 7.63 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.40 (ddd, J = 8.1, 7.0, 1.0 Hz, 1H, H-Ar), 5.68 (s, 1H, CH(OCH3)2), 4.54 (t, J = 6.7 Hz, 2H, OCH2), 3.45 (s, 6H, CH(OCH3)2), 1.93-1.84 (m, 2H, OCH2CH2), 1.56–1.37 (m, 4H, CH2CH2CH3), 0.96 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.76 (C), 146.50 (C), 136.06 (CH), 129.64 (CH), 127.96 (C), 126.85 (CH), 124.66 (CH), 123.99 (CH), 121.95 (C), 99.16 (CH), 66.17 (CH2), 53.90 (2C, CH3), 28.66 (CH2), 28.32 (CH2), 22.47 (CH2), 14.10 (CH3).
3-Dimethoxymethyl-2-methoxy-quinoline (3b)
Compound was obtained with NaH (330 mg, 5.16 mmol), MeOH (210 µL, 5.16 mmol), 2 (613 mg, 2.58 mmol), and NMP (3 mL). Column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) afforded a yellow oil (552 mg, 98% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.22 (s, 1H, H-Ar), 7.85 (dd, J = 8.4, 1.2 Hz, 1H, H-Ar), 7.76 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.63 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.39 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.69 (s, 1H, CH(OCH3)2), 4.12 (s, 3H, OCH3), 3.40 (s, 6H, CH(OCH3)2); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.82 (C), 146.33 (C), 136.38 (CH), 129.76 (CH), 127.98 (C), 126.84 (CH), 124.67 (CH), 124.18 (CH), 121.61 (C), 98.59 (CH), 53.77 (2C, CH3), 53.35 (CH3).
3-Dimethoxymethyl-2-octyloxy-quinoline (3c)
Compound was obtained with NaH (212 mg, 5.30 mmol), 1-octanol (845 µL, 5.30 mmol), 2 (630 mg, 2.65 mmol), and NMP (6 mL). Column chromatography on silica gel (EtOAc/pentane, 7:93 v/v) afforded a colorless oil (817 mg, 93% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.28 (s, 1H, H-Ar), 7.89 (d, J = 8.3 Hz, 1H, H-Ar), 7.80 (d, J = 7.9 Hz, 1H, H-Ar), 7.64 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.39 (dd, J = 7.9, 7.0 Hz, 1H, H-Ar), 5.75 (s, 1H, CH(OCH3)2), 3.48 (s, 6H, CH(OCH3)2), 4.61 (t, J = 6.6 Hz, 2H, OCH2), 2.00–1.82 (m, 2H, OCH2CH2), 1.63–1.24 (m, 10H, (CH2)5CH3), 1.02–0.97 (m, 3H, CH3); 13C NMR: (75 MHz, CDCl3) δ (ppm): 160.17 (C), 146.93 (C), 136.51 (CH), 130.05 (CH), 128.37 (C), 127.30 (CH), 125.08 (CH), 124.41 (CH), 122.40 (C), 99.53 (CH), 66.60 (CH2), 54.20 (2C, CH3), 32.30 (CH2), 29.82 (CH2), 29.76 (CH2), 29.41 (CH2), 26.59 (CH2), 23.13 (CH2), 14.57 (CH3).
3-Dimethoxymethyl-2-(3-methoxy-propoxy)-quinoline (3d)
Compound was obtained with NaH (336 mg, 8.40 mmol), 3-methoxypropanol (804 µL, 8.40 mmol), 2 (1.0 g, 4.20 mmol), and NMP (5 mL). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded a white solid (1.16 g, 95% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H, H-Ar), 7.80 (d, J = 8.4 Hz, 1H, H-Ar), 7.68 (d, J = 8.0 Hz, 1H, H-Ar), 7.52 (dd, J = 8.4, 7.2 Hz, 1H, H-Ar), 7.28 (dd, J = 8.0, 7.2 Hz, 1H, H-Ar), 5.61 (s, 1H, CH(OCH3)2), 4.59 (t, J = 6.4 Hz, 2H, OCH2), 3.51 (t, J = 6.3 Hz, 2H, CH2OCH3), 3.37 (s, 6H, CH(OCH3)2), 3.30 (s, 3H, OCH3), 2.16–2.00 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.80 (C), 146.81 (C), 136.55 (CH), 129.96 (CH), 128.25 (C), 127.29 (CH), 125.03 (CH), 124.40 (CH), 122.23 (C), 99.29 (CH), 69.82 (CH2), 63.45 (CH2), 58.88 (CH3), 53.83 (2C, CH3), 29.66 (CH2).
3-Dimethoxymethyl-2-(4,4,4-trifluoro-butoxy)-quinoline (3e)
Compound was obtained with NaH (336 mg, 8.40 mmol), 4,4,4-trifluorobutanol (850 µL, 8.40 mmol), 2 (1.0 g, 4.20 mmol), and NMP (5 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a white solid (1.28 g, 93% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.31 (s, 1H, H-Ar), 7.98 (d, J = 8.3 Hz, 1H, H-Ar), 7.79 (d, J = 8.0 Hz, 1H, H-Ar), 7.64 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.40 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.71 (s, 1H, CH(OCH3)2), 4.63 (t, J = 6.0 Hz, 2H, OCH2), 3.49 (s, 6H, CH(OCH3)2), 2.53–2.27 (m, 2H, CH2CF3), 2.24–2.11 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.54 (C), 146.73 (C), 136.94 (CH), 130.22 (CH), 128.41 (C), 127.72 (q, J = 276.1 Hz, CF3), 127.34 (CH), 125.23 (CH), 124.72 (CH), 122.14 (C), 99.33 (CH), 64.53 (CH2), 53.86 (2C, CH3), 31.18 (q, J = 29.0 Hz, CH2CF3), 22.28 (CH2).
3-Dimethoxymethyl-2-(5-cyclopropylpentyloxy)-quinoline (3f)
Compound was obtained with NaH (809 mg, 20.23 mmol), 5-cyclopropylpentan-1-ol 12 (1.6 g, 12.50 mmol), 2 (2.82 g, 11.90 mmol), and NMP (15 mL). Column chromatography on silica gel (EtOAc/cyclohexane, 10:90 v/v) afforded a colorless oil (3.46 g, 88% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.22 (s, 1H, H-Ar), 7.84 (d, J = 8.3 Hz, 1H, H-Ar), 7.76 (d, J = 7.9 Hz, 1H, H-Ar), 7.66–7.59 (m, 1H, H-Ar), 7.42–7.34 (m, 1H, H-Ar), 5.68 (s, 1H, CH(OCH3)2), 4.54 (t, J = 6.6 Hz, 2H, OCH2), 3.44 (s, 6H, CH(OCH3)2), 1.94–1.82 (m, 2H, OCH2CH2), 1.61–1.45 (m, 4H, CH2CH2CH2CH), 1.31–1.19 (m, 2H, CH2CH), 0.77–0.61 (m, 1H, CH), 0.46–0.37 (m, 2H, CH2 cyclo), 0.06 to −0.01 (m, 2H, CH2 cyclo); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.76 (C), 146.51 (C), 136.08 (CH), 129.64 (CH), 127.96 (C), 126.87 (CH), 124.67 (CH), 123.99 (CH), 122.02 (C), 99.17 (CH), 68.18 (CH2), 53.86 (2C, CH3), 34.73 (CH2), 29.44 (CH2), 29.04 (CH2), 26.03 (CH2), 10.87 (CH cyclo), 4.42 (2C, CH2 cyclo).
3-Dimethoxymethyl-2-(5-cyclohexylpentyloxy)-quinoline (3g)
Compound was obtained with NaH (276 mg, 6.92 mmol), 5-cyclohexylpentan-1-ol 12g (1.18 g, 6.92 mmol), 2 (1.49 g, 6.29 mmol), and NMP (15 mL). Column chromatography on silica gel (EtOAc/cyclohexane, 5:95 v/v) afforded a colorless oil (1.61 g, 70% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.20 (s, 1H, H-Ar), 7.81 (d, J = 8.3 Hz, 1H, H-Ar), 7.77–7.71 (m, 1H, H-Ar), 7.65–7.56 (m, 1H, H-Ar), 7.41–7.32 (m, 1H, H-Ar), 5.65 (s, 1H, CH(OCH3)2), 4.51 (t, J = 6.6 Hz, 2H, OCH2), 3.42 (s, 6H, CH(OCH3)2), 1.91-0.77 (m, 19H, Cyclohexyl(CH2)4CH2O); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.76 (C), 146.48 (C), 136.05 (CH), 129.63 (CH), 127.96 (C), 126.84 (CH), 124.65 (CH), 123.98 (CH), 121.99 (C), 99.15 (CH), 66.19 (CH2), 53.87 (2C, CH3), 37.65 (CH), 37.50 (CH2), 33.47 (CH2), 29.02 (CH2), 26.77 (CH2), 26.65 (CH2), 26.46 (CH2).
General procedure for the preparation of 4a–4g
To a solution of 3a–3g (or 21a, 21b) in tetrahydrofuran (THF)/water, was added p-toluenesulfonic acid (PTSA) and the resulting solution was refluxed during 4 h. After cooling to room temperature, the reaction was quenched with a saturated aqueous solution of Na2CO3 and EtOAc was added. The organic layer was separated and the aqueous phase was extracted with EtOAc. The collected organic phases were washed with water and then with brine and dried over MgSO4. After evaporation to dryness, the pure product was obtained.
2-Pentyloxy-quinoline-3-carbaldehyde (4a)
Compound was obtained with 3a (3.19 g, 11.0 mmol), PTSA (314 mg, 1.65 mmol), and THF/H2O (100 mL, 9:1 v/v). A yellow solid (2.51 g, 94% yield) was obtained.
1H-NMR: (400 MHz, CDCl3) δ (ppm): 10.52 (s, 1H, CHO), 8.43 (s, 1H, H-Ar), 7.80 (d, J = 8.4 Hz, 1H, H-Ar), 7.78 (d, J = 8.4 Hz, 1H, H-Ar), 7.63 (dd, J = 8.4, 7.8 Hz, 1H, H-Ar), 7.33 (dd, J = 8.4, 7.8 Hz, 1H, H-Ar), 4.49 (t, J = 6.7 Hz, 2H, OCH2), 1.86–1.76 (m, 2H, OCH2CH2), 1.47–1.31 (m, 4H, CH2CH2CH3), 0.87 (t, J = 7.1 Hz, 3H, CH3): 13C-NMR: (100 MHz, CDCl3) δ (ppm): 189.46 (CHO), 161.21 (C), 149.08 (C), 139.55 (CH), 132.44 (CH), 129.74 (C), 127.23 (CH), 124.86 (CH), 124.26 (CH), 120.02 (C), 66.17 (CH2), 28.55 (CH2), 28.35 (CH2), 22.47 (CH2), 14.05 (CH3).
2-Methoxy-quinoline-3-carbaldehyde (4b)
Compound was obtained with 3b (552 mg, 2.54 mmol), PTSA (72 mg, 0.38 mmol), and THF/H2O (25 mL, 9:1 v/v). A white solid (417 mg, 87% yield) was obtained.
M.p.: 114–116°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 10.47 (s, 1H, CHO), 8.59 (s, 1H, H-Ar), 7.89–7.83 (m, 2H, H-Ar), 7.74 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H, H-Ar), 7.44 (ddd, J = 8.5, 7.0, 1.2 Hz, 1H, H-Ar), 4.19 (s, 3H, OCH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 189.41 (CHO), 161.21 (C), 148.95 (C), 140.04 (CH), 132.59 (CH), 129.75 (C), 127.26 (CH), 125.04 (CH), 124.37 (CH), 120.01 (C), 53.85 (CH3).
2-Octyloxy-quinoline-3-carbaldehyde (4c)
Compound was obtained with 3c (800 mg, 2.41 mmol), PTSA (70 mg, 0.36 mmol), and THF/H2O (21 mL, 9:1 v/v). A colorless oil (580 mg, 84% yield) was obtained.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.45 (s, 1H, CHO), 8.42 (s, 1H, H-Ar), 7.84–7.73 (m, 2H, H-Ar), 7.61 (dd, J = 8.4, 7.0 Hz, 1H, H-Ar), 7.34 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 4.51 (t, J = 6.6 Hz, 2H, OCH2), 1.91–1.72 (m, 2H, OCH2CH2), 1.70–1.11 (m, 10H, (CH2)5CH3), 0.84–0.80 (m, 3H, CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 189.93 (CHO), 162.85 (C), 148.82 (C), 140.06 (CH), 132.91 (CH), 130.18 (C), 127.61 (CH), 125.32 (CH), 124.53 (CH), 120.45 (C), 67.14 (CH2), 32.23 (CH2), 29.76 (CH2), 29.66 (CH2), 29.26 (CH2), 26.59 (CH2), 23.07 (CH2), 14.52 (CH3).
2-(3-Methoxy-propoxy)-quinoline-3-carbaldehyde (4d)
Compound was obtained with 3d (1.16 g, 3.98 mmol), PTSA (116 mg, 0.60 mmol), and THF/H2O (38 mL, 9:1 v/v). A white solid (890 mg, 91% yield) was obtained.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.49 (s, 1H, CHO), 8.55 (s, 1H, H-Ar), 7.89–7.75 (m, 2H, H-Ar), 7.70 (dd, J = 8.4, 7.4 Hz, 1H, H-Ar), 7.40 (dd, J = 7.9, 7.4 Hz, 1H, H-Ar), 4.69 (t, J = 6.3 Hz, 2H, OCH2), 3.62 (t, J = 6.3 Hz, 2H, CH2OCH3), 3.39 (s, 3H, OCH3), 2.31–2.02 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 189.61 (CHO), 161.30 (C), 149.36 (C), 140.01 (CH), 132.87 (CH), 130.10 (C), 127.67 (CH), 125.34 (CH), 124.68 (CH), 120.31 (C), 69.83 (CH2), 64.05 (CH2), 59.13 (CH3), 29.59 (CH2).
2-(4,4,4-Trifluoro-butoxy)-quinoline-3-carbaldehyde (4e)
Compound was obtained with 3e (1.28 g, 3.88 mmol), PTSA (111 mg, 0.58 mmol), and THF/H2O (38 mL, 9:1 v/v). A white solid (1.04 g, 95% yield) was obtained.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.27 (s, 3H, CHO), 8.38 (s, 1H, H-Ar), 7.73–7.50 (m, 3H, H-Ar), 7.28 (dd, J = 7.9, 7.0 Hz, 1H, H-Ar), 4.50 (t, J = 6.0 Hz, 2H, OCH2), 2.47–2.18 (m, 2H, CH2CF3), 2.18–1.99 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 188.69 (CHO), 160.62 (C), 148.93 (C), 135.76, 132.76 (CH), 129.94 (CH), 127.53 (q, J = 271.2 Hz, CF3), 127.48 (CH), 125.36 (C), 124.59 (CH), 119.99 (C), 68.14 (CH2), 31.11 (q, J = 29.2 Hz, CH2CF3), 22.03 (CH2).
2-(5-Cyclopropylpentyloxy)-quinoline-3-carbaldehyde (4f)
Compound was obtained with 3f (3.4 g, 10.32 mmol), PTSA (294 mg, 1.54 mmol), and THF/H2O (80 mL, 5:3 v/v). A pale yellow oil (2.92 g, 99% yield) was obtained.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.51 (s, 1H, CHO), 8.58 (s, 1H, H-Ar), 7.84 (d, J = 8.2 Hz, 1H, H-Ar), 7.77–7.69 (m, 1H, H-Ar), 7.46–7.38 (m, 1H, H-Ar), 7.42–7.34 (m, 1H, H-Ar), 4.60 (t, J = 6.6 Hz, 2H, OCH2), 1.97–1.83 (m, 2H, OCH2CH2), 1.62–1.45 (m, 4H, CH2CH2CH2CH), 1.31–1.20 (m, 2H, CH2CH), 0.76–0.61 (m, 1H, CH), 0.46–0.36 (m, 2H, CH2 cyclo), 0.06 to −0.01 (m, 2H, CH2 cyclo); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 189.46 (CHO), 161.23 (C), 149.11 (C), 139.57 (CH), 132.45 (CH), 129.75 (C), 127.25 (CH), 124.88 (CH), 124.30 (CH), 120.05 (C), 66.63 (CH2), 34.66 (CH2), 29.43 (CH2), 28.93 (CH2), 26.05 (CH2), 10.84 (CH cyclo), 4.42 (2C, CH2 cyclo).
2-(5-Cyclohexylpentyloxy)-quinoline-3-carbaldehyde (4g)
Compound was obtained with 3g (2.44 g, 6.67 mmol), PTSA (190 mg, 1.00 mmol), and THF/H2O (100 mL, 6:4 v/v). A pale yellow solid (2.05 g, 96% yield) was obtained.
1H NMR: (300 MHz, CDCl3) δ (ppm): 10.50 (s, 1H, CHO), 8.58 (s, 1H, H-Ar), 7.86–7.80 (m, 2H, H-Ar), 7.78–7.68 (m, 1H, H-Ar), 7.46–7.37 (m, 1H, H-Ar), 4.57 (t, J = 6.6 Hz, 2H, OCH2), 1.95–0.76 (m, 19H, Cyclohexyl(CH2)4CH2O); 13C NMR: (75 MHz, CDCl3) δ (ppm): 189.49 (CHO), 161.24 (C), 149.11 (C), 139.57 (CH), 132.45 (CH), 129.75 (C), 127.25 (CH), 124.88 (CH), 124.30 (CH), 120.05 (C), 66.64 (CH2), 37.64 (CH), 37.43 (CH2), 33.45 (CH2), 28.90 (CH2), 26.75 (CH2), 26.62 (CH2), 26.48 (CH2), 26.44 (CH2).
General procedure for the preparation of 5a–5g
Propargyl bromide was slowly added to a suspension of activated Mg and HgCl2 in Et2O to maintain a gentle reflux. After the end of the addition, stirring was continued until all the Mg was consumed. Et2O was added and the reaction mixture was cooled to −78°C. A solution of 4a–4g (or 22a, 22b) in Et2O was then added dropwise and the reaction mixture was left warming up slowly to room temperature. A saturated aqueous solution of NH4Cl was added to quench the reaction, the organic layer was separated, and the aqueous phase was extracted with Et2O. The collected organic phases were washed with a saturated solution of NH4Cl, dried over MgSO4, and evaporated to dryness. The crude product was purified by column chromatography on silica gel.
1-(2-Pentyloxy-quinolin-3-yl)-but-3-yn-1-ol (5a)
Compound was obtained with Mg (302 mg, 12.6 mmol), HgCl2 (34 mg, 0.13 mmol), propargyl bromide (1.52 mL, 13.7 mmol), 4a (2.55 g, 10.5 mmol) in 13 mL Et2O, and Et2O (30 mL). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded an off-white solid (2.90 g, 96% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.10 (s, 1H, H-Ar), 7.82 (dd, J = 8.3, 1.2 Hz, 1H, H-Ar), 7.74 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.60 (ddd, J = 8.3, 6.9, 1.4 Hz, 1H, H-Ar), 7.36 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H, H-Ar), 5.13–5.10 (m, 1H, CHOH), 4.45 (t, J = 6.6 Hz, 2H, OCH2), 2.98 (d, J = 5.8 Hz, 1H, CHOH), 2.91 (ddd, J = 16.8, 5.0, 2.7 Hz, 1H, CH2C≡C), 2.69 (ddd, J = 16.8, 7.0, 2.7 Hz, 1H, CH2C≡C), 2.10 (t, J = 2.7 Hz, 1H, C≡CH), 2.00–1.80 (m, 2H, OCH2CH2), 1.61–1.35 (m, 4H, CH2CH2CH3), 0.99 (t, J = 7.0 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.03 (C), 145.84 (C), 135.00 (CH), 129.35 (CH), 127.65 (C), 126.83 (CH), 126.10 (CH), 125.02 (CH), 124.20 (C), 80.56 (C), 71.18 (CH), 68.26 (CH), 66.24 (CH2), 28.64 (CH2), 28.44 (CH2), 27.17 (CH2), 22.45 (CH2), 14.06 (CH3).
1-(2-Methoxy-quinolin-3-yl)-but-3-yn-1-ol (5b)
Compound was obtained with Mg (65 mg, 2.68 mmol), HgCl2 (8 mg, 0.03 mmol), propargyl bromide (315 µL, 2.9 mmol), 4b (417 mg, 2.23 mmol) in 1.5 mL Et2O, and Et2O (3 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a white solid (393 mg, 78% yield).
M.p.: 146–148°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.84 (dd, J = 8.4, 1.2 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.61 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.39 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.09–5.15 (m, 1H, CHOH), 4.11 (s, 3H, ArOCH3), 2.92 (d, J = 5.5 Hz, 1H, CHOH), 2.9 (ddd, J = 16.8, 4.8, 2.7 Hz, 1H, CH2C≡C), 2.67 (ddd, J = 16.8, 7.1, 2.7 Hz, 1H, CH2C≡C), 2.07 (t, J = 2.7 Hz, 1H, C≡CH); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.17 (C), 145.76 (C), 135.05 (CH), 129.42 (CH), 127.66 (C), 126.85 (CH), 125.98 (CH), 125.09 (CH), 124.32 (C), 80.50 (C), 71.26 (CH), 68.13 (CH), 53.57 (CH3), 27.18 (CH2).
1-(2-Octyloxy-quinolin-3-yl)-but-3-yn-1-ol (5c)
Compound was obtained with Mg (56 mg, 2.30 mmol), HgCl2 (6 mg, 0.02 mmol), propargyl bromide (280 µL, 2.50 mmol), 4c (550 mg, 1.93 mmol) in 3 mL Et2O, and Et2O (17 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a white solid (484 mg, 77% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.85 (d, J = 8.3 Hz, 1H, H-Ar), 7.73 (d, J = 8.0 Hz, 1H, H-Ar), 7.61 (dd, J = 8.3, 7.1 Hz, 1H, H-Ar), 7.39 (dd, J = 8.0, 7.1 Hz, 1H, H-Ar), 5.20–5.08 (m, 1H, CHOH), 4.52 (t, J = 6.6 Hz, 2H, OCH2), 3.01 (s, 1H, CHOH), 2.94 (ddd, J = 16.7, 5.0, 2.6 Hz, 1H, CH2C≡C), 2.69 (ddd, J = 16.7, 7.0, 2.6 Hz, 1H, CH2C≡C), 2.06 (t, J = 2.6 Hz, 1H, C≡CH), 1.92–1.77 (m, 2H, OCH2CH2), 1.58–1.20 (m, 10H, (CH2)5CH3), 0.94–0.90 (m, 3H, CH3); 13C NMR: (75 MHz, CDCl3) δ (ppm): 159.03 (C), 145.78 (C), 135.06 (CH), 129.39 (CH), 127.66 (C), 126.80 (CH), 126.03 (CH), 125.01 (CH), 124.23 (C), 80.50 (C), 71.23 (CH), 68.36 (CH), 66.34 (CH2), 31.83 (CH2), 29.34 (CH2), 29.25 (CH2), 28.96 (CH2), 27.19 (CH2), 26.27 (CH2), 22.68 (CH2), 14.13 (CH3).
1-[2-(3-Methoxy-propoxy)-quinolin-3-yl]-but-3-yn-1-ol (5d)
Compound was obtained with Mg (95 mg, 3.92 mmol), HgCl2 (11 mg, 0.04 mmol), propargyl bromide (445 µL, 4.25 mmol), 4d (801 mg, 3.26 mmol) in 5 mL Et2O, and Et2O (33 mL). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded a white solid (830 mg, 89% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 7.99 (s, 1H, H-Ar), 7.72 (d, J = 8.4 Hz, 1H, H-Ar), 7.66 (d, J = 8.0 Hz, 1H, H-Ar), 7.52 (dd, J = 8.4, 7.0 Hz, 1H, H-Ar), 7.30 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.02–4.92 (m, 1H, CHOH), 4.56 (t, J = 6.1 Hz, 2H, OCH2), 4.12 (d, J = 6.6 Hz, 1H, CHOH), 3.44 (t, J = 6.0 Hz, 2H, CH2OCH3), 3.31 (s, 3H, OCH3), 2.82 (ddd, J = 16.7, 5.8, 2.6 Hz, 1H, CH2C≡C), 2.68 (ddd, J = 16.7, 6.8, 2.6 Hz, 1H, CH2C≡C), 2.13–2.01 (m, 2H, OCH2CH2), 2.00 (t, J = 2.6 Hz, 1H, C≡CH); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.00 (C), 145.81 (C), 135.31 (CH), 129.36 (CH), 127.63 (C), 126.85 (CH), 125.95 (CH), 125.05 (CH), 124.25 (C), 80.65 (C), 71.06 (CH), 70.59 (CH2), 69.01 (CH), 64.13 (CH2), 58.77 (CH3), 29.21 (CH2), 26.85 (CH2).
1-[2-(4,4,4-Trifluoro-butoxy)quinolin-3-yl]-but-3-yn-1-ol (5e)
Compound was obtained with Mg (103 mg, 4.24 mmol), HgCl2 (12 mg, 0.04 mmol), propargyl bromide (480 µL, 4.59 mmol), 4e (1.0 g, 3.53 mmol) in 5 mL Et2O, and Et2O (34 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded an off-white solid (990 mg, 87% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H, H-Ar), 7.83 (d, J = 8.4 Hz, 1H, H-Ar), 7.77 (d, J = 8.0 Hz, 1H, H-Ar), 7.63 (dd, J = 8.4, 7.0 Hz, 1H, H-Ar), 7.41 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.22–5.11 (m, 1H, CHOH), 4.61 (t, J = 6.0 Hz, 2H, OCH2), 2.91 (ddd, J = 16.7, 4.7, 2.6 Hz, 1H, CH2C≡C), 2.77 (d, J = 5.2 Hz, 1H, CHOH), 2.67 (ddd, J = 16.7, 7.1, 2.6 Hz, 1H, CH2C≡C), 2.42-2.23 (m, 2H, CH2CF3), 2.08 (t, J = 2.6 Hz, 1H, C≡CH), 2.22–2.06 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.76 (C), 146.00 (C), 135.60 (CH), 129.95 (CH), 128.12 (C), 127.59 (q, J = 271.4 Hz, CF3), 127.21 (CH), 126.68 (CH), 125.58 (CH), 124.88 (C), 80.82 (C), 71.74 (CH), 67.74 (CH2), 64.76 (CH), 31.27 (q, J = 29.1 Hz, CH2CF3), 27.71 (CH2), 22.21 (CH2).
1-[2-(5-Cyclopropylpentyloxy)quinolin-3-yl]-but-3-yn-1-ol (5f)
Compound was obtained with Mg (344 mg, 14.16 mmol), HgCl2 (29 mg, 0.11 mmol), propargyl bromide (1.69 mL, 15.19 mmol), 4f (2.87 g, 10.12 mmol) in 5 mL Et2O, and Et2O (20 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a still impure 5f as a yellow oil (1.61 g), used without any further purification for the next step. HRMS: calcd. for C18H22NO2 (M+) 323.18853; Found 323.1872 (4 ppm).
1-[2-(5-Cyclohexylpentyloxy)quinolin-3-yl]-but-3-yn-1-ol (5g)
Compound was obtained with Mg (184 mg, 7.57 mmol), HgCl2 (17 mg, 0.064 mmol), propargyl bromide (903 µL, 8.13 mmol), 4g (1.76 g, 5.41 mmol) in 5 mL Et2O, and Et2O (20 mL). Column chromatography on silica gel (EtOAc/cyclohexane, 15:85 v/v) afforded a white solid (1.42 g, 72% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.09 (s, 1H, H-Ar), 7.82 (d, J = 8.3 Hz, 1H, H-Ar), 7.77–7.69 (m, 1H, H-Ar), 7.65–7.55 (m, 1H, H-Ar), 7.42–7.33 (m, 1H, H-Ar), 5.15–5.06 (m, 1H, CHOH), 4.52 (t, J = 7.2 Hz, 2H, OCH2), 2.97–2.86 (m, 1H, CH2C≡CH), 2.95 (d, J = 5.9 Hz, 1H, CHOH), 2.61 (ddd, J = 16.7, 7.0, 2.6 Hz, 1H, CH2C≡CH), 2.06, (t, J = 2.6 Hz, 1H, C≡CH), 1.89–0.78 (m, 19H, Cyclohexyl(CH2)4CH2O); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.03 (C), 145.85 (C), 135.00 (CH), 129.34 (CH), 127.64 (C), 126.84 (CH), 125.99 (CH), 125.01 (CH), 124.19 (C), 80.50 (C), 71.23 (CH), 68.41 (CH), 66.25 (CH2), 37.63 (CH), 37.43 (CH2), 33.45 (CH2), 28.99 (CH2), 27.19 (CH2), 26.76 (CH2), 26.58 (CH2), 26.51 (CH2), 26.45 (CH2).
General procedure for the preparation of 6a–6g
To a stirred solution at 0°C of 5a–5g (or 23a, 23b) in dimethylformamide (DMF) was added imidazole followed by t-butyldimethylsilyl chloride (TBDMSCl). The cooling bath was then removed and the reaction was stirred overnight. It was then quenched with brine and Et2O was added. The organic layer was separated and the aqueous phase was extracted with Et2O. The collected organic phases were washed with water and then with brine, dried over MgSO4, and evaporated to dryness. The crude product was purified by column chromatography on silica gel.
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-pentyloxy-quinoline (6a)
Compound was obtained with 5a (3.0 g, 10.5 mmol), imidazole (1.77 g, 26.3 mmol), TBDMSCl (2.3 g, 15.8 mmol), and DMF (56 mL). Column chromatography on silica gel (EtOAc/pentane, 2:98 v/v) afforded a colorless oil (4.20 g, 91% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.16 (s, 1H, H-Ar), 7.80 (dd, J = 8.3, 1.2 Hz, 1H, H-Ar), 7.73 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.57 (ddd, J = 8.3, 6.9, 1.5 Hz, 1H, H-Ar), 7.36 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H, H-Ar), 5.21–5.18 (m, 1H, CHOTBDMS), 4.45 (t, J = 6.6 Hz, 1H, OCH2), 2.70 (ddd, J = 16.7, 3.8, 2.6 Hz, 1H, CH2C≡C), 2.52 (ddd, J = 16.7, 6.9, 2.6 Hz, 1H, CH2C≡C), 1.93 (t, J = 2.6 Hz, 1H, C≡CH), 1.90–1.80 (m, 2H, OCH2CH2), 1.53–1.35 (m, 4H, CH2CH2CH3), 0.94 (t, J = 6.9 Hz, 3H, CH3), 0.93 (s, 9H, tBuSi), 0.15 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 158.84 (C), 145.84 (C), 135.16 (CH), 129.04 (CH), 128.11 (C), 127.63 (CH), 126.77 (CH), 125.20 (CH), 123.90 (C), 81.87 (C), 69.87 (CH), 67.70 (CH), 66.01 (CH2), 28.64 (CH2), 28.63 (CH2), 28.45 (CH2), 25.85 (3C, CH3), 22.45 (CH2), 18.36 (C), 14.09 (CH3), −4.77 (CH3), −4.85 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-methoxy-quinoline (6b)
Compound was obtained with 5b (390 mg, 1.72 mmol), imidazole (293 mg, 4.30 mmol), TBDMSCl (310 mg, 2.06 mmol), and DMF (2 mL). Column chromatography on silica gel (EtOAc/pentane, 3:97 v/v) afforded a colorless oil (414 mg, 70% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.17 (s, 1H, H-Ar), 7.84 (dd, J = 8.4, 1.1 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.6 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H, H-Ar), 7.38 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H, H-Ar), 5.20 (dd, J = 6.9, 3.8 Hz, 1H, CHOTBDMS), 4.10 (s, 3H, OCH3), 2.71 (ddd, J = 16.7, 3.8, 2.6 Hz, 1H, CH2C≡C), 2.53 (ddd, J = 16.7, 6.9, 2.6 Hz, 1H, CH2C≡C), 1.94 (t, J = 2.6 Hz, 1H, C≡CH), 0.95 (s, 9H, tBuSi), 0.15 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.02 (C), 145.75 (C), 135.20 (CH), 129.11 (CH), 128.00 (C), 127.65 (CH), 126.79 (CH), 125.30 (CH), 124.03 (C), 81.58 (C), 69.94 (CH), 67.67 (CH), 53.47 (CH3), 28.58 (CH2), 25.86 (3C, CH3), 18.35 (C), −4.76 (CH3), −4.84 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-octyloxy-quinoline (6c)
Compound was obtained with 5c (480 mg, 1.47 mmol), imidazole (250 mg, 3.67 mmol), TBDMSCl (295 mg, 1.92 mmol), and DMF (4 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a colorless oil (453 mg, 70% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H, H-Ar), 7.82 (d, J = 8.4 Hz, 1H, H-Ar), 7.76 (d, J = 8.0 Hz, 1H, H-Ar), 7.59 (dd, J = 8.4, 7.0 Hz, 1H, H-Ar), 7.38 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.28–5.20 (m, 1H, CHOTBDMS), 4.52 (t, J = 6.6 Hz, 2H, OCH2), 2.73 (ddd, J = 16.6, 5.0, 2.6 Hz, 1H, CH2C≡C), 2.56 (ddd, J = 16.6, 6.8, 2.6 Hz, 1H, CH2C≡C), 1.94 (t, J = 2.6 Hz, 1H, C≡CH), 1.91–1.80 (m, 2H, OCH2CH2), 1.61–1.24 (m, 10H, (CH2)5CH3), 0.97 (s, 9H, tBuSi), 0.89–0.86 (m, 3H, CH3), 0.18 (s, 3H, CH3Si), 0.01 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.84 (C), 145.76 (C), 135.23 (CH), 129.06 (CH), 128.11 (C), 127.62 (CH), 126.71 (CH), 125.18 (CH), 123.90 (C), 81.53 (C), 77.21 (CH), 69.88 (CH), 67.70 (CH), 31.81 (CH2), 29.33 (CH2), 29.28 (CH2), 28.95 (CH2), 28.62 (CH2), 26.23 (CH2), 25.86 (3C, CH3), 22.68 (CH2), 18.35 (C), 14.10 (CH3), −4.77 (CH3), −4.86 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-(3-methoxy-propoxy)-quinoline (6d)
Compound was obtained with 5d (801 mg, 2.84 mmol), imidazole (484 mg, 7.10 mmol), TBDMSCl (568 mg, 3.69 mmol), and DMF (7.5 mL). Column chromatography on silica gel (EtOAc/pentane, 15:85 v/v) afforded a colorless oil (1.04 g, 92% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.19 (s, 1H, H-Ar), 7.89 (d, J = 8.1 Hz, 1H, H-Ar), 7.82 (d, J = 8.4 Hz, 1H, H-Ar), 7.62 (dd, J = 8.4, 6.8 Hz, 1H, H-Ar), 7.39 (dd, J = 8.1, 6.8 Hz, 1H, H-Ar), 5.28–5.22 (m, 1H, CHOTBDMS), 4.67 (t, J = 6.3 Hz, 2H, OCH2), 3.62 (t, J = 6.4 Hz, 2H, CH2OCH3), 3.41 (s, 3H, OCH3), 2.75 (ddd, J = 16.3, 4.5, 2.6 Hz, 1H, CH2C≡C), 2.58 (ddd, J = 16.3, 6.8, 2.6 Hz, 1H, CH2C≡C), 2.23–2.07 (m, 2H, OCH2CH2), 2.00 (t, J = 2.6 Hz, 1H, C≡CH), 1.01 (s, 9H, tBuSi), 0.21 (s, 3H, CH3Si), 0.08 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.52 (C), 145.78 (C), 135.18 (CH), 129.06 (CH), 127.96 (C), 127.58 (CH), 126.83 (CH), 125.21 (CH), 123.98 (C), 81.48 (C), 70.05 (CH), 69.59 (CH2), 67.73 (CH), 63.03 (CH2), 58.66 (CH3), 29.26 (CH2), 28.66 (CH2), 25.85 (3C, CH3), 18.32 (C), −4.77 (CH3), −4.83 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-(4,4,4-trifluoro-butoxy)-quinoline (6e)
Compound was obtained with 5e (960 mg, 2.97 mmol), imidazole (505 mg, 7.42 mmol), TBDMSCl (594 mg, 3.86 mmol), and DMF (8 mL). Column chromatography on silica gel (EtOAc/pentane, 15:85 v/v) afforded a colorless oil (1.22 g, 94% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.27 (s, 1H, H-Ar), 7.88 (d, J = 8.3 Hz, 1H, H-Ar), 7.81 (d, J = 7.9 Hz, 1H, H-Ar), 7.61 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.43 (dd, J = 7.9, 7.0 Hz, 1H, H-Ar), 5.33–5.26 (m, 1H, CHOTBDMS), 4.53 (t, 2H, J = 6.1 Hz, OCH2), 2.90–2.52 (m, 2H, CH2C≡C), 2.50–2.27 (m, 2H, CH2CF3), 2.27–2.12 (m, 2H, OCH2CH2), 2.03 (t, J = 2.6 Hz, 1H, C≡CH), 1.04 (s, 9H, tBuSi), 0.27 (s, 3H, CH3Si), 0.12 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.28 (C), 145.77 (C), 135.63 (CH), 129.32 (CH), 127.93 (C), 127.72 (CH), 127.21 (q, J = 276.1 Hz, CF3), 126.97 (CH), 125.45 (CH), 124.31 (C), 81.30 (C), 70.24 (CH), 67.89 (CH), 64.17 (CH2), 31.01 (q, J = 29.2 Hz, CH2CF3), 28.91 (CH2), 25.87 (3C, CH3), 22.01 (CH2), 18.38 (C), −4.78 (CH3), −4.83 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-(5-cyclopropylpentyloxy)-quinoline (6f)
Compound was obtained with 5f (1.5 g, 4.65 mmol), imidazole (792 mg, 11.62 mmol), TBDMSCl (912 mg, 6.05 mmol), and DMF (20 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90, then 30:70 v/v) afforded a not-pure yellow oil (1.22 g) used without any further purification for the next step.
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-(5-cyclohexylpentyloxy)-quinoline (6g)
Compound was obtained with 5g (1.5 g, 4.10 mmol), imidazole (698 mg, 10.25 mmol), TBDMSCl (1.23 g, 8.2 mmol), and DMF (10 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80, then 30:70 v/v) afforded a colorless oil (1.66 g, 85% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.17 (s, 1H, H-Ar), 7.83 (d, J = 8.3 Hz, 1H, H-Ar), 7.75 (d, J = 8.0 Hz, 1H, H-Ar), 7.63–7.55 (m, 1H, H-Ar), 7.41–7.33 (m, 1H, H-Ar), 5.23 (dd, J = 6.4, 3.7 Hz, 1H, CHOH), 4.51 (t, J = 6.4 Hz, 2H, OCH2), 2.74 (ddd, J = 16.6, 3.7, 2.5 Hz, 1H, CH2C≡CH), 2.55 (ddd, J = 16.6, 6.7, 2.5 Hz, 1H, CH2C≡CH), 1.94 (t, J = 2.5 Hz, 1H, C≡CH), 1.91-0.79 (m, 19H, Cyclohexyl(CH2)4CH2O), 0.97 (s, 9H, tBuSi), 0.15 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.87 (C), 145.89 (C), 135.18 (CH), 129.04 (CH), 128.12 (C), 127.63 (CH), 126.82 (CH), 125.23 (CH), 123.89 (C), 81.59 (C), 69.92 (CH), 67.73 (CH), 66.02 (CH2), 37.65 (CH), 37.52 (CH2), 33.49 (CH2), 29.04 (CH2), 28.67 (CH2), 26.80 (CH2), 26.64 (CH2), 26.60 (CH2), 26.48 (CH2), 25.91 (3C, CH3), 18.39 (C), −4.71 (CH3), −4.79 (CH3).
General procedure for the preparation of 7a–7g
To a stirred solution of 6a–6g (or 24a, 24b) in THF at −78°C was added dropwise n-BuLi 1.6 M in THF. Stirring was continued for an additional 30 min, and hexamethylphosphoramide (HMPA) and trimethyl 4-bromoorthobutyrate were then added. The reaction was stirred overnight while the temperature was slowly raised to room temperature. A saturated solution of NH4Cl was added to quench the reaction, the organic layer was separated, and the aqueous phase was extracted with Et2O. The collected organic phases were washed with a saturated solution of NH4Cl, dried over MgSO4, and filtered over Celite. The filtrate was evaporated to dryness and the crude product was purified by column chromatography on silica gel.
8-(t-Butyl-dimethyl-silanyloxy)-8-(2-pentyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (7a)
Compound was obtained with 6a (749 mg, 1.88 mmol), n-BuLi (2.3 mL, 2.26 mmol), trimethyl 4-bromoorthobutyrate (395 µL, 2.26 mmol), THF (2 mL), and HMPA (2 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a pale yellow oil (636 mg, 68% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.14 (s, 1H, H-Ar), 7.81 (d, J = 8.4 Hz, 1H, H-Ar), 7.74 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.58 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H, H-Ar), 7.36 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H, H-Ar), 5.17 (ddd, J = 6.7, 3.9, 0.8 Hz, 1H, CHOTBDMS), 4.50 (t, J = 6.6 Hz, 2H, OCH2), 3.67 (s, 3H, CO2CH3), 2.67 (ddd, J = 16.5, 3.9, 2.3 Hz, 1H, CH2C≡C), 2.49 (ddd, J = 16.5, 6.7, 2.3 Hz, 1H, CH2C≡C), 2.39 (t, J = 7.6 Hz, 2H, CH2CO2CH3), 2.21–2.15 (m, 2H, C≡CCH2), 1.89–1.80 (m, 2H, OCH2CH2), 1.77 (tt, J = 7.6, 7.0 Hz, 2H, CH2CH2CO2CH3), 1.52–1.37 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3), 0.94 (s, 9H, tBuSi), 0.13 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.78 (C), 159.91 (C), 145.76 (C), 135.08 (CH), 128.92 (CH), 128.48 (C), 127.56 (CH), 126.72 (CH), 125.24 (CH), 123.82 (C), 80.47 (C), 78.17 (C), 68.04 (CH), 65.95 (CH2), 51.49 (CH3), 32.78 (CH2), 28.86 (CH2), 28.62 (CH2), 28.45 (CH2), 25.82 (3C, CH3), 24.05 (CH2), 22.44 (CH2), 18.35 (C), 18.27 (CH2), 14.10 (CH3), −4.81 (CH3), −4.91 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-(2-methoxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (7b)
Compound was obtained with 6b (150 mg, 0.44 mmol), n-BuLi (365 µL, 0.53 mmol), trimethyl 4-bromoorthobutyrate (92 µL, 0.53 mmol), THF (0.5 mL), and HMPA (0.5 mL). Column chromatography on silica gel (EtOAc/pentane, 2:98 v/v) afforded a pale yellow oil (75 mg, 39% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.15 (s, 1H, H-Ar), 7.82 (d, J = 8.5 Hz, 1H, H-Ar), 7.75 (dd, J = 8.1, 1.5 Hz, 1H, H-Ar), 7.59 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H, H-Ar), 7.37 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H, H-Ar), 5.16 (dd, J = 6.8, 3.9 Hz, 1H, CHOTBDMS), 4.08 (s, 3H, OCH3), 3.65 (s, 3H, CO2CH3), 2.65 (ddd, J = 16.5, 3.9, 2.4 Hz, 1H, CH2C≡C), 2.47 (ddd, J = 16.5, 6.8, 2.4 Hz, 1H, CH2C≡C), 2.39 (t, J = 7.5 Hz, 2H, CH2CO2CH3), 2.20–2.13 (m, 2H, C≡CCH2), 1.77–1.70 (m, 2H, CH2CH2CO2CH3), 0.95 (s, 9H, tBuSi), 0.13 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.78 (C), 159.10 (C), 145.68 (C), 135.15 (CH), 129.00 (CH), 128.41 (C), 127.59 (CH), 126.75 (CH), 125.34 (CH), 123.98 (C), 80.56 (C), 78.15 (C), 67.94 (CH), 53.46 (CH3), 51.51 (CH3), 32.81 (CH2), 28.85 (CH2), 25.97 (CH2), 25.81 (3C, CH3), 24.04 (CH2), 18.33 (C), −4.79 (CH3), −4.89 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-(2-octyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (7c)
Compound was obtained with 6c (443 mg, 1.00 mmol), n-BuLi (755 µL, 1.20 mmol), trimethyl 4-bromoorthobutyrate (250 µL, 1.30 mmol), THF (3 mL), and HMPA (3 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a colorless oil (410 mg, 76% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H, H-Ar), 7.84 (d, J = 8.3 Hz, 1H, H-Ar), 7.76 (d, J = 8.0 Hz, 1H, H-Ar), 7.58 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.39 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.28–5.16 (m, 1H, CHOTBDMS), 4.52 (t, J = 6.5 Hz, 2H, OCH2), 3.67 (s, 3H, CO2CH3), 2.80–2.34 (m, 2H, CH2C≡C), 2.40 (t, J = 7.5 Hz, 2H, CH2CO2CH3), 2.28–2.14 (m, 2H, C≡CCH2), 1.93–1.64 (m, 4H, CH2CH2CO2CH3, OCH2CH2), 1.61–1.22 (m, 10H, (CH2)5CH3), 1.00 (s, 9H, tBuSi), 0.95–0.92 (m, 3H, CH3), 0.18 (s, 3H, CH3Si), 0.04 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.64 (C), 158.86 (C), 145.80 (C), 135.09 (CH), 128.90 (CH), 128.43 (C), 127.54 (CH), 126.78 (CH), 125.24 (CH), 123.82 (C), 80.48 (C), 78.13 (C), 68.06 (CH), 65.92 (CH2), 51.39 (CH3), 32.73 (CH2), 31.83 (CH2), 29.33 (CH2), 28.95 (CH2), 28.87 (CH2), 26.26 (CH2), 25.81 (3C, CH3), 24.07 (CH2), 22.70 (CH2), 20.96 (CH2), 18.33 (C), 18.26 (CH2), 14.17 (CH3), −4.81 (CH3), −4.91 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-[2-(3-methoxy-propoxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (7d)
Compound was obtained with 6d (960 mg, 2.40 mmol), n-BuLi (1.8 mL, 2.88 mmol), trimethyl 4-bromoorthobutyrate (570 µL, 3.12 mmol), THF (8 mL), and HMPA (8 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a pale yellow oil (460 mg, 38% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.15 (s, 1H, H-Ar), 7.80 (d, J = 8.3 Hz, 1H, H-Ar), 7.72 (d, J = 7.9 Hz, 1H, H-Ar), 7.59 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.32 (dd, J = 7.9, 7.0 Hz, 1H, H-Ar), 5.29–5.13 (m, 1H, CHOTBDMS), 4.59 (t, J = 6.3 Hz, 2H, OCH2), 3.62 (s, 3H, CO2CH3), 3.58 (t, J = 6.3 Hz, 2H, CH2OCH3), 3.38 (s, 3H, OCH3), 2.39 (t, J = 7.4 Hz, 1H, CH2CO2CH3), 2.78–2.27 (m, 2H, CH2C≡C), 2.21–2.02 (m, 4H, C≡CCH2CH2), 1.81–1.62 (m, 2H, OCH2CH2), 0.98 (s, 9H, tBuSi), 0.17 (s, 3H, CH3Si), –0.01 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.54 (C), 158.56 (C), 145.73 (C), 135.10 (CH), 128.93 (CH), 128.30 (C), 127.51 (CH), 126.83 (CH), 125.25 (CH), 123.91 (C), 80.54 (C), 78.11 (C), 69.58 (CH2), 68.10 (CH), 62.96 (CH2), 58.62 (CH3), 51.36 (CH3), 32.68 (CH2), 29.29 (CH2), 28.89 (CH2), 25.80 (3C, CH3), 24.04 (CH2), 18.30 (C), 18.22 (CH2), −4.81 (CH3), –4.91 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-[2-(4,4,4-trifluoro-butoxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (7e)
Compound was obtained with 6e (1.18 g, 2.69 mmol), n-BuLi (2.02 mL, 3.23 mmol), trimethyl 4-bromoorthobutyrate (640 µL, 3.50 mmol), THF (9 mL), and HMPA (9 mL). Column chromatography on silica gel (EtOAc/pentane, 12:88 v/v) afforded a pale yellow oil (1.29 g, 89% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H, H-Ar), 7.81 (d, J = 8.3 Hz, 1H, H-Ar), 7.76 (d, J = 8.0 Hz, 1H, H-Ar), 7.60 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.39 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.22–5.13 (m, 1H, CHOTBDMS), 4.61 (t, J = 6.4 Hz, 2H, OCH2), 3.69 (s, 3H, CO2CH3), 2.68 (ddd, J = 16.5, 4.1, 2.2 Hz, 1H, CH2C≡C), 2.51 (ddd, J = 16.5, 6.6, 2.2 Hz, 1H, CH2C≡C), 2.38 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.45–2.24 (m, 2H, CH2CF3), 2.24–2.09 (m, 4H, C≡CCH2, OCH2CH2), 1.82–1.70 (m, 2H, CH2CH2CO2CH3), 0.96 (s, 9H, t-BuSi), 0.16 (s, 3H, CH3Si), 0.02 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.66 (C), 158.27 (C), 145.56 (C), 135.53 (CH), 129.12 (CH), 128.19 (C), 127.58 (CH), 126.79 (CH), 125.39 (CH), 127.10 (q, J = 275.5 Hz, CF3), 124.17 (C), 80.72 (C), 77.87 (C), 68.18 (CH), 64.09 (CH2), 51.43 (CH3), 32.77 (CH2), 30.36 (q, J = 29.4 Hz, CH2CF3), 29.04 (CH2), 25.78 (3C, CH3), 24.04 (CH2), 21.97 (CH2), 18.30 (C), 18.22 (CH2), −4.85 (CH3), −4.92 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-[2-(5-cyclopropylpentyloxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (7f)
Compound was obtained with 6f (1.31 g, 1.94 mmol), n-BuLi (1.45 mL, 2.32 mmol), trimethyl 4-bromoorthobutyrate (473 µL, 2.59 mmol), THF (5 mL), and HMPA (5 mL). Column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) afforded a not-pure yellow oil (560 mg) used without any further purification for the next step.
8-(t-Butyl-dimethyl-silanyloxy)-8-[2-(5-cyclohexylpentyloxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (7g)
Compound was obtained with 6g (1.37 g, 2.85 mmol), n-BuLi (1.95 mL, 3.13 mmol), trimethyl 4-bromoorthobutyrate (594 µL, 3.25 mmol), THF (5 mL), and HMPA (5 mL). Column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) afforded a not-pure yellow oil (950 mg) used without any further purification for the next step.
General procedure for the preparation of 8a–8g
Tetrabutylammonium fluoride (TBAF) 1 M in THF was added to a solution of 7a–7g (or 25a, 25b) in THF and the resulting solution was stirred for 2 h at 45°C. After cooling to room temperature, the solvent was evaporated and the crude product was dissolved in EtOAc, washed with water, dried over MgSO4, and evaporated. The crude product was purified by flash chromatography to afford the pure product.
8-Hydroxy-8-(2-pentyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (8a)
Compound was obtained with 7a (689 mg, 1.39 mmol), TBAF (1.94 mL, 1.94 mmol), and THF (5 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a white solid (368 mg, 69% yield).
M.p.: 53–55°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.10 (s, 1H, H-Ar), 7.81 (dd, J = 8.3, 1.2 Hz, 1H, H-Ar), 7.74 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.59 (ddd, J = 8.3, 7.0, 1.4 Hz, 1H, H-Ar), 7.37 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.06 (ddd, J = 6.9, 5.8, 4.9 Hz, 1H, CHOH), 4.53–4.47 (m, 2H, OCH2), 3.66 (s, 3H, CO2CH3), 3.04 (d, J = 5.8 Hz, 1H, CHOH), 2.88 (ddd, J = 16.6, 4.9, 2.4 Hz, 1H, CH2C≡C), 2.62 (ddd, J = 16.6, 6.9, 2.4 Hz, 1H, CH2C≡C), 2.36 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.22 (tt, J = 6.9, 2.4 Hz, 2H, C≡CCH2), 1.88–1.81 (m, 2H, OCH2CH2), 1.77 (tt, J = 7.4, 6.9 Hz, 2H, CH2CH2CO2CH3), 1.51–1.37 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.74 (C), 159.11 (C), 145.77 (C), 134.85 (CH), 129.19 (CH), 127.58 (C), 126.79 (CH), 126.47 (CH), 125.07 (CH), 124.10 (C), 82.23 (C), 76.72 (C), 68.44 (CH), 66.16 (CH2), 51.60 (CH3), 32.80 (CH2), 28.64 (CH2), 28.43 (CH2), 27.61 (CH2), 23.92 (CH2), 22.44 (CH2), 18.23 (CH2), 14.06 (CH3); HRMS: calcd. for C22H26NO3 [M – .OCH3]+ 352.19127; Found 352.1914 (0 ppm).
8-Hydroxy-8-(2-methoxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (8b)
Compound was obtained with 7b (149 mg, 0.34 mmol), TBAF (470 µL, 0.47 mmol), and THF (1.4 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a colorless oil (91 mg, 82% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.13 (s, 1H, H-Ar), 7.85 (dd, J = 8.4, 1.0 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.61 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.39 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H, H-Ar), 5.09–5.04 (m, 1H, CHOH), 4.11 (s, 3H, OCH3), 3.67 (s, 3H, CO2CH3), 2.99 (d, J = 3.8 Hz, 1H, CHOH), 2.85 (ddd, J = 16.6, 4.8, 2.4 Hz, 1H, CH2C≡C), 2.60 (ddd, J = 16.6, 7.1, 2.4 Hz, 1H, CH2C≡C), 2.38 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.23 (tt, J = 6.9, 2.4 Hz, 2H, C≡CCH2), 1.78 (tt, J = 7.4, 6.9 Hz, 2H, CH2CH2CO2CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.75 (C), 159.27 (C), 145.68 (C), 134.90 (CH), 129.28 (CH), 127.61 (C), 126.79 (CH), 126.46 (CH), 125.17 (CH), 124.23 (C), 82.29 (C), 76.71 (C), 68.23 (CH), 53.55 (CH3), 51.63 (CH3), 32.81 (CH2), 27.67 (CH2), 23.89 (CH2), 18.21 (CH2).
8-Hydroxy-8-(2-octyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (8c)
Compound was obtained with 7c (400 mg, 0.74 mmol), TBAF (1.04 mL, 1.04 mmol), and THF (4 mL). Column chromatography on silica gel (EtOAc/pentane, 50:50 v/v) afforded a white solid (251 mg, 80% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.81 (d, J = 8.2 Hz, 1H, H-Ar), 7.74 (d, J = 7.8 Hz, 1H, H-Ar), 7.60 (dd, J = 8.2, 7.0 Hz, 1H, H-Ar), 7.38 (dd, J = 7.8, 7.0 Hz, 1H, H-Ar), 5.11–5.02 (m, 1H, CHOH), 4.52 (t, J = 6.5 Hz, 2H, OCH2), 3.66 (s, 3H, CO2CH3), 3.10 (d, J = 5.7 Hz, 1H, CHOH), 2.89 (ddd, J = 16.5, 4.4, 2.3 Hz, 1H, CH2C≡C), 2.63 (ddd, J = 16.5, 6.6, 2.3 Hz, 1H, CH2C≡C), 2.35 (t, J = 7.3 Hz, 2H, CH2CO2CH3), 2.29–2.17 (m, 2H, C≡CCH2), 1.93–1.71 (m, 4H, CH2CH2CO2CH3, OCH2CH2), 1.59–1.21 (m, 10H, (CH2)5CH3), 0.95–0.89 (m, 3H, CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.69 (C), 159.11 (C), 145.78 (C), 134.84 (CH), 129.16 (CH), 127.57 (C), 126.79 (CH), 126.54 (CH), 125.08 (CH), 124.08 (C), 82.20 (C), 79.99 (C), 68.39 (CH), 66.16 (CH2), 51.57 (CH3), 32.81 (CH2), 31.82 (CH2), 29.34 (CH2), 29.25 (CH2), 28.95 (CH2), 27.61 (CH2), 26.25 (CH2), 23.95 (CH2), 22.67 (CH2), 18.23 (CH2), 14.11 (CH3); HRMS: calcd. for C18H24NO2 [M – .C8H11O2]+ 286.18070; Found 286.1806 (0 ppm); Anal. calcd. for C26H35NO4: C, 73.38; H, 8.29; N, 3.29; Found: C, 73.44; H, 8.26; N, 3.43%.
8-Hydroxy-8-[2-(3-methoxy-propoxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (8d)
Compound was obtained with 7d (440 mg, 0.88 mmol), TBAF (1.23 mL, 1.23 mmol), and THF (3.8 mL). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded a white solid (204 mg, 60% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.06 (s, 1H, H-Ar), 7.80 (d, J = 8.3 Hz, 1H, H-Ar), 7.71 (d, J = 8.0 Hz, 1H, H-Ar), 7.59 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.42 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.10–4.93 (m, 1H, CHOH), 4.60 (t, J = 6.4 Hz, 2H, OCH2), 3.77–3.48 (m, 6H, CH2OCH3, CO2CH3, CHOH), 3.40 (s, 3H, OCH3), 2.99–2.78 (m, 1H, CH2C≡C), 2.78–2.53 (m, 1H, CH2C≡C), 2.33 (t, J = 7.1 Hz, 2H, CH2CO2CH3), 2.28–1.99 (m, 4H, C≡CCH2CH2), 1.84–1.62 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.74 (C), 159.00 (C), 145.71 (C), 135.34 (CH), 129.16 (CH), 127.58 (C), 126.80 (CH), 126.68 (CH), 125.12 (CH), 124.13 (C), 81.86 (C), 70.36 (C), 68.61 (CH), 63.79 (CH2), 58.72 (CH3), 51.55 (CH3), 32.74 (CH2), 29.18 (CH2), 27.30 (CH2), 25.70 (CH2), 23.94 (CH2), 18.21 (CH2); HRMS: calcd. for C14H16NO3 [M – .C8H11O2]+ 246.11302; Found 246.1132 (0 ppm); Anal. calcd. for C22H27NO5: C, 68.55; H, 7.06; N, 3.63; Found: C, 68.55; H, 7.30; N, 3.66%.
8-Hydroxy-8-[2-(4,4,4-trifluoro-butoxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (8e)
Compound was obtained with 7e (1.15 g, 2.14 mmol), TBAF (2.99 mL, 2.99 mmol), and THF (9.2 mL). Column chromatography on silica gel (EtOAc/pentane, 40:60 v/v) afforded a white solid (600 mg, 66% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.16 (s, 1H, H-Ar), 7.68 (d, J = 8.4 Hz, 1H, H-Ar), 7.50 (d, J = 8.1 Hz, 1H, H-Ar), 7.49 (dd, J = 8.4, 7.0 Hz, 1H, H-Ar), 7.28 (dd, J = 8.1, 7.0 Hz, 1H, H-Ar), 5.04–4.93 (m, 1H, CHOH), 4.52 (t, J = 6.3 Hz, 2H, OCH2), 3.59 (s, 3H, CO2CH3), 3.00 (d, 1H, J = 5.0 Hz, CHOH), 2.80–2.68 (m, 2H, CH2C≡C), 2.53–2.41 (m, 2H, CH2CF3), 2.21 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.30–1.91 (m, 4H, C≡CCH2CH2), 1.72–1.58 (m, 2H, OCH2CH2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.72 (C), 158.41 (C), 145.57 (C), 132.57 (CH), 129.32 (CH), 128.92 (C), 127.25 (q, J = 276.2 Hz, CF3), 126.82 (CH), 126.56 (CH), 125.29 (CH), 124.35 (C), 82.35 (C), 76.77 (C), 67.69 (CH), 64.23 (CH2), 51.55 (CH3), 32.78 (CH2), 30.93 (q, J = 29.4 Hz, CH2CF3), 27.80 (CH2), 23.99 (CH2), 21.92 (CH2), 18.18 (CH2); HRMS: calcd. for C14H13F3NO2 [M – .C8H11O2]+ 284.08984; Found 284.0901 (0 ppm); Anal. calcd. for C22H23F3NO4: C, 62.40; H, 5.71; N, 3.31; Found: C, 62.41; H, 5.86; N, 3.20%.
8-Hydroxy-8-[2-(5-cyclopropylpentyloxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (8f)
Compound was obtained with 7f (530 mg, 0.98 mmol), TBAF (1.2 mL, 1.28 mmol), and THF (9.2 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 then 20:80 v/v) afforded a white solid (360 mg).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.83 (d, J = 8.3 Hz, 1H, H-Ar), 7.75 (d, J = 7.9 Hz, 1H, H-Ar), 7.63–7.57 (m, 1H, H-Ar), 7.41–7.35 (m, 1H, H-Ar), 5.12–5.00 (m, 1H, CHOH), 4.53 (t, J = 6.4 Hz, 2H, OCH2), 3.67 (s, 3H, CO2CH3), 3.00 (s, 1H, CHOH), 2.98–2.62 (m, 2H, CH2C≡C), 2.38 (t, J = 7.3 Hz, 2H, CH2CO2CH3), 2.30–2.17 (m, 2H, C≡CCH2), 1.95–1.70 (m, 4H, OCH2CH2, C≡CCH2CH2), 1.60–1.42 (m, 4H, CH2CH2CH2CH), 1.32–1.28 (m, 2H, CH2CH), 0.78–0.60 (m, 1H, CH), 0.48–0.35 (m, 2H, CH2 cyclo), 0.06 to −0.01 (m, 2H, CH2 cyclo); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.70 (C), 159.12 (C), 145.78 (C), 134.86 (CH), 129.18 (CH), 127.58 (C), 126.80 (CH), 126.47 (CH), 125.08 (CH), 124.09 (C), 82.27 (C), 68.47 (CH), 66.16 (CH2), 51.59 (CH3), 37.64, 37.44, 33.45, 32.83 (CH2), 28.99, 27.62 (CH2), 26.76, 26.60, 26.55, 26.46, 23.95 (CH2), 18.25 (CH2); HRMS: calcd. for C18H22NO2 [M – .C8H8O2]+ 284.16505; Found 284.1648 (0 ppm); Anal. calcd. for C26H33NO4: C, 73.73; H, 7.85; N, 3.31; Found: C, 73.59; H, 7.80; N, 3.30%.
8-Hydroxy-8-[2-(5-cyclohexylpentyloxy)-quinolin-3-yl]-oct-5-ynoic acid methyl ester (8g)
Compound was obtained with 7g (920 mg, 1.5 mmol), TBAF (2 mL, 2.06 mmol), and THF (10 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 then 20:80 v/v) afforded a white solid (620 mg, 47% yield for the last two steps).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.83 (d, J = 8.3 Hz, 1H, H-Ar), 7.78–7.73 (m, 1H, H-Ar), 7.64–7.57 (m, 1H, H-Ar), 7.42–7.35 (m, 1H, H-Ar), 5.10–5.04 (m, 1H, CHOH), 4.53 (t, J = 6.4 Hz, 2H, OCH2), 3.67 (s, 3H, OCH3), 3.03 (s, 1H, CHOH), 2.90 (ddt, J = 16.5, 4.8, 2.3 Hz, 1H, CH2 C≡CCH2), 2.64 (ddt, J = 16.5, 6.8, 2.3 Hz, 1H, CH2 C≡C CH2), 2.38 (t, J = 7.3 Hz, 2H, CH2CO2CH3), 2.28–2.18 (m, 2H, C≡CCH2), 1.91–0.80 (m, 21H, Cyclohexyl(CH2)4CH2O, CH2CH2CO2CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.70 (C), 159.12 (C), 145.78 (C), 134.86 (CH), 129.18 (CH), 127.58 (C), 126.80 (CH), 126.47 (CH), 125.08 (CH), 82.27 (C), 68.47 (CH), 66.16 (CH2), 51.59 (CH3), 37.64 (CH), 37.44 (CH2), 33.45 (CH2), 32.83 (CH2), 28.99 (CH2), 27.62 (CH2), 26.76 (CH2), 26.60 (CH2), 26.56 (CH2), 26.45 (CH2), 23.95 (CH2), 18.25 (CH2); HRMS: calcd. for C19H20NO4 [M – .C10H19]+ 326.1392; Found 326.1393 (0 ppm); Anal. calcd. for C29H39NO4: C, 74.81; H, 8.44; N, 3.01; Found: C, 75.04; H, 8.58; N, 3.14%.
General procedure for the preparation of 9a–9g
To a stirred solution of 8a–8g (or 26a, 26b) in MeOH/water (9:1 v/v) was added LiOH·H2O, and the resulting suspension was stirred during 48 h. Oxalic acid was then added and the suspension was stirred for an additional 15 min. Solvents were evaporated and the crude product was dissolved in EtOAc, and washed with a minimum amount of water. The collected organic phases were washed with a small amount of water, dried over MgSO4, and evaporated. The crude product was purified by flash chromatography. The acid was then dissolved in MeOH and NaOH was added. The resulting suspension was stirred until all the NaOH was consumed. The solvent was then evaporated under reduced pressure to give the corresponding sodium salt.
Sodium 8-hydroxy-8-(2-pentyloxy-quinolin-3-yl)-oct-5-ynoate (9a)
Acid was prepared with 8a (100 mg, 0.26 mmol), LiOH·H2O (38 mg, 0.90 mmol), oxalic acid (123 mg, 1.36 mmol), and MeOH/water (5.9 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a white solid (90 mg, 92% yield). Salt was prepared with the acid (90 mg, 0.24 mmol), NaOH (10 mg, 0.24 mmol), and MeOH (1 mL). A white solid was obtained (93 mg, 99%). Anal. calcd. for C22H26NNaO4: C, 71.52; H, 7.37; N, 3.79; Found: C, 71.63; H, 7.32; N, 3.55%.
Sodium 8-hydroxy-8-(2-methoxy-quinolin-3-yl)-oct-5-ynoate (9b)
Acid was prepared with 8b (91 mg, 0.23 mmol), LiOH·H2O (41 mg, 0.98 mmol), oxalic acid (132 mg, 1.46 mmol), and MeOH/water (8.3 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc/pentane, 30:70 v/v) afforded a white solid (70 mg, 99% yield). Salt was prepared with the acid (70 mg, 0.22 mmol), NaOH (9 mg, 0.22 mmol), and MeOH (1 mL). A white solid was obtained (75 mg, 100%). HRMS: Calcd for C11H10NO2 [M – .C8H11O2]+ 188.07115; Found 188.0708 (2 ppm).
Sodium 8-hydroxy-8-(2-octyloxy-quinolin-3-yl)-oct-5-ynoate (9c)
Acid was prepared with 8c (111 mg, 0.26 mmol), LiOH·H2O (38 mg, 0.91 mmol), oxalic acid (117 mg, 1.30 mmol), and MeOH/water (6 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc) afforded a white solid (107 mg, 95% yield).
1H-NMR: (200 MHz, CDCl3) δ (ppm): 8.22 (s, 1H, H-Ar), 8.10 (s, 1H, CO2H), 7.93 (d, J = 8.3 Hz, 1H, H-Ar), 7.80 (d, J = 8.0 Hz, 1H, H-Ar), 7.65 (dd, J = 8.3, 7.0 Hz, 1H, H-Ar), 7.45 (dd, J = 8.0, 7.0 Hz, 1H, H-Ar), 5.19–5.05 (m, 1H, CHOH), 4.59 (t, J = 6.7 Hz, 2H, OCH2), 3.00–2.58 (m, 2H, CH2C≡C), 2.40 (t, J = 7.4 Hz, 2H, CH2CO2H), 2.31–2.15 (m, 2H, C≡CCH2), 1.99–1.68 (m, 4H, CH2CH2CO2CH3, OCH2CH2), 1.60–1.16 (m, 10H, (CH2)5CH3), 0.96–0.90 (m, 3H, CH3).
Salt was prepared with the acid (107 mg, 0.25 mmol), NaOH (10 mg, 0.24 mmol), and MeOH (1 mL). An off-white hygroscopic solid was obtained (108 mg, 100%).
Sodium 8-hydroxy-8-[2-(3-methoxy-propoxy)-quinolin-3-yl]-oct-5-ynoate (9d)
Acid was prepared with 8d (110 mg, 0.28 mmol), LiOH·H2O (41.9 mg, 0.98 mmol), oxalic acid (128 mg, 1.14 mmol), and MeOH/water (5 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc) afforded a pale yellow oil (98 mg, 95% yield).
1H-NMR: (200 MHz, CO(CD3)2) δ (ppm): 8.30 (s, 1H, H-Ar), 7.83 (d, J = 8.3 Hz, 1H, H-Ar), 7.80 (d, J = 8.0 Hz, 1H, H-Ar), 7.62 (dd, J = 8.3, 6.9 Hz, 1H, H-Ar), 7.41 (dd, J = 8.0, 6.9 Hz, 1H, H-Ar), 5.23–5.12 (m, 1H, CHOH), 4.59 (t, J = 6.4 Hz, 2H, OCH2), 3.59 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.33 (s, 3H, OCH3), 2.96–2.51 (m, 2H, CH2C≡C), 2.37 (t, J = 7.0 Hz, 1H, CH2CO2H), 2.28–2.02 (m, 4H, C≡CCH2CH2), 1.80–1.58 (m, 2H, OCH2CH2).
Salt was prepared with the acid (98 mg, 0.26 mmol), NaOH (10 mg, 0.25 mmol), and MeOH (0.8 mL). An off-white and hygroscopic solid was obtained (102 mg, 100%).
Sodium 8-hydroxy-8-[2-(4,4,4-trifluoro-butoxy)-quinolin-3-yl]-oct-5-ynoate (9e)
Acid was prepared with 8e (234 mg, 0.55 mmol), LiOH·H2O (81 mg, 1.93 mmol), oxalic acid (253 mg, 2.75 mmol), and MeOH/water (9 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc) afforded a pale yellow oil (197 mg, 87% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.62 (s, 1H, CO2H), 8.38 (s, 1H, H-Ar), 7.87 (d, J = 8.4 Hz, 1H, H-Ar), 7.80 (d, J = 8.0 Hz, 1H, H-Ar), 7.62 (dd, J = 8.4, 7.2 Hz, 1H, H-Ar), 7.45 (dd, J = 8.0, 7.2 Hz, 1H, H-Ar), 5.31–5.18 (m, 1H, CHOH), 4.59 (t, J = 6.1 Hz, 2H, OCH2), 2.97–2.60 (m, 2H, CH2C≡C), 2.32 (t, J = 7.4 Hz, 2H, CH2CO2H), 2.60–2.25 (m, 2H, CH2CF3), 2.23–2.05 (m, 4H, C≡CCH2CH2), 1.80–1.58 (m, 2H, OCH2CH2).
Salt was prepared with the acid (197 mg, 0.48 mmol), NaOH (19 mg, 0.47 mmol), and MeOH (1.5 mL). A white hygroscopic solid was obtained (207 mg, 100%).
Ethyl 5-cyclopropylpentanoate (11)
Trifluoroacetic acid (2.95 mL, 38.4 mmol) in 20 mL of dichloromethane (DCM) was added dropwise very slowly to a solution of diethylzinc at 0°C (1 M in hexane, 38.4 mL, 38.4 mmol) diluted in 20 mL of DCM. After this addition the solution was stirred during 20 min before adding diiodoethane (3.09 mL, 38.4 mmol) in 15 mL of DCM. Stirring was continued for 15 min, then ethyl hept-6-enoate in 15 mL of DCM was added. The cooling bath was then removed and the mixture was stirred overnight. Satured solution of NH4Cl was added and the collected organic layer was dried over MgSO4 and evaporated under vacuum to give a yellow oil. The crude product was purified by column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) to afford a colorless oil (3.23 g, 99% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.29 (t, J = 7.6 Hz, 2H, CH2CO), 1.71–1.59 (m, 2H, CH2CH2CO), 1.48–1.36 (m, 2H, CH2CH2CH2CO), 1.25 (t, J = 7.1 Hz, 3H, CH2CH3), 1.24–1.16 (m, 2H, CH2CH), 0.72–0.57 (m, 1H, CH), 0.42–0.35 (m, 2H, CH2 cyclo), 0.02 to −0.04 (m, 2H, CH2 cyclo); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.83 (C), 60.12 (CH2), 34.42 (CH2), 34.33 (CH2), 29.17 (CH2), 24.83 (CH2), 14.23 (CH3), 10.67 (CH cyclo), 4.35 (2C, CH2 cyclo).
5-Cyclopropylpentan-1-ol (12f)
To a suspension of LiAlH4 (1.38 g, 36.4 mmol) in diethylether at 0°C was added dropwise a solution of ester 11 (3.1 g, 18.2 mmol) in 2 mL of diethylether. After addition, stirring was continued for 1 h. The reaction was then quenched by a minimum amount of water and MgSO4 was added. The resulting mixture was filtered through cotton and evaporated under vacuum to give a colorless oil (2.31 g, 99% yield) used without any further purification.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 3.64 (t, J = 6.6 Hz, 2H, CH2OH), 1.64–1.50 (m, 2H, CH2CH2OH), 1.49–1.30 (m, 4H, CH2CH2CH2CH2OH), 1.26–1.13 (m, 2H, CH2CH), 0.72–0.57 (m, 1H, CH), 0.43–0.32 (m, 2H, CH2 cyclo), 0.02 to −0.06 (m, 2H, CH2 cyclo); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 64.01 (CH2), 34.70 (CH2), 32.84 (CH2), 29.45 (CH2), 25.61 (CH2), 10.81 (CH cyclo), 4.37 (2C, CH2 cyclo).
5-Cyclohexylpentan-1-ol (12g)
To a suspension of LiAlH4 (617 g, 16.27 mmol) at 0°C in THF was added dropwise a solution of 5-cyclohexylpentanoic acid (3.1 g, 18.2 mmol) in 2 mL of THF. After addition, stirring was continued for 1 h. The reaction was then quenched by a minimum amount of water and MgSO4 was added. The resulting mixture was filtered through cotton and evaporated under vacuum to give a colorless oil (1.278 g, 92% yield) used without any further purification for the next step.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 3.66 (t, J = 6.6 Hz, 2H, CH2OH), 1.85–0.72 (m, 20H, Cyclohexyl(CH2)4CH2OH); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 63.09 (CH2), 37.60 (CH), 37.48 (CH2), 33.44 (CH2), 32.83 (CH2), 26.75 (CH2), 26.65 (CH2), 26.44 (CH2), 26.05 (CH2).
8-Hydroxy-8-(2-pentyloxy-quinolin-3-yl)-oct-5-ynoic acid (2-hydroxy-ethyl)-amide (14a)
Acid (90 mg, 0.24 mmol) was prepared from 8a (100 mg, 0.26 mmol) following the general procedure described for the preparation of 9a–9g and was then solubilized in CH2Cl2 (1 mL) and triethylamine was added (34 µL, 0.24 mmol). Then the mixture was cooled to 0°C, BOPCl (bis(2-oxo-3-oxazolidinyl)phosphinic chloride) was added (61 mg, 0.24 mmol), and the cooling bath was removed. Stirring was continued for 20 min before triethylamine (34 µL, 0.24 mmol) and ethanolamine (16 µL, 0.27 mmol) were added. Stirring was continued for 1 h at room temperature and water was then added to quench the reaction. The organic layer was separated and the aqueous phase was extracted with EtOAc. The collected organic phases were washed with water, dried over MgSO4, and the filtrate was evaporated to dryness. The crude product was purified by column chromatography on silica gel (EtOAc/pentane, 50:50 v/v) to afford a colorless oil (50 mg, 50% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.13 (s, 1H, H-Ar), 7.84 (d, J = 8.4 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.61 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H, H-Ar), 7.39 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H, H-Ar), 6.10 (s, 1H, NH), 5.16 (ddd, J = 6.9, 4.9, 0.8 Hz, 1H, CHOH), 4.55–4.50 (m, 2H, OCH2), 3.69 (t, J = 5.0 Hz, 2H, NHCH2), 3.39–3.34 (m, 2H, CH2OH), 3.13–3.05 (m, 1H, CHOH), 2.85 (ddt, J = 16.7, 4.9, 2.4 Hz, 1H, CH2C≡C), 2.68 (ddt, J = 16.7, 6.9, 2.4 Hz, 1H, CH2C≡C), 2.41 (t, J = 7.3 Hz, 1H, CH2OH), 2.27–2.18 (m, 4H, C≡CCH2, CH2CONHCH2), 1.90–1.81 (m, 2H, OCH2CH2), 1.81–1.72 (m, 2H, CH2CH2CONH), 1.50–1.42 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.79 (C), 159.20 (CH), 145.77 (C), 135.14 (CH), 129.42 (CH), 127.59 (C), 126.76 (CH), 126.70 (CH), 125.03 (CH), 124.34 (C), 82.17 (C), 77.24 (C), 68.62 (CH2), 62.61 (CH), 45.82 (CH2), 42.42 (CH2), 34.77 (CH2), 28.64 (CH2), 28.42 (CH2), 27.46 (CH2), 24.12 (CH2), 22.46 (CH2), 17.91 (CH2), 14.08 (CH3); HRMS: calcd. for C15H18NO2 [M – .C9H14NO2]+ 244.13375; Found 244.1349 (4 ppm).
General procedure for the preparation of 15a, 15b
n-BuLi 1.6M in THF was added dropwise to a solution at −78°C of 6a, 6b in THF. After the end of the addition, stirring was continued for 15 min then paraformaldehyde was added. The cooling bath was then removed and the mixture was stirred overnight. Water was added to quench the reaction and EtOAc was added. The organic layer was separated and the aqueous phase was extracted with EtOAc. The collected organic phases were washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was purified by column chromatography on silica gel.
5-(t-Butyl-dimethyl-silanyloxy)-5-(2-pentyloxy-quinolin-3-yl)-pent-2-yn-1-ol (15a)
Compound was obtained with 6a (3.08 g, 7.75 mmol), n-BuLi (5.8 mL, 9.24 mmol), paraformaldehyde (462 mg, 15.4 mmol), and THF (16 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a pale yellow oil (2.59 g, 78% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.11 (s, 1H, H-Ar), 7.84 (d, J = 8.4 Hz, 1H, H-Ar), 7.76 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.60 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.37 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.21–5.17 (m, 1H, CHOTBDMS), 4.50 (t, J = 6.6 Hz, 2H, OCH2), 4.22 (dt, J = 5.6, 2.1 Hz, 2H, CH2OH), 2.75 (ddt, J = 16.7, 3.7, 2.1 Hz, 1H, CH2C≡C), 2.54 (ddt, J = 16.7, 7.2, 2.1 Hz, 1H, CH2C≡C), 1.92–1.81 (m, 2H, OCH2CH2), 1.64 (broad s, 1H, CH2OH), 1.54–1.39 (m, 4H, CH2CH2CH3), 0.98–0.93 (m, 12H, tBuSi, CH3), 0.16 (s, 3H, CH3Si), 0.03 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 158.87 (C), 145.79 (C), 135.08 (CH), 129.09 (CH), 128.31 (C), 127.58 (CH), 126.78 (CH), 125.21 (CH), 123.94 (C), 83.75 (C), 79.93 (C), 67.88 (CH), 66.05 (CH2), 51.38 (CH2), 29.04 (CH2), 28.64 (CH2), 28.45 (CH2), 25.85 (3C, CH3), 22.46 (CH2), 18.38 (C), 14.10 (CH3), −4.73 (CH3), −4.86 (CH3).
5-(t-Butyldimethylsilanyloxy)-5-(2-methoxy-quinolin-3-yl)-pent-2-yn-1-ol (15b)
Compound was obtained with 6b (228 mg, 0.67 mmol), n-BuLi (616 µL, 0.98 mmol), paraformaldehyde (30 mg, 1.00 mmol), and THF (1.3 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a colorless oil (179 mg, 72% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.16 (s, 1H, H-Ar), 7.84 (d, J = 8.4 Hz, 1H, H-Ar), 7.74 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.60 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.38 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.20 (dd, J = 7.4, 4.2 Hz, 1H, CHOTBDMS), 4.22 (t, J = 2.1 Hz, 2H, CH2OH), 4.10 (s, 3H, OCH3), 2.74 (ddt, J = 16.7, 4.2, 2.1 Hz, 1H, CH2C≡C), 2.53 (ddt, J = 16.7, 7.4, 2.1 Hz, 1H, CH2C≡C), 0.96 (s, 9H, tBuSi), 0.15 (s, 3H, CH3Si), 0.01 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 159.03 (C), 145.70 (C), 135.09 (CH), 129.17 (CH), 128.22 (C), 127.60 (CH), 126.77 (CH), 125.29 (CH), 124.01 (C), 83.88 (C), 79.90 (C), 67.79 (CH), 53.53 (CH3), 51.43 (CH2), 29.00 (CH2), 25.83 (3C, CH3), 18.36 (C), −4.73 (CH3), −4.86 (CH3).
General procedure for the preparation of 16a, 16b
To a heterogeneous solution of 15a, 15b in toluene and an aqueous solution of NaOH (25% w/v) were added tetrabutylammonium bromide and t-butyl bromoacetate. The resulting reaction mixture was stirred at room temperature for 4 h then water was added to quench the reaction. The organic layer was separated and the aqueous phase was extracted with EtOAc. The collected organic phases were washed with brine, dried over MgSO4, and evaporated to dryness. The crude product was purified by column chromatography on silica gel.
tert-Butyl 2-(5-(tert-butyldimethylsilyloxy)-5-(2-(pentyloxy)quinolin-3-yl)pent-2-ynyloxy)acetate (16a)
Compound was obtained with 15a (309 mg, 0.72 mmol), t-butyl bromoacetate (126 µL, 0.87 mmol), tetrabutylammonium bromide (16 mg, 0.05 mmol), toluene (2.2 mL), and aqueous NaOH (180 µL, 25% m/v). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a colorless oil (354 mg, 90% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.15 (s, 1H, H-Ar), 7.80 (dd, J = 8.4, 1.2 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.4 Hz, 1H, H-Ar), 7.59 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H, H-Ar), 7.37 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.20 (dd, J = 7.3, 3.9 Hz, 1H, CHOTBDMS), 4.64 (t, J = 2.1 Hz, 2H, C≡CCH2), 4.50 (t, J = 6.6 Hz, 2H, OCH2), 2.75 (ddt, J = 16.7, 3.9, 2.1 Hz, 1H, CH2C≡C), 2.54 (ddt, J = 16.7, 7.3, 2.1 Hz, 1H, CH2C≡C), 2.05 (s, 2H, CH2CO2tBu), 1.88–1.81 (m, 2H, OCH2CH2), 1.47 (s, 9H, CO2tBu), 1.46–1.41 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.2 Hz, 3H, CH3), 0.95 (s, 9H, tBuSi), 0.14 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 170.33 (C), 158.79 (C), 145.82 (C), 135.10 (CH), 129.07 (CH), 128.16 (C), 127.62 (CH), 126.74 (CH), 125.19 (CH), 123.89 (C), 84.91 (C), 82.89 (C), 67.69 (CH), 66.01 (CH2), 52.75 (C), 29.02 (CH2), 28.61 (CH2), 28.44 (CH2), 27.78 (3C, CH3), 25.81 (3C, CH3), 22.45 (CH2), 20.75 (3C, CH3, Ot-Bu), 18.34 (C), 14.10 (CH3), −4.78 (CH3), −4.96 (CH3).
tert-Butyl 2-(5-(tert-butyldimethylsilyloxy)-5-(2-methoxyquinolin-3-yl)pent-2-ynyloxy)acetate (16b)
Compound was obtained with 15b (110 mg, 0.30 mmol), t-butyl bromoacetate (52 µL, 0.44 mmol), tetrabutylammonium bromide (8 mg, 0.03 mmol), toluene (0.9 mL), and aqueous NaOH (70 µL, 25% m/v). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a colorless oil (98 mg, 68% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.16 (s, 1H, H-Ar), 7.85 (dd, J = 8.4, 1.2 Hz, 1H, H-Ar), 7.76 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.60 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.38 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.20 (dd, J = 6.9, 3.8 Hz, 1H, CHOTBDMS), 4.25 (t, J = 2.1 Hz, 2H, C≡CCH2), 4.11 (s, 3H, OCH3), 4.02 (d, J = 1.2 Hz, 2H, CH2CO2tBu), 2.76 (ddt, J = 16.7, 3.8, 2.1 Hz, 1H, CH2C≡C), 2.56 (ddt, J = 16.7, 6.9, 2.1 Hz, 1H, CH2C≡C), 1.45 (s, 9H, CO2tBu), 0.96 (s, 9H, tBuSi), 0.14 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 169.27 (C), 158.99 (C), 145.73 (C), 135.13 (CH), 129.14 (CH), 128.10 (C), 127.60 (CH), 126.79 (CH), 125.30 (CH), 124.07 (C), 85.04 (C), 81.64 (C), 67.64 (CH), 66.40 (CH2), 58.60 (CH2), 53.50 (CH3), 28.95 (CH2), 28.11 (3C, CH3), 25.82 (3C, CH3), 18.31 (C), −4.76 (CH3), −4.89 (CH3).
Preparation of 17a, 17b follows the general procedure described for compounds 8a–8g
tert-Butyl 2-(5-hydroxy-5-(2-pentyloxyquinolin-3-yl)pent-2-ynyloxy)acetate (17a)
Compound was obtained with 16a (414 mg, 0.76 mmol), TBAF (1.07 mL, 1.07 mmol), and THF (3 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a colorless oil (236 mg, 72% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.09 (s, 1H, H-Ar), 7.82 (dd, J = 8.4, 1.2 Hz, 1H, H-Ar), 7.74 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.60 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.38 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H-Ar), 5.20 (ddd, J = 7.0, 5.8, 4.8 Hz, 1H, CHOH), 4.54–4.50 (m, 2H, OCH2), 4.27 (t, J = 2.1 Hz, 2H, C≡CCH2), 2.99 (d, J = 5.8 Hz, 1H, CHOH), 2.95 (ddt, J = 16.7, 4.8, 2.1 Hz, 1H, CH2C≡C), 2.72 (ddt, J = 16.7, 7.0, 2.1 Hz, 1H, CH2C≡C), 1.90–1.80 (m, 2H, OCH2CH2), 1.47 (s, 9H, CO2tBu), 1.52–1.37 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 169.22 (C), 159.01 (C), 145.79 (C), 134.93 (CH), 129.34 (CH), 127.60 (C), 126.80 (CH), 126.14 (CH), 125.02 (CH), 124.22 (C), 81.84 (C), 78.04 (C), 68.39 (CH), 66.73 (CH2), 66.25 (CH2), 58.70 (CH2), 28.63 (CH2), 28.43 (CH2), 28.08 (CH2), 27.60 (3C, CH3), 22.44 (CH2), 14.07 (CH3).
tert-Butyl 2-(5-hydroxy-5-(2-methoxyquinolin-3-yl)pent-2-ynyloxy)acetate (17b)
Compound was obtained with 16b (98 mg, 0.20 mmol), TBAF (285 µL, 0.29 mmol), and THF (0.8 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a colorless oil (48 mg, 64% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.12 (s, 1H, H-Ar), 7.84 (dd, J = 8.4, 1.1 Hz, 1H, H-Ar), 7.75 (dd, J = 8.0, 1.5 Hz, 1H, H-Ar), 7.61 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H, H-Ar), 7.39 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H, H-Ar), 5.12 (dd, J = 6.9, 4.7 Hz, 1H, CHOH), 4.26 (t, J = 2.1 Hz, 2H, C≡CCH2), 4.11 (s, 3H, OCH3), 3.99 (s, 2H, CH2CO2t-Bu), 2.94 (ddt, J = 16.8, 4.7, 2.1 Hz, 1H, CH2C≡C), 2.68 (ddt, J = 16.8, 6.9, 2.1 Hz, 1H, CH2C≡C), 1.47 (s, 9H, CO2t-Bu); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 169.24 (C), 159.18 (C), 145.71 (C), 134.98 (CH), 129.40 (CH), 129.38 (C), 127.63 (CH), 126.82 (CH), 125.13 (CH), 124.33 (C), 83.80 (C), 81.86 (C), 68.11 (CH), 66.75 (CH2), 58.71 (CH2), 53.58 (CH3), 28.09 (CH2), 27.66 (3C, CH3).
Preparation of 18a, 18b follows the general procedure described for compounds 9a–9g
Sodium [5-hydroxy-5-(2-pentyloxy-quinolin-3-yl)-pent-2-ynyloxy]-acetate (18a)
Acid was prepared with 17a (226 mg, 0.53 mmol), NaOH (53 mg, 1.33 mmol), oxalic acid (178 mg, 1.98 mmol), and MeOH/water (12 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc/pentane, 90:10 v/v) afforded a white solid (165 mg, 84% yield). Salt was prepared with the acid (165 mg, 0.44 mmol), NaOH (17 mg, 0.43 mmol), and MeOH (1 mL). A white solid was obtained (173 mg, 99%); HRMS: calcd. for C15H18NO2 244.13375; Found 244.1325 (5 ppm).
Sodium [5-hydroxy-5-(2-methoxy-quinolin-3-yl)-pent-2-ynyloxy]-acetate (18b)
Acid was prepared with 17b (48 mg, 0.13 mmol), NaOH (13 mg, 0.33 mmol), oxalic acid (44 mg, 0.49 mmol), and MeOH/water (3 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc/pentane, 90:10 v/v) afforded a white solid (22 mg, 54% yield). Salt was prepared with the acid (22 mg, 0.07 mmol), NaOH (3 mg, 0.07 mmol), and MeOH (0.5 mL). A white hygroscopic solid was obtained (23 mg, 99%).
2-Chloro-benzo[h]quinoline-3-carbaldehyde (19b)
A solution of N-acetyl-1-naphthylamine (1.0 g, 5.4 mmol) in POCl3 (9.5 mL) and DMF (1.0 mL) was refluxed for 6 h. After cooling to room temperature, the solution was slowly poured into crushed ice. The resulting brown solid was filtered, washed with water, and solubilized in EtOAc. After filtration, the desired product, a yellow solid (540 mg, 40% yield), was obtained by recrystallization of the crude product in EtOAc/pentane.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.63 (s, 1H, CHO), 9.34–9.21 (m, 1H, H-Ar), 8.77 (s, 1H, H-Ar), 8.02–7.73 (m, 5H, H-Ar); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 189.44 (CHO) 150.08 (C), 149.28 (C), 138.84 (CH), 135.03 (C), 130.28 (C), 129.93 (CH), 129.36 (CH), 128.02 (CH), 127.85 (CH), 126.64 (CH), 125.73 (C), 125.09 (C), 125.03 (CH).
Preparation of 20a, 20b follows the general procedure described for 2
2-Chloro-3-dimethoxymethyl-6-methoxy-quinoline (20a)
Compound was obtained with 2-chloro-6-methoxyquinoline-3-carbaldehyde (1.99 g, 9.00 mmol), trimethyl orthoformate (1.18 mL, 10.8 mmol), NH4NO3 (36 mg, 0.45 mmol), and MeOH (9 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a pale yellow solid (2.22 g, 98% yield).
M.p.: 95–96°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.31 (s, 1H, H-Ar), 7.92 (d, J = 9.4 Hz, 1H, H-Ar), 7.39 (dd, J = 9.4, 2.8 Hz, 1H, H-Ar), 7.12 (d, J = 2.8 Hz, 1H, H-Ar), 5.70 (s, 1H, CH(OCH3)2), 3.93 (s, 3H, ArOCH3), 3.44 (s, 6H, CH(OCH3)2); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 158.27 (C), 146.74 (C), 143.52 (C), 136.02 (CH), 129.63 (CH), 129.41 (C), 127.90 (C), 123.55 (CH), 105.50 (CH), 100.52 (CH), 55.61 (CH3), 53.92 (2C, CH3).
2-Chloro-3-dimethoxymethyl-benzo[h]quinoline (20b)
Compound was obtained with 19b (1.21 g, 5.00 mmol), trimethyl orthoformate (660 µL, 6.00 mmol), NH4NO3 (20 mg, 0.25 mmol), MeOH (12 mL), and THF (4 mL). Column chromatography on silica gel (EtOAc) afforded a brown solid (1.20 g, 80% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.27–9.13 (m, 1H, H-Ar), 8.42 (s, 1H, H-Ar), 7.93–7.62 (m, 5H, H-Ar), 5.83 (s, 1H, CH(OCH3)2), 3.51 (s, 6H, CH(OCH3)2); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 148.75 (C), 146.58 (C), 137.35 (CH), 134.34 (C), 130.57 (CH), 130.14 (CH), 129.25 (CH), 128.78 (CH), 128.22 (CH), 127.72 (CH), 125.35 (C), 125.21 (CH), 125.16 (CH), 100.90 (CH), 54.28 (2C, CH3).
Preparation of 21a, 21b follows the general procedure described for compounds 3a–3g
3-Dimethoxymethyl-6-methoxy-2-pentyloxy-quinoline (21a)
Compound was obtained with NaH (456 mg, 11.4 mmol), 1-pentanol (1.24 mL, 11.4 mmol), 20a (2.38 g, 9.49 mmol), and NMP (7.5 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a white solid (2.06 g, 68% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.12 (s, 1H, H-Ar), 7.72 (d, J = 9.1 Hz, 1H, H-Ar), 7.27 (dd, J = 9.1, 2.9 Hz, 1H, H-Ar), 7.07 (d, J = 2.9 Hz, 1H, H-Ar), 5.64 (s, 1H, CH(OCH3)2), 4.47 (t, J = 6.7 Hz, 2H, OCH2), 3.88 (s, 3H, ArOCH3), 3.43 (s, 6H, CH(OCH3)2), 1.86–1.82 (m, 2H, OCH2CH2), 1.46–1.42 (m, 4H, CH2CH2CH3), 0.94 (t, J = 7.2 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 158.47 (C), 156.02 (C), 141.90 (C), 135.12 (CH), 128.16 (CH), 125.17 (C), 122.06 (C), 121.29 (CH), 106.55 (CH), 99.22 (CH), 65.99 (CH2), 55.48 (CH3), 53.89 (2C, CH3), 28.69 (CH2), 28.33 (CH2), 22.48 (CH2), 14.09 (CH3).
3-Dimethoxymethyl-2-pentyloxy-benzo[h]quinoline (21b)
Compound was obtained with NaH (118 mg, 2.95 mmol), 1-pentanol (310 µL, 2.95 mmol), 20b (425 mg, 1.47 mmol), and NMP (2 mL). Column chromatography on silica gel (EtOAc/pentane, 8:92 v/v) afforded a yellow oil (399 mg, 80% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.22 (d, J = 7.8 Hz, 1H, H-Ar), 8.57 (s, 1H, H-Ar), 7.89 (d, J = 8.4 Hz, 1H, H-Ar), 7.81–7.62 (m, 4H, H-Ar), 5.84 (s, 1H, CH(OCH3)2), 4.75 (t, J = 6.7 Hz, 2H, OCH2), 3.51 (s, 6H, CH(OCH3)2), 2.09–1.93 (m, 2H, OCH2CH2), 1.72–1.41 (m, 4H, CH2CH2CH3), 1.08 (t, J = 7.4 Hz, 3H, CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 159.28 (C), 144.25 (C), 136.16 (CH), 133.93 (C), 130.56 (C), 127.61 (CH), 126.14 (CH), 125.34 (CH), 124.63 (CH), 124.43 (CH), 121.44 (C), 121.03 (C), 98.98 (CH), 66.18 (CH2), 60.26, 53.56 (2C, CH3), 28.65 (CH2), 28.40 (CH2), 22.50 (CH2), 14.10 (CH3).
Preparation of 22a, 22b follows the general procedure described for compounds 4a–4g
6-Methoxy-2-pentyloxy-quinoline-3-carbaldehyde (22a)
Compound was obtained with 21a (2.06 g, 6.46 mmol), PTSA (184 mg, 0.97 mmol), and THF/H2O (43 mL, 9:1 v/v). A yellow solid (1.75 g, 99% yield) was obtained.
M.p.: 87–90°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 10.49 (s, 1H, CHO), 8.48 (s, 1H, H-Ar), 7.74 (d, J = 9.1 Hz, 1H, H-Ar), 7.38 (dd, J = 9.1, 2.8 Hz, 1H, H-Ar), 7.11 (d, J = 2.8 Hz, 1H, H-Ar), 4.54 (t, J = 6.6 Hz, 2H, OCH2), 3.96 (s, 3H, ArOCH3), 1.90–1.86 (m, 2H, OCH2CH2), 1.48–1.44 (m, 4H, CH2CH2CH3), 0.92 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 189.72 (CHO), 160.82 (C), 157.20 (C), 145.50 (C), 138.80 (CH), 129.12 (CH), 125.39 (C), 125.32 (CH), 120.43 (C), 107.69 (CH), 66.23 (CH2), 55.81 (CH3), 28.74 (CH2), 28.51 (CH2), 22.56 (CH2), 14.10 (CH3).
2-Pentyloxy-benzo[h]quinoline-3-carbaldehyde (22b)
Compound was obtained with 21b (341 mg, 1.00 mmol), PTSA (39 mg, 0.15 mmol), and THF/H2O (10 mL, 9:1 v/v). A yellow solid (249 mg, 85% yield) was obtained.
1H-NMR: (300 MHz, CDCl3) δ (ppm): 10.36 (s, 1H, CHO), 8.82 (d, J = 7.5 Hz, 1H, H-Ar), 8.27 (s, 1H, H-Ar), 7.65, (d, J = 8.3 Hz, 1H, H-Ar), 7.58–7.30 (m, 4H, H-Ar), 4.53 (t, J = 6.6 Hz, 2H, OCH2), 2.02–1.79 (m, 2H, OCH2CH2), 1.64–1.38 (m, 4H, CH2CH2CH3), 1.02 (t, J = 6.9 Hz, 3H, CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 188.91 (CHO), 161.01 (C), 147.66 (C), 137.99 (CH), 134.74 (C), 129.88, 129.01, 127.66, 126.45, 125.38, 125.37, 125.15, 121.06 (CH), 118.53 (C), 66.62 (CH2), 28.47 (CH2), 28.39 (CH2), 22.52 (CH2), 14.10 (CH3).
Preparation of 23a, 23b follows the general procedure described for compounds 5a–5g
1-(6-Methoxy-2-pentyloxy-quinolin-3-yl)-but-3-yn-1-ol (23a)
Compound was obtained with Mg (193 mg, 7.94 mmol), HgCl2 (21 mg, 0.08 mmol), propargyl bromide (745 µL, 8.61 mmol), 22a (1.81 g, 6.62 mmol) in 10 mL Et2O, and Et2O (17 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a white solid (2.05 g, 99% yield).
M.p.: 61–64°C; 1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.01 (s, 1H, H-Ar), 7.72 (d, J = 9.1 Hz, 1H, H-Ar), 7.26 (dd, J = 9.1, 2.8 Hz, 1H, H-Ar), 7.05 (d, J = 2.8 Hz, 1H, H-Ar), 5.09 (ddd, J = 5.1, 7.0, 5.8 Hz, 1H, CHOH), 4.49–4.45 (m, 2H, OCH2), 3.88 (s, 3H, ArOCH3), 3.03 (d, J = 5.8 Hz, 1H, CHOH), 2.90 (ddd, J = 16.8, 5.1, 2.8 Hz, 1H, CH2C≡C), 2.68 (ddd, J = 16.8, 7.0, 2.8 Hz, 1H, CH2C≡C), 2.06 (t, J = 2.8 Hz, 1H, C≡CH), 1.85–1.81 (m, 2H, OCH2CH2), 1.49–1.43 (m, 4H, CH2CH2CH3), 0.94 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 158.43 (C), 156.90 (C), 141.86 (C), 134.72 (CH), 128.73 (CH), 126.68 (C), 126.14 (C), 121.51 (CH), 106.83 (CH), 80.98 (C), 71.43 (CH), 68.82 (CH), 66.36 (CH2), 55.76 (CH3), 28.81 (CH2), 28.58 (CH2), 27.28 (CH2), 22.55 (CH2), 14.11 (CH3).
1-(2-Pentyloxy-benzo[h]quinolin-3-yl)-but-3-yn-1-ol (23b)
Compound was obtained with Mg (28 mg, 1.15 mmol), HgCl2 (3 mg, 0.01 mmol), propargyl bromide (140 µL, 1.25 mmol), 22b (282 mg, 0.96 mmol) in 3 mL Et2O, and Et2O (7 mL). Column chromatography on silica gel (EtOAc/pentane, 20:80 v/v) afforded a yellow solid (290 mg, 90% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.17 (d, J = 7.6 Hz, 1H, H-Ar), 8.14 (s, 1H, H-Ar), 7.88, (d, J = 7.2 Hz, 1H, H-Ar), 7.79–7.58 (m, 4H, H-Ar), 5.26-5.12 (m, 1H, CHOH), 4.64 (t, J = 6.6 Hz, 2H, OCH2), 3.28 (d, J = 5.3 Hz, 1H, CHOH), 2.98 (ddd, J = 16.8, 7.0, 2.6 Hz, 1H, CH2C≡C), 2.74 (ddd, J = 16.8, 4.9, 2.6 Hz, 1H, CH2C≡C), 2.12 (t, J = 2.6 Hz, 1H, C≡CH), 2.00–1.81 (m, 2H, OCH2CH2), 1.63–1.40 (m, 4H, CH2CH2CH3), 1.03 (t, J = 6.9 Hz, 3H, CH2CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.41 (C), 143.40 (C), 135.00 (CH), 133.70 (C), 130.49, 127.63, 127.54, 126.19, 125.17 (2C), 124.74, 124.24, 121.80, 80.65 (C), 71.09 (CH), 68.12 (CH), 66.21 (CH2), 28.60 (CH2), 28.47 (CH2), 27.07 (CH2), 22.48 (CH2), 14.10 (CH3).
Preparation of 24a, 24b follows the general procedure described for compounds 6a–6g
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-6-methoxy-2-pentyloxy-quinoline (24a)
Compound was obtained with 23a (2.05 g, 6.60 mmol), imidazole (1.12 g, 16.5 mmol), TBDMSCl (1.19 g, 7.92 mmol), and DMF (7 mL). Column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) afforded a colorless oil (2.76 g, 98% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.05 (s, 1H, H-Ar), 7.71 (d, J = 9.1 Hz, 1H, H-Ar), 7.23 (dd, J = 9.1, 2.8 Hz, 1H, H-Ar), 7.05 (d, J = 2.8 Hz, 1H, H-Ar), 5.26 (ddd, J = 7.1, 3.8, 0.9 Hz, 1H, CHOTBDMS), 4.45 (t, J = 6.6 Hz, 2H, OCH2), 3.88 (s, 3H, ArOCH3), 2.70 (ddd, J = 16.7, 7.1, 2.6 Hz, 1H, CH2C≡C), 2.68 (ddd, J = 16.7, 3.8, 2.6 Hz, 1H, CH2C≡C), 1.92 (t, J = 2.6 Hz, 1H, C≡CH), 1.86–1.80 (m, 2H, OCH2CH2), 1.50–1.43 (m, 4H, CH2CH2CH3), 0.94 (t, J = 7.1 Hz, 3H, CH3), 0.93 (s, 9H, t-BuSi), 0.13 (s, 3H, CH3Si), −0.01 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 157.52 (C), 156.03 (C), 141.23 (C), 134.23 (CH), 128.09 (CH), 125.72 (2C, C), 120.62 (CH), 106.38 (CH), 81.63 (C), 69.83 (CH), 67.69 (CH), 65.82 (CH2), 55.54 (CH3), 28.67 (CH2), 28.65 (CH2), 28.46 (CH2), 25.92 (3C, CH3), 22.46 (CH2), 18.37 (C), 14.09 (CH3), −4.79 (CH3), −4.85 (CH3).
3-[1-(t-Butyl-dimethyl-silanyloxy)-but-3-ynyl]-2-pentyloxy-benzo[h]quinoline (24b)
Compound was obtained with 23b (285 mg, 0.85 mmol), imidazole (145 mg, 2.13 mmol), TBDMSCl (171 mg, 1.10 mmol), and DMF (3 mL). Column chromatography on silica gel (EtOAc/pentane, 15:85 v/v) afforded a colorless oil (370 mg, 97% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.26 (d, J = 7.5 Hz, 1H, H-Ar), 8.30 (s, 1H, H-Ar), 7.95 (d, J = 7.2 Hz, 1H, H-Ar), 7.81–7.62 (m, 4H, H-Ar), 5.47–5.36 (m, 1H, CHOTBDMS), 4.74 (t, 2H, J = 6.5 Hz, OCH2), 2.98–2.60 (m, 2H, CH2C≡C), 2.13–1.92 (m, 3H, OCH2CH2, C≡CH), 1.72–1.43 (m, 4H, CH2CH2CH3), 1.09-1.03 (m, 12H, t-BuSi, CH3), 0.28 (s, 3H, CH3Si), 0.11 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 158.21 (C), 143.37 (C), 135.31 (CH), 133.67 (C), 130.63, 127.59, 127.37, 127.19, 126.22, 125.30, 124.52, 124.19, 121.97 (C), 81.55 (C), 69.86 (CH), 67.64 (CH), 65.99 (CH2), 28.61 (CH2), 28.51 (CH2), 25.82 (3C, CH3), 22.45 (CH2), 20.91 (CH2), 18.30 (C), 14.11 (CH3), −4.82 (CH3), −4.90 (CH3).
Preparation of 25a, 25b follows the general procedure described for compounds 7a–7g
8-(t-Butyl-dimethyl-silanyloxy)-8-(6-methoxy-2-pentyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (25a)
Compound was obtained with 24a (2.86 g, 6.68 mmol), n-BuLi (10.9 mL, 10.0 mmol), trimethyl 4-bromoorthobutyrate (1.40 mL, 7.7 mmol), THF (8 mL), and HMPA (8 mL). Column chromatography on silica gel (EtOAc/pentane, 5:95 v/v) afforded a colorless oil (2.29 g, 65% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.100 (s, 1H, H-Ar), 7.75 (d, J = 7.9 Hz, 1H, H-Ar), 7.73 (dd, J = 7.9, 1.0 Hz, 1H, H-Ar), 7.09 (s, 1H, H-Ar), 5.20–5.17 (m, 1H, CHOTBDMS), 4.46 (t, J = 6.6 Hz, 2H, OCH2), 3.92 (s, 3H, ArOCH3), 3.67 (s, 3H, CO2CH3), 2.67 (ddd, J = 16.5, 4.4, 2.2 Hz, 1H, CH2C≡C), 2.49 (ddd, J = 16.5, 6.6, 2.2 Hz, 1H, CH2C≡C), 2.39 (t, J = 7.7 Hz, 2H, CH2CO2CH3), 2.22–2.16 (m, 2H, C≡CCH2), 1.88–1.81 (m, 2H, OCH2CH2), 1.77 (tt, J = 7.7, 7.1 Hz, 2H, CH2CH2CO2CH3), 1.52–1.38 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3), 0.94 (s, 9H, tBuSi), 0.14 (s, 3H, CH3Si), 0.02 (s, 3H, CH3Si); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.78 (C), 157.58 (C), 155.98 (C), 141.15 (C), 134.19 (CH), 128.59 (CH), 128.04 (C), 128.03 (CH), 125.76 (CH), 120.48 (C), 80.44 (C), 78.22 (C), 68.05 (CH), 65.78 (CH2), 55.54 (CH3), 51.48 (CH3), 32.82 (CH2), 28.89 (CH2), 28.68 (CH2), 28.48 (CH2), 25.86 (CH2), 25.92 (3C, CH3), 23.30 (CH2), 19.21 (CH2), 18.30 (C), 14.11 (CH3), −4.75 (CH3), −4.90 (CH3).
8-(t-Butyl-dimethyl-silanyloxy)-8-(2-pentyloxy-benzo[h]quinolin-3-yl)-oct-5-ynoic acid methyl ester (25b)
Compound was obtained with 24b (265 mg, 0.59 mmol), n-BuLi (480 µL, 0.77 mmol), trimethyl 4-bromoorthobutyrate (160 µL, 0.88 mmol), THF (2 mL), and HMPA (2 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a pale yellow oil (160 mg, 50% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.12 (d, J = 7.5 Hz, 1H, H-Ar), 8.27 (s, 1H, H-Ar), 7.92 (d, J = 7.3 Hz, 1H, H-Ar), 7.74–7.61 (m, 4H, H-Ar), 5.33–5.25 (m, 1H, CHOTBDMS), 4.69 (t, J = 6.6 Hz, 2H, OCH2), 3.65 (s, 3H, CO2CH3), 2.80–2.52 (m, 2H, CH2C≡C), 2.39 (t, J = 7.5 Hz, 2H, CH2CO2CH3), 2.28–2.16 (m, 2H, C≡CCH2), 2.05–1.91 (m, 2H, OCH2CH2), 1.82–1.71 (m, 2H, CH2CH2CO2CH3), 1.51–1.39 (m, 4H, CH2CH2CH3), 1.00 (m, 12H, t-BuSi, CH3), 0.18 (s, 3H, CH3Si), 0.02 (s, 3H, CH3Si); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.76 (C), 158.39 (C), 143.32 (C), 135.41 (CH), 133.72 (C), 130.73, 127.68, 127.50, 127.39, 126.18, 125.41, 124.50, 124.25, 122.07, 80.49 (C), 78.22 (C), 67.99 (CH), 66.07 (CH2), 51.43 (CH3), 32.79 (CH2), 28.90 (CH2), 28.69 (CH2), 28.58 (CH2), 25.86 (3C, CH3), 24.07 (CH2), 22.52 (CH2), 18.37 (CH2), 18.30 (C), 14.15 (CH3), −4.76 (CH3), −4.87 (CH3).
Preparation of 26a, 26b follows the general procedure described for compounds 8a–8g
8-Hydroxy-8-(6-methoxy-2-pentyloxy-quinolin-3-yl)-oct-5-ynoic acid methyl ester (26a)
Compound was obtained with 25a (2.08 g, 3.95 mmol), TBAF (5.50 mL, 5.50 mmol), and THF (14.6 mL). Column chromatography on silica gel (EtOAc/pentane, 10:90 v/v) afforded a yellow oil (351 mg, 22% yield).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.05 (s, 1H, H-Ar), 7.71 (d, J = 9.1 Hz, 1H, H-Ar), 7.24 (dd, J = 9.1, 2.8 Hz, 1H, H-Ar), 7.09 (d, J = 2.8 Hz, 1H, H-Ar), 5.17 (dd, J = 6.6, 4.6 Hz, 1H, CHOH), 4.45 (t, J = 6.6 Hz, 2H, OCH2), 3.91 (s, 3H, ArOCH3), 3.65 (s, 3H, CO2CH3), 2.65 (ddd, J = 16.5, 4.6, 2.2 Hz, 1H, CH2C≡C), 2.48 (ddd, J = 16.5, 6.6, 2.2 Hz, 1H, CH2C≡C), 2.38 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.21–2.14 (m, 2H, C≡CCH2), 1.87–1.79 (m, 2H, O-CH2CH2), 1.75 (tt, J = 7.4, 7.1 Hz, 2H, CH2CH2CO2CH3), 1.51–1.36 (m, 4H, CH2CH2CH3), 0.95 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 173.74 (C), 157.81 (C), 156.14 (C), 141.15 (C), 134.61 (CH), 129.01 (CH), 128.11 (CH), 126.61 (CH), 120.78 (C), 82.13 (C), 77.40 (C), 68.61 (CH), 66.00 (CH2), 55.51 (CH3), 51.90 (CH3), 32.83 (CH2), 31.62 (CH2), 28.71 (CH2), 28.48 (CH2), 23.98 (CH2), 22.69 (CH2), 18.27 (CH2), 14.16 (CH3).
8-Hydroxy-8-(2-pentyloxy-benzo[h]quinolin-3-yl)-oct-5-ynoic acid methyl ester (26b)
Compound was obtained with 25b (135 mg, 0.29 mmol), TBAF (410 µL, 0.41 mmol), and THF (1.5 mL). Column chromatography on silica gel (EtOAc/pentane, 25:75 v/v) afforded a white solid (79 mg, 62% yield).
1H-NMR: (300 MHz, CDCl3) δ (ppm): 9.12 (d, J = 7.5 Hz, 1H, H-Ar), 8.19 (s, 1H, H-Ar), 7.90 (d, J = 7.3 Hz, 1H, H-Ar), 7.74–7.62 (m, 4H, H-Ar), 5.19–5.10 (m, 1H, CHOH), 4.67 (t, J = 6.7 Hz, 2H, OCH2), 3.62 (s, 3H, CO2CH3), 3.08 (s, 1H, CHOH), 3.00–2.86 (m, 1H, CH2C≡C), 2.76–2.62 (m, 1H, CH2C≡C), 2.36 (t, J = 7.4 Hz, 2H, CH2CO2CH3), 2.28–2.19 (m, 2H, C≡CCH2), 2.01–1.87 (m, 2H, OCH2CH2), 1.84–1.72 (m, 2H, CH2CH2CO2CH3), 1.60–1.39 (m, 4H, CH2CH2CH3), 0.98 (t, J = 7.2 Hz, CH3); 13C-NMR: (75 MHz, CDCl3) δ (ppm): 173.70 (C), 158.67 (C), 143.44 (C), 135.11 (CH), 133.78 (C), 130.65, 127.68, 127.54, 126.23, 125.69, 125.26, 124.79, 124.29, 121.96, 82.20 (C), 77.08 (C), 68.51 (CH), 66.27 (CH2), 51.54 (CH3), 32.80 (CH2), 28.70 (CH2), 28.55 (CH2), 27.64 (CH2), 23.94 (CH2), 22.52 (CH2), 18.25 (CH2), 14.11 (CH3); HRMS: calcd. for C27H31NO4 [M]+ 433.22531; Found 433.2253 (0 ppm). Anal. calcd. for C27H31NO4: C, 74.80; H, 7.21; N, 3.23; Found: C, 74.89; H, 7.38; N, 3.37%.
Preparation of 27a, 27b follows the general procedure described for compounds 9a–9g
Sodium 8-hydroxy-8-(6-methoxy-2-pentyloxy-quinolin-3-yl)-oct-5-ynoate (27a)
Acid was prepared with 26a (218 mg, 0.53 mmol), LiOH·H2O (78 mg, 1.86 mmol), oxalic acid (251 mg, 2.79 mmol), and MeOH/water (13 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc) afforded a white solid (94 mg, 44% yield). Salt was prepared with the acid (94 mg, 0.23 mmol), NaOH (9 mg, 0.23 mmol), and MeOH (1 mL). An off-white solid was obtained (98 mg, 99%).
1H-NMR: (400 MHz, CDCl3) δ (ppm): 8.05 (s, 1H, H-Ar), 7.67 (d, J = 9.2 Hz, 1H, H-Ar), 7.36 (d, J = 2.8 Hz, 1H, H-Ar), 7.27 (dd, J = 9.2, 2.8 Hz, 1H, H-Ar), 5.00 (t, J = 5.3 Hz, 1H, CHOH), 4.46–4.36 (m, 2H, ArOCH2), 3.85 (s, 3H, ArOCH3), 2.72–2.67 (m, 1H, CH2C≡C), 2.51–2.45 (m, 1H, CH2C≡C), 2.25 (t, J = 7.4 Hz, 2H, CH2CO2H), 2.11 (t, J = 7.1 Hz, 2H, C≡CCH2), 1.83–1.76 (m, 2H, O-CH2CH2), 1.58 (tt, J = 7.4 7.1 Hz, 2H, CH2CH2CO2H), 1.50–1.37 (m, 4H, CH2CH2CH3), 0.94 (t, J = 7.1 Hz, 3H, CH3); 13C-NMR: (100 MHz, CDCl3) δ (ppm): 174.52 (C), 157.59 (C), 155.98 (C), 140.59 (C), 134.46 (CH), 129.01 (CH), 127.95 (C), 125.89 (C), 120.78 (CH), 106.97 (CH), 81.20 (C), 78.31 (C), 66.06 (CH), 65.61 (CH2), 55.71 (CH3), 32.85 (CH2), 28.49 (CH2), 28.30 (CH2), 27.66 (CH2), 24.39 (CH2), 22.27 (CH2), 17.96 (CH2), 14.32 (CH3).
Sodium 8-hydroxy-8-(2-pentyloxy-benzo[h]quinolin-3-yl)-oct-5-ynoate (27b)
Acid was prepared with 26b (81 mg, 0.19 mmol), LiOH·H2O (27 mg, 0.65 mmol), oxalic acid (86 mg, 0.93 mmol), and MeOH/water (4 mL, 9:1 v/v). Column chromatography on silica gel (EtOAc) afforded a white solid (51 mg, 65% yield).
1H-NMR: (200 MHz, CDCl3) δ (ppm): 9.13 (d, J = 7.5 Hz, 1H, H-Ar), 8.20 (s, 1H, H-Ar), 7.94 (d, J = 7.3 Hz, 1H, H-Ar), 7.80–7.62 (m, 4H, H-Ar), 5.23–5.10 (m, 1H, CHOH), 4.87 (t, J = 6.7 Hz, 2H, OCH2), 3.05–2.65 (m, 2H, CH2C≡C), 2.45 (t, J = 7.2 Hz, 2H, CH2CO2H), 2.39–2.23 (m, 2H, C≡CCH2), 2.05–1.72 (m, 4H, CH2CH2CO2H, OCH2CH2), 1.68–1.40 (m, 4H, CH2CH2CH3), 1.00 (t, 3H, J = 7.1 Hz, CH3).
Salt was prepared with the acid (51 mg, 0.12 mmol), NaOH (5 mg, 0.12 mmol), and MeOH (1 mL). A white hygroscopic solid was obtained (52 mg, 100%).
Pharmacological in vitro assays
Binding assays were performed in 96-well plate format, using a classical filtration assay with a human full length PPARγ construct (GST-PPAR LBD (25 µg/mL)) expressed in bacteria with some modifications regarding the conditions of the experiments. The membrane-associated PPARγ was used as the biological source as previously described. Binding buffer consisted of 10 mM Tris/HCl, pH 8.2, containing 50 mM KCl and 1 mM dithiothreitol. Membrane preparations (5 µg/mL) were incubated for 180 min at 4°C in the presence of [3H]rosiglitazone (BRL49653, Amersham) (4 nM) and the tested compounds. Nonspecific binding was defined using an excess of unlabeled rosiglitazone (10 µM). Incubation was terminated by the addition of ice-cold 50 mM Tris/HCl buffer pH 7.4, followed by rapid filtration under reduced pressure through Whatman GF/C filter plates presoaked with ice-cold buffer, followed by three successive washes with the same buffer. Radioactivity was measured in a TopCount apparatus (Packard). The receptor preparation used during these experiments presented a Bmax of 49 pmol/mg protein and a Kd of 5.58 nM for [3H]rosiglitazone. The compounds were solubilized in pure dimethylsuilfoxide (DMSO) and diluted to the appropriate working concentrations (100 µM to 0.1 nM). For each compound tested, plots of ligand concentration versus DPM of bound radioligand were constructed, and apparent Ki values were estimated from nonlinear least-squares fit of the data assuming simple competitive binding. The details of this assay have been reported elsewhereCitation15.
Compounds were screened for functional potency in a transient transfection assay performed on Cos-7 cells, where a previously established chimeric receptor system was used to allow comparison of the relative transcriptional activity on the same target gene. Cos-7 cells were transiently transfected with luciferase reporter plasmid (pG5-TK-pGL3) in the presence of pGal4hPPARγ or pGal4hPPARα (these vectors expressed chimeric proteins containing the Gal4 DNA-binding domain fused to the human PPARγ or PPARα ligand binding domain coding sequence) expression vectors. Plasmid pGal4hPPARs and pG5-TK-pGL3 were constructed as described previouslyCitation16. Cells were seeded in 60 mm dishes at a density of 5.5 × 105 cells/dish in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and incubated at 37°C for 24 h prior to transfection. Cells were transfected in an OptiMEM without FCS for 3 h at 37°C, using polyethylenimine (PEI), with reporter and expression plasmids. The plasmid pBluescript (Stratagene, La Jolla, CA) was used as carrier DNA to set the final amount of DNA to 5.5 µg/dish. The pCMV-β-galactosidase expression plasmid was cotransfected as a control for transfection efficiency. Transfection was stopped by the addition of DMEM supplemented with 10% FCS and cells were then incubated at 37°C. After 16 h, cells were trypsinized and seeded in 96-well plates at the density of 2 × 104 cells/well and incubated for 6 h in 10% FCS containing DMEM. Cells were then incubated for 16 h in DMEM containing 0.2% FCS and increasing concentrations of the compound tested (10 µM to 10 nM) or vehicle (DMSO). At the end of the experiment, cells were washed once with ice-cold phosphate buffered saline (PBS) and the luciferase activity was measured and normalized to internal control β-galactosidase activity as described previouslyCitation16. Compounds that elicited on average at least 80% activation of PPAR(s) versus rosiglitazone (PPARγ) or WY 14,643 (PPARα) (positive controls) were considered full agonists. EC50 values were estimated using Prism software (GraphPad). All transactivation and binding experiments were performed once. For each concentration tested, the measurements were made in triplicate.
Results and discussion
The activity of the esters 8 and 26, the amide 14a, and the sodium salts 9, 18, and 27 was tested in vitro on both subtypes PPARα and PPARγ, and the results are given in .
Table 1. In vitro activity of S 70655 analogs in cell-based transactivation assay and binding assay against human PPARα/Gal4 and PPARγ/Gal4 receptors.
During this work, our internal reference was S 70655 (9a), that is, in vitro, a full agonist on the PPARα subtype and a partial agonist on PPARγ, but which presented no activity in vivo. In the first part of this work, we tested different lipophilic chains. As we can see from , when the length of this chain was diminished (9b and 8e/9e) or when a methoxy group was introduced at the end of the chain (8d or 9d), no or poor activity was observed. These results indicated the need for a more hindered and/or lipophilic moiety at this position. For that purpose, we first introduced an elongated side chain, such as the octyloxy chain (8c/9c). This afforded very interesting compounds with a SPPARM-type agonist activity (specific PPAR modulator): high affinity for the PPARα subtype with a partial-agonist profile. On the other hand, the introduction of a cycle at the end of the pentyloxy chain of S 70655, such as cyclopropyl or cyclohexyl groups (8f and 8g), led to agonists with a strong affinity on PPARα (full agonist profile) and still the desired partial activity on PPARγ. These new compounds presented the desired in vitro profile and are under further active study.
All the new S 70655 analogs involving modifications on the acid chain afforded only inactive molecules. Even the replacement of a single CH2 by an oxygen atom (18a, 18b) led to a complete loss of activity toward the two PPARs, indicating the high sensitivity of this part of the molecule to structural modifications. On the other hand, modifications of the quinoline core gave less potent, but still active, molecules.
Conclusions
The synthesis and biological studies of the new analogs of our lead S 70655 have confirmed the potentialities of this family of quinolines as dual PPAR agonists. The SAR studies have indicated the high sensitivity of the upper acid chain to modifications as well as the strong effect of the length and size of the lipophilic side chain. They afforded new derivatives, such as 8c, 8g, 9c, which are dual agonists with a high PPARα activity in vitro. Development of this family of new quinoline analogs of 8-HETE is under active study in our groupsCitation17,Citation18.
Declaration of interest
The authors report no conflicts of interest.
Related Research Data
References
- Zimmet P, Magliano D, Matsuzawa Y, Alberti G, Shaw J. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb 2005;12:295–300.
- Stumvoll M, Golstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333–46.
- Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321:405–12.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart J-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998;98:2088–93.
- Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor γ in diabetes and metabolism. Trends Pharmacol Sci 2004;25:331–6.
- Göttlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc Natl Acad Sci USA 1992;89:4653–7.
- Leff T, Reed JE. The antidiabetic PPARγ ligands: an update on compounds in development. Curr Med Chem Imunol Endocr Metab Agents 2002;2:33–47.
- Henke BR. Peroxisome proliferator-activated receptor α/γ dual agonists for the treatment of type 2 diabetes. J Med Chem 2004;47:4118–27.
- Rubenstrunk A, Hanf R, Hum DW, Fruchart J-C, Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta 2007;1771:1065–81.
- Fiévet C, Fruchart J-C, Staels B. PPARα and PPARγ dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr Opin Pharmacol 2006;6:606–14.
- Gross B, Staels B. PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab 2007;21:687–710.
- Caijo F, Mosset P, Grée R, Audinot-Bouchez V, Boutin J, Renard P, et al. Synthesis of new carbo- and heterocyclic analogues of 8-HETE and evaluation of their activity towards the PPARs. Bioorg Med Chem Lett 2005;15:4421–6.
- Caijo F, Mosset P, Grée R, Audinot-Bouchez V, Boutin J, Renard P, et al. Synthesis of aromatic analogs of 8(S)-HETE and their biological evaluation as activators of the PPAR nuclear receptors. Eur J Org Chem 2006:2181–96.
- Kerry MA, Boyd GW, Mackay SP, Meth-Cohn O, Platt L. The synthesis of benzo[h]quinolines as topoisomerase inhibitors. J Chem Soc Perkin Trans 1 1999:2315–21.
- Ferry G, Bruneau V, Beauverger P, Goussard M, Rodriguez M, Lamamy VO, et al. Binding of prostaglandins to human PPARγ: tool assessment and new natural ligands. Eur J Pharmacol 2001;417:77–89.
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 1998;393:790–3.
- Grée R, Liutkus M, Caijo F, Brioche J, Jennequin T, Dacquet C, et al. Preparation of heterocycles, in particular 3-[(alkynyl)(hydroxy)methyl]-2-phenylalkoxyquinolines, pharmaceutical compositions containing them, and their use in the treatment of type II diabetes and obesity. PCT Int. Appl. WO/2008/081096. Geneva: WIPO, 2008.
- Liutkus M, Dacquet C, Audinot-Bouchez V, Boutin J, Caignard D-H, Ktorza A, et al. Synthesis of a novel series of 8-HETE analogs and their biological evaluation towards the PPAR nuclear receptors. Lett Drug Des Discov 2008;5:503–11.