Abstract
Affecting hepatic cytochrome (CYP) activity is one of the major concerns in drug–drug interaction. Thus the testing of drug candidates on their impact on these enzymes is an essential step in early drug discovery. We tested a collection of 480 in-house phthalimide derivatives against different CYP450s using a high throughput inhibition assay. In initial tests with the isoform CYP2C19 about 57.5% of the tested phthalimide derivatives showed significantly enhanced inhibitory effects against this enzyme. In addition similar patterns of phthalimide inhibition for CYP2C9 and CYP2C19 were found, whereas the unrelated isoforms CYP2D6 and CYP3A4 were not specifically affected. Also less than 10% of randomly chosen substances inhibited CYP2C9. Analyses of structure-function relationships revealed that the substituent at the nitrogen atom in the isoindole ring is of crucial impact for the activity of CYP2C9/19.
Introduction
The pharmacological and agrochemical properties of phthalimide (, Structure 1) derived substances are widely used for more than half a century. The best known among all these substances would be undoubtedly thalidomide (, Structure 2). This substance was discovered in 1954 by Grünenthal chemicals and first introduced to the German market where it became a top-selling sedative and agent against morning sickness under the name Contergan. A few years later thalidomide turned out to cause drastic teratogenic effects and had to be withdrawn. Up until then it left a trace of tens of thousands of newborn children worldwide with malformations, especially of their limbs. About 25 years after the ‘Contergan scandal’ (as this incidence is called in Germany) thalidomide encountered a revival as a therapeutic drug against erythema nodosum leprosum (ENL), a severe form of leprosy. Afterwards, thalidomide was revealed to be effective also against a number of different other inflammatory and autoimmune diseases as well as against different forms of cancer. As a result of these therapeutic successes the FDA approved thalidomide for the application in the USA under a special monitoring programme. For a review of thalidomide’s history see Silverman [Citation1].
The broad range of therapeutic applications of thalidomide has been attributed to a variety of action mechanisms [Citation2, Citation3]. Its efficacy seems to stem primarily from its anti-angiogenic properties [Citation4] combined with its inhibitory activity on tumour necrosis factor α (TNF-α) [Citation5]. Although much is known about the therapeutic properties of this drug, its molecular mode of action still remains to be resolved. One question is, is it thalidomide itself or a metabolite causing the observed therapeutic effects? It has been shown that about 12 breakdown products evolve physiologically which are pharmaceutically not effective and also do not contribute to its teratogenicity [Citation6]. All these metabolites are mostly resulting from non-enzymatic processes and they are quickly eliminated in the urine [Citation7].
In contrast, it has been shown that thalidomide is activated by biotransformation. It reacts to its hydroxylation products catalysed by different cytochrome P450 mono-oxygenases (CYP), especially by the isoform CYP2C19 [Citation8, Citation9]. CYPs comprise a superfamily of enzymes involved in the detoxification of a large number of endogenous and exogenous compounds. Hepatic isoforms of these enzymes are especially involved in the metabolism of a number of drugs. Braun et al. [Citation10] provided the first pieces of evidence that thalidomide is activated by CYP enzymes. The CYP-catalysed hydroxylation seems to activate thalidomide to its therapeutic active form, although the enzymatic activity is rather low [Citation11]. Also the nature of the pharmacologically effective metabolite is still a matter of debate, but there are two lines of evidence which support the essential impact of CYP2C19 on thalidomide’s properties. Firstly, in pharmacogenetic studies the response of extensive metabolisers of CYP2C19 on thalidomide against multiple myeloma was significantly higher than that of poor metabolisers [Citation9, Citation12]. Secondly, there are recent reports in which a correlation between specific inhibition of CYP2C19 and the activity of thalidomide on myeloma cell lines and on angiogenesis could be shown [Citation13, Citation14]. There are further CYP isoforms involved in the metabolism and efficacy of thalidomide, such as CYP2C9 and CYP1A1, which are involved in its hydroxylation [Citation3]. In other cases thalidomide affects the activity of these enzymes itself and therefore influences drug metabolism. Recently, an activation of CYP3A4 and CYP3A5 by thalidomide was reported causing enhanced clearance of cyclosporine A and midazolam [Citation15].
Despite the revival of thalidomide as a remedy there is still the challenge to develop analogues with reduced side effects. Beyond the teratogenicity described above there are a number of side effects accompanying treatment with thalidomide such as peripheral neuropathy and thromboembolism [Citation16]. Analogues of the second generation, such as lenalidomide and actimid, are currently in therapeutic use. Further substances that have been developed are under investigation at different test phases [Citation2, Citation16]. Most approaches for the development of such analogues are based on phthalimide derivatives. To optimise the therapeutic properties mostly substituents either in the phthaloyl ring or at the amino group are varied. In contrast to the observed interactions between thalidomide and CYPs neither metabolisation of nor inhibition by its analogue lenalidomide by CYPs has been reported recently [Citation17].
On the search for novel phthalimide derivatives with agrochemical and pharmaceutical properties candidate compounds were synthesised in multicomponent reactions in our laboratories and then purified for screening purposes. Due to the aforementioned interaction of most members of this compound class with CYPs we tested the inhibitory potential of our focused compound library on isoforms of this enzyme. Surprisingly, the majority of the tested substances inhibited specifically the closely related isoforms CYP2C19 and CYP2C9 at physiological concentrations. Further analyses revealed that this inhibition strongly depends on the substitution at the nitrogen atom of the phthalimide. The presented data provide valuable insights into the interaction of phthalimides with CYP enzymes.
Materials and methods
Synthesis and purification of phthalimide derivatives
An improved one-pot procedure for the three-component coupling reaction of aldehydes, amides and dienophiles (AAD reaction) was utilised to build up the skeletal structure of the investigated tetrahydro-isoindole-1,3-dione compounds [Citation18–20]. With this methodology at hand, all structures summarised in were synthesised using a fully automated system for combinatorial library synthesis. Sample preparation (e.g. addition of liquids, solids, and reactant stock solutions), reaction processing (e.g. heating, mixing), and work-up steps such as cooling and diluting were performed by a laboratory automation system based on a Zymate XP robot (Caliper Life Science, Hopkinton, MA). The reactions were carried out in 2 mL screw-cap vials. Starting materials and solvents were used as received from commercial suppliers [Citation21].
Table 1. Basic structure of phthalimide with positions of substitutions and structures of the substitutes. Numbers of tested compounds with indicated substitution are given in parentheses below the structures.
The desired products were isolated using a semi-preparative high performance liquid chromatography (HLPC) with mass-tagged fraction collection system. Methanol HPLC Gradient Grade (Roth, Karlsruhe, Germany), formic acid (98%, Sigma-Aldrich, Steinheim, Germany), DMSO (99.8%, Biotech Quality, Roth) and purified water from a Milli-Q Synthesis A10 system (Millipore, Schwalbach, Germany) were used for the liquid chromatography (LC) eluent. The atmospheric pressure ionisation (API) mass spectrometry experiments were performed using an Agilent 1946C quadrupole mass spectrometer (Agilent, Waldbronn, Germany). The MS was used with an electrospray ionisation (API-ES) source. Semi-preparative HPLC separation was performed on an Agilent Technologies 1100 Series HPLC system with a Zorbax Eclipse XDB-C8, 9.4 × 250 mm, 5 micron, column (Agilent) at 45°C at a flow rate of 4 mL/min within a 45-minute gradient, and H2O and MeOH as eluents. The gradient was adjusted according to the retention properties of the particular analyte. For the fraction collection, an analytical fraction collector (G1364C, Agilent) coupled to an active splitter with a variable splitting option (G1968D, Agilent) was used. According to the preselected trigger mass for each analyte, as [M+H]+ or [M+Na]+ quasimolecular ions, fractions were collected in microtitre plates. API-ES MS conditions (positive ion mode) were as follows: nebuliser and drying gas, nitrogen; nebuliser pressure, 35 psig; drying gas flow, 10 L/min; drying gas temperature, 350°C; capillary voltage, 4 kV.
LC high-resolution mass spectrometry experiments for the preselection of semi-preparative HPLC parameters and the final purity confirmation were performed on an Agilent Series 1200 HPLC system and an Agilent 1969A time-of-flight mass spectrometer. The TOF-MS conditions (positive ion mode) with a dual sprayer API-ES source were as follows: nebuliser and drying gas, nitrogen; nebuliser pressure, 40 psig; drying gas flow, 10 L/min; drying gas temperature, 350°C; capillary voltage, 4 kV; fragmenter voltage, 175 V; skimmer voltage, 60 V; octopole voltage, 250 V; mass reference (m/z), 121.05087 and 922.00979 in positive ion mode. HPLC separation was performed on a Zorbax Eclipse XDB-C8, 4.6 × 50 mm, 3.5 micron, column (Agilent) at 25°C at a flow rate of 0.5 mL/min within a 10-min gradient from 10 to 100% methanol in 0.1% aqueous formic acid.
The collected substances were, for further application, dissolved in DMSO to obtain 10 mmol/L stock solutions. Therefore, collected fractions underwent a purity analysis via HPLC-MS. Fractions with a purity of more than 90% were combined, evaporated to dryness, balanced out, and dissolved in DMSO.
Automated CYP inhibition assay
The inhibitory potential on four different CYP enzymes (CYP2C19, CYP2C9, CYP3A4, and CYP2D6) of candidate phthalimide derivatives was tested with the P450-Glo™ screening system (Promega, Madison) according to the manufacturer’s protocol. All compounds were tested in triplicate in a final concentration of 10 µM containing 0.1% DMSO in the final reaction mix. The same reaction mix including DMSO without additional compounds was used as positive control. As specific inhibitors, 10 µM troglitazone, 80 µM sulphaphenazole, 5 µM ketoconazole and 2 µM quinidine were applied for CYP2C19, CYP2C9, CYP3A4 and CYP2D6, respectively. Beside these negative control reactions another negative control series was performed to analyse the chemical decay of the substrate. Therefore, positive control reactions were run without the addition of CYP enzyme. In a single automated run 80 phthalimide compounds were analysed in three 96-well plates. In each of these plates all control reactions were also performed in quadruplicate for the control of intra-day and inter-day variations and also for determination of Z-factor. Z-factors of all assays were between 0.54 and 0.84 and therefore fulfilled the requirements of good assay conditions according to [Citation22].
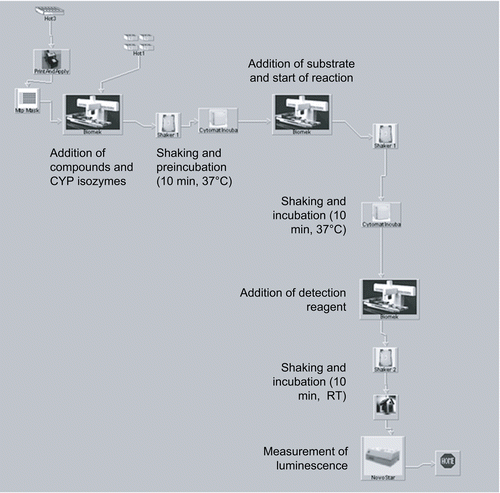
All assays were performed in an automated environment. The development of an automation assay procedure was done on a Beckman Coulter core facility consisting of a central robot (ORCA) and a Biomek 2000 pipetting station providing a single-channel and an 8-channel pipetting tool (Beckman Coulter, Fullerton, CA). The ORCA was positioned on a central rail to supply peripheral devices with all types of required labware. The whole asset includes an incubator (Cytomat, Kendro, Waltham, MA), two readers (FLUOStar and NOVOStar, BMG, Offenburg, Germany), several shaking units (Variomag Teleshake, H+P, Oberschleißheim, Germany), a barcode printer and reader (Microplate Print & Apply, Beckman Coulter) and the required amount of labware stacking and storage positions. After manual optimisation of the assay process, the conversion into an automated procedure was done by programming a superordinated system scheduling software (Sagian Automated Method Interface SAMI, Version 3.5, Beckman Coulter), that rules and administrates the incorporated instrumentation as slave devices (design and schedule of the automated assay process are visualised in ).
Figure 1. Examples for derivatives of phthalimide (1) thalidomide (2), fosmet (3), captan (4) and indoprofen (5).
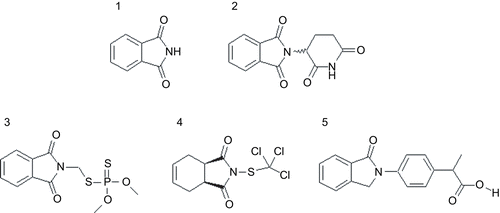
Figure 2. Automation scheme of CYP inhibition assays.
Data handling took place via an in-house developed web-based laboratory information management system (LIMS) [Citation23]. The LIMS provides flexible process documentation and the process data exchanges within the robotic process control system SAMI-3.5 as well as data storage are done in real time guaranteeing data safety and quality. After completion of particular assays, luminescence data were exported with a specific export tool (LIMS-Excel-Export) and calculated in EXCEL. Calculations of scatterplots were made with Spotfire Decision Site 9.0.
Automated cell viability assay
293 human embryonic kidney (HEK) cells were cultivated in Dulbeccos’s MEM with 10% foetal calf serum, and 2 mM L-glutamine (PAA, Cölbe, Germany). For viability tests the cells were automatically seeded in 96-well microtitre plates (Greiner bio-one, Frickenhausen, Germany) with 30,000 cells/180 µL complete medium with a Biomek 2000 Laboratory Automation Workstation (Beckman Coulter). The compounds were added to a final concentration of 10 µM and the cells were incubated overnight at 37°C and 5% CO2 in a humidified atmosphere. The viability of the cells was determined with the EZ4U-assay (Biomedica, Vienna, Austria). For that purpose 20 µL of substrate per well was added and the plates were incubated for 3 h at 37°C and 5% CO2. Absorption was measured in a Fluostar plate reader (BMG Labtech, Offenburg, Germany) at 450 nm. All assays were performed in quadruplicates.
Results and discussion
Inhibitory effects of phthalimide derivatives on CYP2C19 and CYP2C9
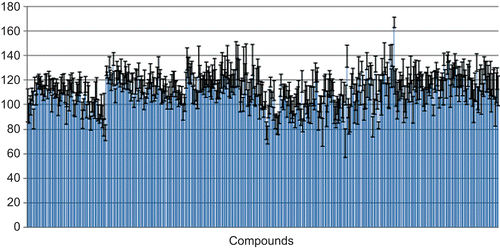
To analyse our in-house synthesised compound library of phthalimide derivatives first 480 compounds were purified and MS verified (see Materials and methods). In the first round of analysis these candidate substances were tested on their properties to affect the viability of HEK cells (see Materials and methods). As can be seen in none of the tested compounds led to a significant decrease of cell viability below 75%. This was the first indication that the synthesised substances are not generally toxic and fulfil one of the requirements for the development of novel pharmacological and agrochemical phthalimide derivatives.
Figure 3. Viability of HEK cells 16 h after treatment with phthalimide derivatives. Values are given in percentage viability compared to untreated control cells (± SE).
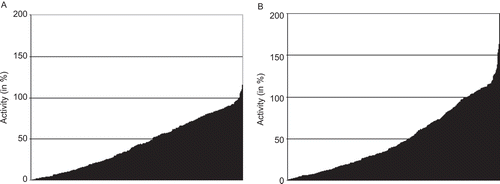
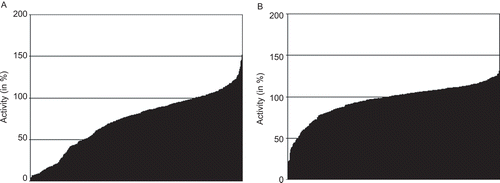
As a next step of analysis we were interested if our library of phthalimide derivatives has any influence on CYP activity. To answer this question all 480 compounds were tested in inhibition assays against the CYP isoenzyme CYP2C19 (see Materials and methods), which has been shown to have a major impact on the efficacy of thalidomide. The results of these analyses are displayed in . As it can be seen in this figure, 277 out of 480 (57.7%) tested compounds decrease the activity of CYP2C19 by more than 50%. To verify the inhibitory effect of the phthalimide derivatives inhibition assays were performed on the closely related CYP isoform CYP2C9. Also in this case more than half of the substances (274 out of 480 tested compounds; 57.1%) led to an inhibition of more than 50% (). Considering the inhibitory effects of phthalimides on two similar CYP enzymes the question arose if in both cases the same compounds caused the inhibition. A scatterplot comparing these enzyme activities after treatment with all 480 phthalimide derivatives shows that this is mostly the case (). The vast majority of the data points accumulate around a regression line with similar activity values. Only some outliers are found at the ends of both axes, which may point to specific inhibitors of either CYP2C9 or CYP2C19.
Figure 4. Inhibition of CYP2C19 and CYP2C9 by 480 phthalimide derivatives (A). Activity of CYP2C19 after treatment with 480 phthalimide derivatives compared to (B) the activity of CYP2C9 after treatment with the same compounds. The substances in the ordinate of both graphics are ordered in ascending activity.
It has been shown previously that particular phthalimide derivatives also affect the activities of different CYPs[24]. Phthalimide forms the basic structure of a number of insecticides (e.g. tetramethrin, dialifos or fosmet), fungicides (e.g. captan, thiochlorfenphim or ditalimfos) and NSAIDs (e.g. indoprofen or chlorthalidone), as shown in . For captan it has been shown previously that it leads to loss of CYP activity in rat liver [Citation25]. Later an impact of this fungicide on the activity of various CYPs was confirmed in mouse liver microsomes [Citation24]. Incubation with captafol caused the same effects on different CYPs in the liver of rats [Citation26, Citation27]. Also indobufen was found to inhibit CYPs in experiments with rats, but only to toxicological tolerable extents [Citation28].
CYP2C9 and CYP2C19 are specifically inhibited by phthalimide derivatives
The described observations opened the question, whether the tested cytochrome proteins are generally affected by any substances at the given concentration or whether their inhibition is confined to certain substance classes such as the phthalimides. To verify this aspect a subset of our in-house compound library consisting of 1200 randomly chosen compounds was used for inhibition assays. Among these compounds there were substances as different as GSK3ß-inhibitors, hymendialsine derivatives or inhibitors of transporters and enzymes. Their inhibitory potential on the activity of CYP2C9 was, like the phthalimide derivatives before, also measured in a final concentration of 10 µM. As it can be seen in a much smaller portion of the tested compounds led to an activity decrease compared to the tested phthalimides (). Just about 10% (124 out of 1200) of the compounds decrease enzymatic activity of CYP2C9 by more than 50%.
Figure 5. Scatter-plot of CYP2C9 and CYP2C19 activities after treatment with phthalimide derivatives.
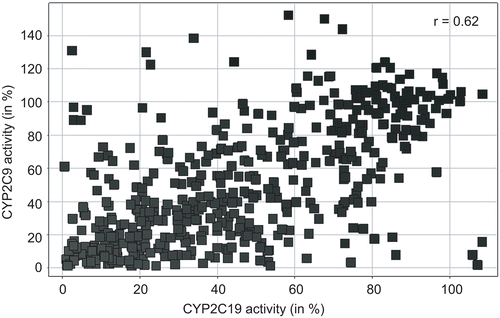
Figure 6. Activity of CYP2C9 after treatment with 1200 randomly chosen substances. The substances in the ordinate of both graphics are ordered in ascending activity.

But the inhibition of CYP2C9 and CYP2C19 by the majority of phthalimide derivatives could also be caused as a general reaction of all CYPs against this substance class. To answer this question two additional CYPs (CYP2D6 and CYP3A4) were tested with our phthalimide library. With CYP3A4 also a member of a different subclass was chosen. The results are displayed in and where it can be seen that, in contrast to the experiments with CYP2C9/19, apparently less than half of the tested compounds led to activity loss. In the case of CYP3A4 () 25.2% of the tested compounds decreased its activity more than 50%. For CYP2D6 () this number is even smaller, where only 25 of the 480 compounds fulfilled this criterion, which equals 5.2%.
Figure 7. Distribution of (A) CYP3A4 and (B) CYP2D6 activities after treatment with 480 phthalimide derivatives. The substances in the ordinate of both graphics are ordered in ascending activity.
These results indicate that the majority of the tested phthalimide derivatives specifically inhibit the isoforms CYP2C9/19, whereas other isoforms are just affected to a lesser extent. These findings are partially in concordance with recently published studies [Citation15] where inhibition of CYP2C19, but not of CYP3A4 and CYP3A5, by thalidomide was observed. But with an IC50 of 270 µM the concentration was rather high. Nevertheless, by the inhibition of hepatic CYPs such as CYP2C9/19 two problems arise: first, assuming biotransformation of phthalimides into pharmacologically active compounds by CYPs, as it was shown for thalidomide (see above), an inhibition of these enzymes would decrease the pharmacologic effects of phthalimides. Second, a decrease of CYP activity would increase the risk of drug–drug interactions, which could lead to loss of efficacy of co-administered drugs or an accumulation of them to toxic levels. In this regard the specific inhibition of particular CYPs by newly synthesised phthalimides has to be tested carefully.
Substitutions at the nitrogen atom of the phthaloyl ring have a major impact on CYP2C9/19 activity
The tested phthalimide compound library originates from multicomponent reactions where five sites of the basic molecule are derivatised with different substituents. The majority of the analysed compounds had different substituents at the positions R1, R2c and R3. With the results of the experiments described above the question arose if certain substitutions determine the power of inhibition on CYP2C9/19. To this all compounds were grouped according to the substituent at a given position. According to this procedure three positional groups with 8–14 substitutional subgroups were formed. Afterwards the proportion of inhibitors within each subgroup was calculated. For the eight substitutions at position R1, named R1-1 to R1-8, 15-94 substances were analysed. For the 10 substitutions at position R2, named R2-1 to R2-10, 23-60 substances were analysed. For the 14 substitutions at position R3, named R3-1 to R3-5, 3-7, 3-8, 3-10 to 3-14, 3-16 and 3-17, 5-62 substances were analysed. The results of these analyses are given in –.
Figure 8. Percentage of phthalimide inhibitors on CYP2C9 and CYP2C19 activity in dependence on different substituents at position (A) R1, (B) R2 and (C) R3. Positions and nomenclature of substituents are given in .
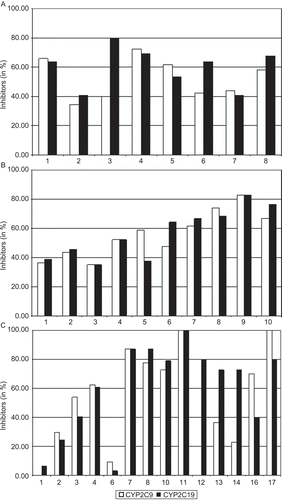
In the inhibitory impact of substitutions at position R1 is summarised. As it can be seen, a substitution at this position leads to a level of inhibitors of more than 10% over the average of 57%. There are some more substitutions with lowered inhibition, but all these effects are quite moderate. There is a similar trend for substitutions at position R2, with the difference that in most cases the effects of the substitutions are more similar on both enzymes than in the first example (). Changing the substituents at position R3 leads to a different picture. As depicted in the differences between particular groups on the enzymatic activity of CYP2C9 and CYP2C19 are much greater than seen before. In the cases of R3-1 and R3-5 less than 10% of the compounds with the given substitutions show inhibitory potential. In contrast, in other cases all tested compounds of a substitution subgroup displayed inhibition. Although just five compounds for the substitutions R3-11 and R3-12 were tested, which lowers the informative value of these substitutions, also all of the 55 compounds with the substituents R3-17 inhibited the activity of CYP2C9.
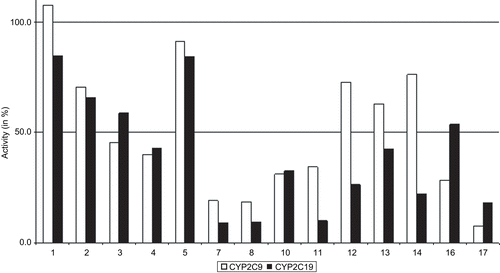
The observation that substitutions at position R3 are of specific impact on the activity of CYP2C9/19 is also supported when the median of activities at this position is considered (). The activity median over all 480 tested phthalimides lies at 40.1% and 42.6% for CYP2C9 and CYP2C19, respectively. As it can be seen in most of the substituents with either the highest or smallest fractions of inhibitors () can also be recognised in this bar chart. This holds especially true for the subgroups 3-1 and 3-5 with the highest median activities and simultaneously almost no inhibitors found among them. In contrast, subgroups 3-7, 3-8 and 3-17 revealed the lowest median of activity and the highest portion of inhibitors. But both isoforms of cytochrome proteins do not always react equally when treated with the same compound of a subgroup. Partially drastic differences between CYP2C9 and CYP2C19 can be recognised when the activity values of the subgroups 3-13, 3-14 and 3-16 are compared. This may be an indication for individual differences between CYP2C9 and CYP2C19 in respect to particular compounds as they have been observed in the scatterplot in .
Figure 9. Median of CYP2C9 and CYP2C19 activities after treatment with phthalimide derivatives in dependence on different substituents at position R3. Positions and nomenclature of substituents are given in .
A closer look at the inhibitory potential of particular derivatives () in combination with their structures () provides first signs for the explanation of the observations. Therefore the substitutions R3-1 to R3-4 are of special interest, because with just a hydrogen bound to the nitrogen at the given position (R3-1) almost no inhibition is observed ( and ), whereas with successive chain length also the inhibition of the compounds grows. There are still many questions remaining, up to now there is no explanation why some of the substitutions affect enzymatic activity of highly related CYP isoforms so differently like R3-14 and 3-16. In previous docking simulations it has been shown that different rings of the thalidomide molecule interact with the haem in CYP3A4 and CYP3A5 and affect these related isoforms differently [Citation15]. Further docking studies, such as have been done before for particular phthalimides and cytochrome proteins [Citation29] are necessary to achieve sufficient answers on most questions concerning the inhibitory potential of phthalimides on CYP activity.
Conclusion
The applied method to synthesise phthalimide derivatives has been shown to be highly efficient due to its high degree of automation in synthesis and purification. The presented results also revealed that the synthesised substances can be utilised for primary pharmacological tests. In such tests the inhibitory potential of a sub-library, consisting of 480 phthalimide compounds, on cytochrome protein activity could be stated on the closely related isoforms CYP2C9 and CYP2C19, whereas other CYP enzymes were not affected to a considerable extent. Furthermore, the enhanced inhibition of CYP2C9/19 seems to depend on the substituent located at the nitrogen atom within the isindole ring.
The influence on drug-metabolising enzymes has a steadily growing impact on the development of novel test substances and lead structures. Therefore the presented results might have a certain impact on the development of further phthalimides, especially of thalidomide analogues, whose inhibitory capacity on different CYP enzymes should be investigated in the future. Furthermore, docking studies should reveal details about the interaction of the different derivatives with the analysed CYP isoforms. Such studies may on the one hand side resolve the different inhibitory activities of particular substitutions and on the other provide guidance for future synthesis of novel pharmacologically and agronomically relevant phthalimide derivatives.
Declaration of interest
The authors wish to thank the German Federal Ministry of Education and Research (BMBF) for financial support of the studies.
References
- Silverman WA. The schizophrenic career of a ‘monster drug’. Pediatrics 2002;110:404–406.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–322.
- Lepper ER, Smith NF, Cox MC, Scripture CD, Figg WD. Thalidomide metabolism and hydrolysis: Mechanisms and implications. Curr Drug Metabol 2006;7:677–685.
- D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA 1994;91:4082–4085.
- Moreira AL, Sampaio EP, Zmuidinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor α by enhancing mRNA degradation. J Exp Med 1993;177:1675–1680.
- Braun AG, Weinreb SL. Teratogen metabolism: Spontaneous decay products of thalidomide and thalidomide analogues are not bioactivated by liver microsomes. Teratog Carcinog Mutagen 1985;5:149–158.
- Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol 2007;39:1489–1499.
- Ando Y, Fuse E, Figg WD. Thalidomide metabolism by the CYP2C subfamily. Clin Cancer Res 2002;8:1964–1973.
- Ando Y, Price DK, Dahut WL, Cox MC, Reed E, Figg WD. Pharmacogenetic associations of CYP2C19 genotype with in vivo metabolisms and pharmacological effects of thalidomide. Cancer Biol Therapy 2002;1:669–673.
- Braun AG, Harding FA, Weinreb SL. Teratogen metabolism: Thalidomide activation is mediated by cytochrome P-450. Toxicol Appl Pharmacol 1986;82:175–179.
- Teo SK, Sabourin PJ, Kook KA, Thomas SD. Metabolism of thalidomide in human microsomes, cloned human cytochrome P-450 isozymes, and Hansen’s disease patients. J Biochem Mol Toxicol 2000;14:140–147.
- Li Y, Hou J, Jiang H, Wang D, Fu W, Yuan Z, Chen Y, Zhou L. Polymorphisms of CYP2C19 gene are associated with the efficacy of thalidomide-based regimens in multiple myeloma cells. Haematologica 2007; 92:1246–1249.
- Li YH, Hou J, Jiang H, Huang HM. Antiangiogenic activity of thalidomide in vitro mediated by cytochrome CYP2C19. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2009;17:102–106.
- Li YH, Hou J, Jiang H, Huang HM, Zhu R. Effect of cytochrome CYP2C19 on the antimyeloma activity of thalidomide in vitro. Zhonghua Xue Ye Xue Za Zhi 2008;29:654–657.
- Okada Y, Murayama N, Yanagida C, Shimizu M, Guengerich FP, Yamazaki H. Drug interactions of thalidomide with midazolam and cyclosporine A: Heterotropic cooperativity of human cytochrome P450 3A5. Drug Metabol Dispos 2009;37:18–23.
- Aragon-Ching JB, Li H, Gardner ER, Figg WD. Thalidomide analogues as anticancer drugs. Recent Pat Anticancer Drug Discov 2007;2:167–174.
- Kumar G, Lau H, Laskin O. Lanelidomide: in vitro evaluation of the metabolism and assessment of P450 cytochrome inhibition and induction. Cancer Chemother Pharmacol 2009;63:1171–1175.
- Neumann H, von Wangelin AJ, Gördes D, Spannenberg A, Beller M. A new multicomponent coupling of aldehydes, amides, and dienophiles: Atom efficient one-pot synthesis of highly substituted cyclohexenes and cyclohexadienes. J Am Chem Soc 2001;123:8398–8399.
- von Wangelin A, Neumann H, Gördes D, Klaus S, Strübing D, Beller M. Multicomponent coupling reactions for organic synthesis: chemoselective reactions with amide-aldehyde mixtures. Chem Eur J 2003;9:4286–4294.
- Klaus S, Hübner S, Neumann H, Strübing D, von Wangelin A, Gördes D, Beller M. Second generation protocol for multicomponent coupling reactions of aldehydes, amides and dienophiles. Adv Synth Catal 2004;346:970–978.
- Wendler C, Krüger-Sundhaus T, Beller M, Gördes D, Thurow K. Single-pot solution phase synthesis optimization using fully automated systems for combinatorial screening. J Assoc Lab Automation 2005;10:237–241.
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73.
- Thurow K, Göde B, Dingerdissen U, Stoll N. Laboratory information management systems for life science applications. Organic Process Research and Development 2004;8:970–982.
- Paolini M, Barillari J, Trespidi S, Valgimigli L, Pedulli GF, Cartelli-Forti G. Captan impairs CYP-catalyzed drug metabolism in the mouse. Chemico-Biol Interact 1999;123:149–170.
- Peebles A, Dalvi RR. Toxicology studies of N-trichlormethylthio-4-cyclohexene-1,2-dicarboximide (Captan): Its metabolism by rat liver drug metabolizing enzyme system. Toxicology 1978;9:341–351.
- Dalvi RR, Mutinga ML. Comparative studies of the effects on liver and liver microsomal drug-metabolizing enzyme system by the fungicides captan, captafol and folpet in rats. Pharmacol Toxicol 1990;66:231–233.
- Rahden-Staron I, Czeczot H, Szumilo M. Induction of rat liver cytochrome P450 isoenzymes CYP1A and CYP2B by different fungicides, nitrofurans and quercetin. Mut Res 2001;498:57–66.
- Thomas H, Strolin-Benedetti M, Dostert P, Oesch F. The effect of indobufen on the activities of selected rat liver phase I and phase II drug metabolizing enzymes, peroxisomal beta-oxidation and hepatic glutathione status. J Pharm Pharmacol 1994;46:833–837.
- Cruciani G, Carosati E, De Boeck B, Ethirajulu K, Mackie C, Howe T, Vianello R. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J Med Chem 2006;48:6970–6979.